Biomolecular Component Analysis of Phospholipids Composition in Live HeLa Cells



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
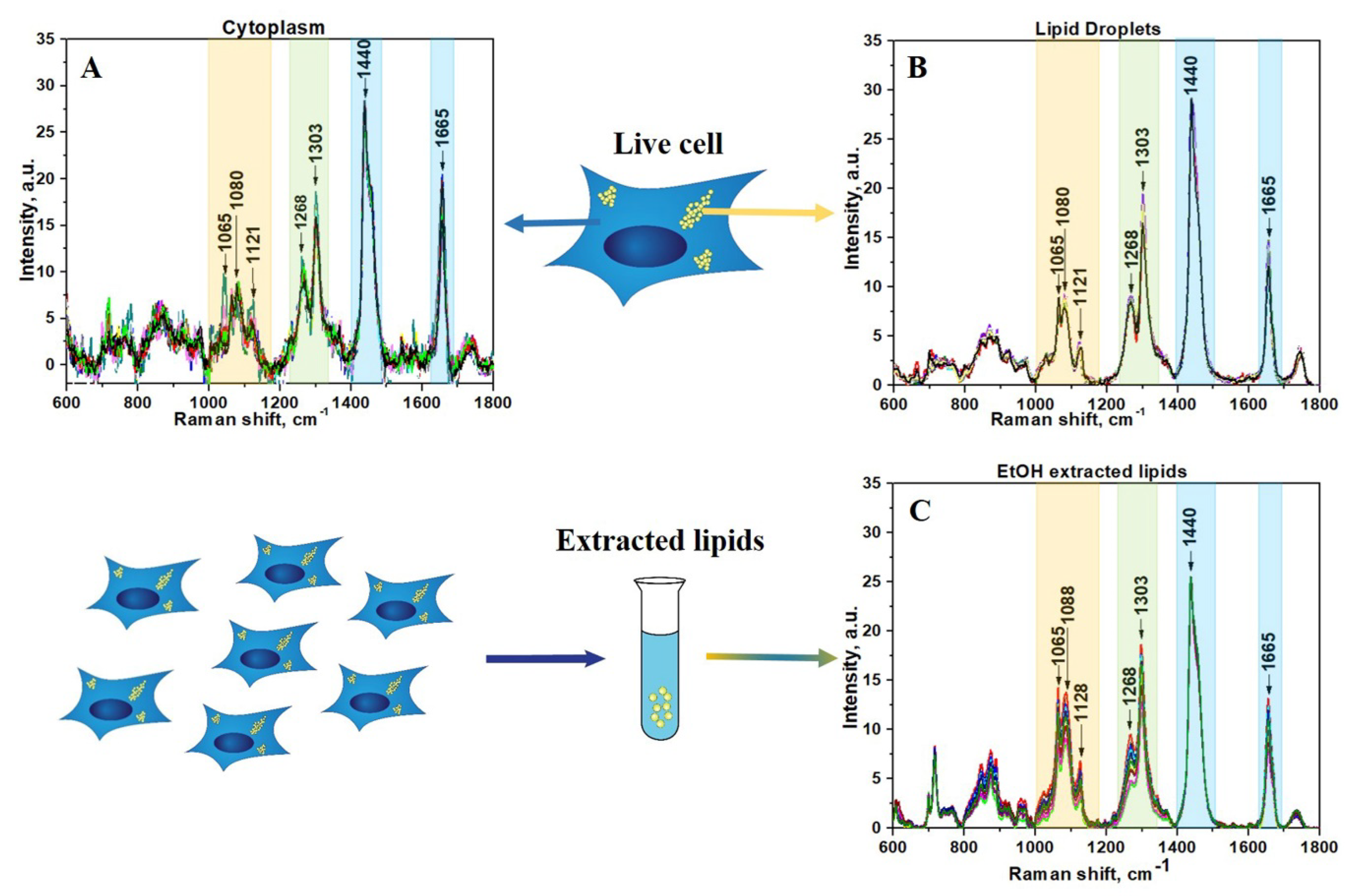
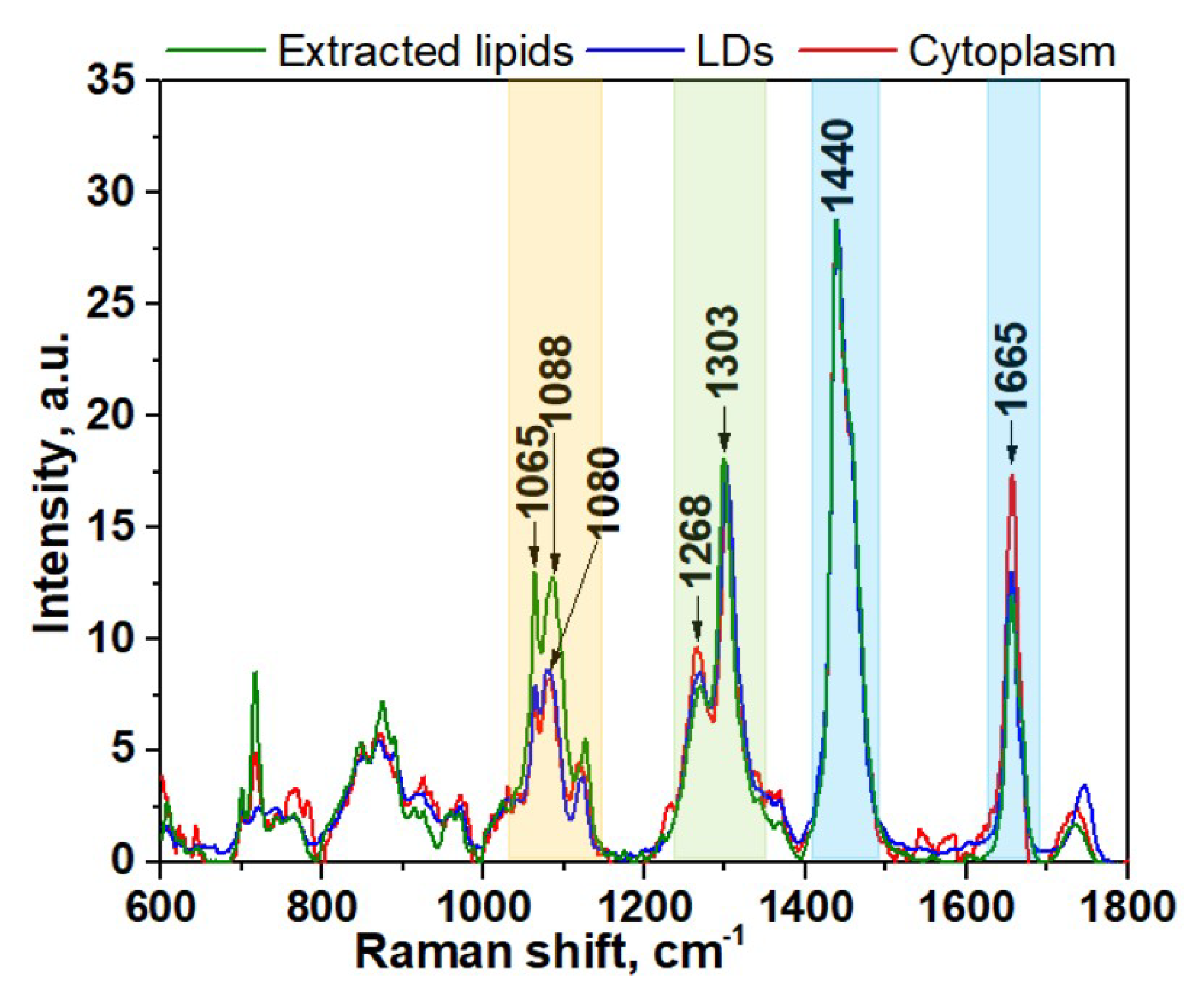
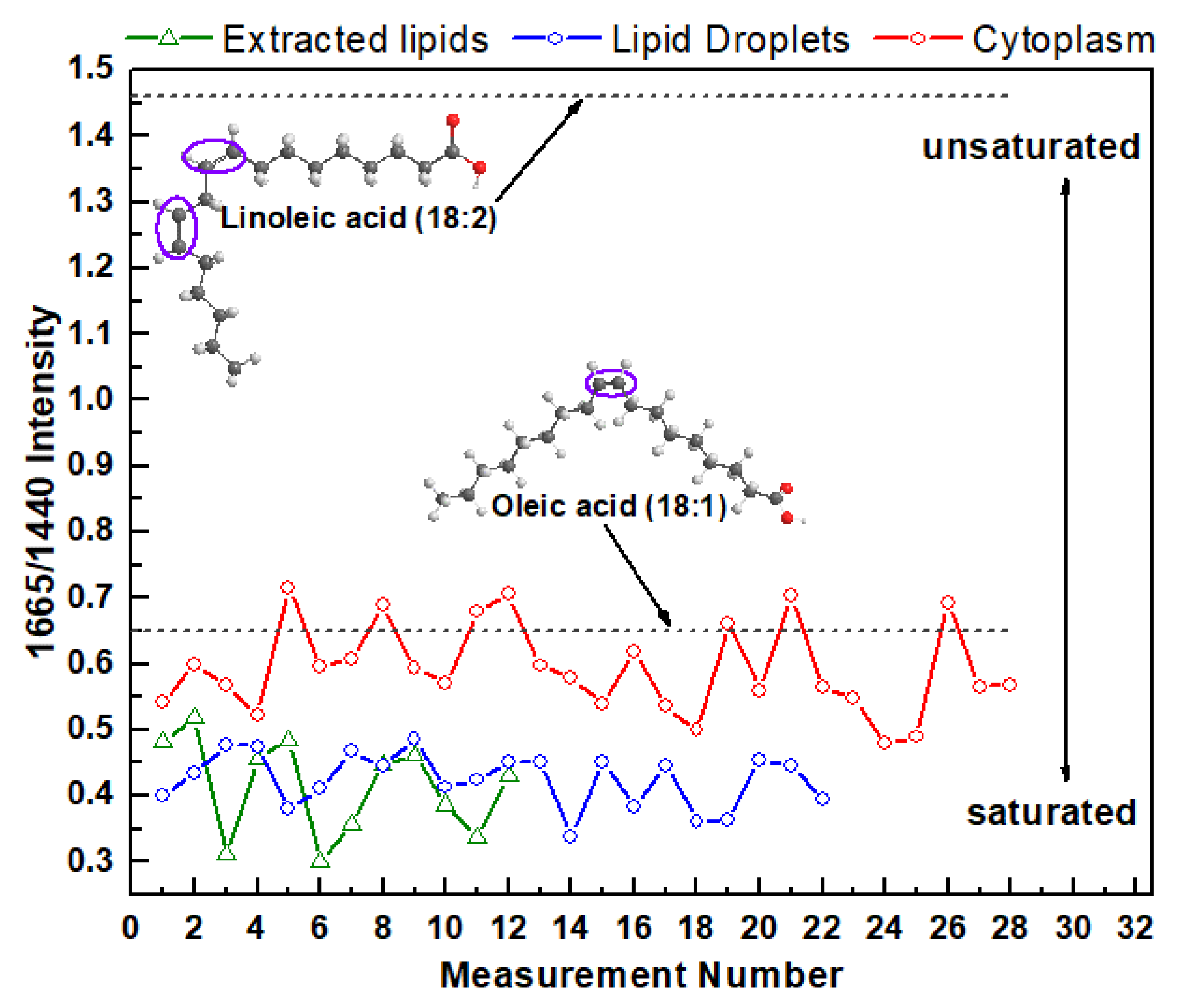
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta 2017, 1862, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant role for lipid droplets in a stem cell niche of drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Lizardo, D.Y.; Parisi, L.R.; Li, N.; Atilla-Gokcumen, G.E. Noncanonical roles of lipids in different cellular fates. Biochemistry 2018, 57, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, R.P.; Laye, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Morishita, R. The roles of lipid and glucose metabolism in modulation of beta-amyloid, tau, and neurodegeneration in the pathogenesis of alzheimer disease. Front. Aging Neurosci. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Aufschnaiter, A.; Kohler, V.; Diessl, J.; Peselj, C.; Carmona-Gutierrez, D.; Keller, W.; Buttner, S. Mitochondrial lipids in neurodegeneration. Cell Tissue Res. 2017, 367, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Peirce, V.; Carobbio, S.; Vidal-Puig, A. The different shades of fat. Nature 2014, 510, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, J.; Kumar, R.; Kuzmin, A.N.; Pliss, A.; Yadav, N.; Balachandar, S.; Wang, J.; Attwood, K.; Prasad, P.N.; Chandra, D. Lipid quantification by raman microspectroscopy as a potential biomarker in prostate cancer. Cancer Lett. 2017, 397, 52–60. [Google Scholar] [CrossRef]
- Deep, G.; Schlaepfer, I.R. Aberrant lipid metabolism promotes prostate cancer: Role in cell survival under hypoxia and extracellular vesicles biogenesis. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniels, V.W.; Machiels, J.; et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [PubMed]
- Jarc, E.; Kump, A.; Malavasic, P.; Eichmann, T.O.; Zimmermann, R.; Petan, T. Lipid droplets induced by secreted phospholipase a2 and unsaturated fatty acids protect breast cancer cells from nutrient and lipotoxic stress. Biochim. Biophys. Acta 2018, 1863, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wei, Y.; Pagliassotti, M.J. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 2006, 147, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Gentile, C.L.; Pagliassotti, M.J. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol. Cell. Biochem. 2009, 331, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, H. Fatty acid metabolism and prospects for targeted therapy of cancer. Eur. J. Lipid Sci. Technol. 2017, 119. [Google Scholar] [CrossRef]
- Liu, Q.; Luo, Q.; Halim, A.; Song, G. Targeting lipid metabolism of cancer cells: A promising therapeutic strategy for cancer. Cancer Lett. 2017, 401, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Arisawa, K.; Mitsudome, H.; Yoshida, K.; Sugimoto, S.; Ishikawa, T.; Fujiwara, Y.; Ichi, I. Saturated fatty acid in the phospholipid monolayer contributes to the formation of large lipid droplets. Biochem. Biophys. Res. Commun. 2016, 480, 641–647. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ros induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef]
- Dowhan, W. Molecular basis for membrane phospholipid diversity: Why are there so many lipids? Annu. Rev. Biochem. 1997, 66, 199–232. [Google Scholar] [CrossRef]
- Le, T.T.; Yue, S.; Cheng, J.-X. Shedding new light on lipid biology with coherent anti-stokes raman scattering microscopy. J. Lipid Res. 2010, 51, 3091–3102. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.; Smith, E.A. Raman imaging in cell membranes, lipid-rich organelles, and lipid bilayers. Annu. Rev. Anal. Chem. 2017, 10, 271–291. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Armando, A.M.; Dennis, E.A. High sensitivity quantitative lipidomics analysis of fatty acids in biological samples by gas chromatography-mass spectrometry. Biochim. Biophys. Acta 2011, 1811, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Park, H.M.; Shon, J.C.; Lee, M.Y.; Liu, K.H.; Kim, J.K.; Lee, S.J.; Lee, C.H. Mass spectrometry-based metabolite profiling in the mouse liver following exposure to ultraviolet b radiation. PLoS ONE 2014, 9, e109479. [Google Scholar] [CrossRef] [PubMed]
- Harkewicz, R.; Dennis, E.A. Applications of mass spectrometry to lipids and membranes. Annu. Rev. Biochem. 2011, 80, 301–325. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Fiehn, O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. TrAC Trends Anal. Chem. 2014, 61, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.C.; Gaskell, S.J. New applications of mass spectrometry in lipid analysis. J. Biol. Chem. 2011, 286, 25427–25433. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Lee, H.; Kim, S.H.; Jin, H.; Bae, J.; Choi, H.K. Discovery of potential biomarkers in human melanoma cells with different metastatic potential by metabolic and lipidomic profiling. Sci. Rep. 2017, 7, 8864. [Google Scholar] [CrossRef] [PubMed]
- Levchenko, S.M.; Kuzmin, A.N.; Pliss, A.; Qu, J.; Prasad, P.N. Macromolecular profiling of organelles in normal diploid and cancer cells. Anal. Chem. 2017, 89, 10985–10990. [Google Scholar] [CrossRef]
- Kuzmin, A.N.; Levchenko, S.M.; Pliss, A.; Qu, J.; Prasad, P.N. Molecular profiling of single organelles for quantitative analysis of cellular heterogeneity. Sci. Rep. 2017, 7, 6512. [Google Scholar] [CrossRef]
- Kuzmin, A.N.; Pliss, A.; Kachynski, A.V. Biomolecular component analysis of cultured cell nucleoli by raman microspectrometry. J. Raman Spectrosc. 2013, 44, 198–204. [Google Scholar] [CrossRef]
- Kuzmin, A.N.; Pliss, A.; Prasad, P.N. Changes in biomolecular profile in a single nucleolus during cell fixation. Anal. Chem. 2014, 86, 10909–10916. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, A.N.; Pliss, A.; Prasad, P.N. Ramanomics: New omics disciplines using micro raman spectrometry with biomolecular component analysis for molecular profiling of biological structures. Biosensors 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- De Gelder, J.; De Gussem, K.; Vandenabeele, P.; Moens, L. Reference database of raman spectra of biological molecules. J. Raman Spectrosc. 2007, 38, 1133–1147. [Google Scholar] [CrossRef]
- Wu, H.; Volponi, J.V.; Oliver, A.E.; Parikh, A.N.; Simmons, B.A.; Singh, S. In vivo lipidomics using single-cell raman spectroscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 3809–3814. [Google Scholar] [CrossRef] [PubMed]
- Schie, I.W.; Huser, T. Methods and applications of raman microspectroscopy to single-cell analysis. Appl. Spectrosc. 2013, 67, 813–828. [Google Scholar] [CrossRef]
- Zinin, P.V.; Misra, A.; Kamemoto, L.; Yu, Q.G.; Hu, N.J.; Sharma, S.K. Visible, near-infrared, and ultraviolet laser-excited raman spectroscopy of the monocytes/macrophages (U937) cells. J. Raman Spectrosc. 2010, 41, 268–274. [Google Scholar] [CrossRef]
- Munchberg, U.; Wagner, L.; Rohrer, C.; Voigt, K.; Rosch, P.; Jahreis, G.; Popp, J. Quantitative assessment of the degree of lipid unsaturation in intact mortierella by raman microspectroscopy. Anal. Bioanal. Chem. 2015, 407, 3303–3311. [Google Scholar] [CrossRef]
- Le, T.T.; Duren, H.M.; Slipchenko, M.N.; Hu, C.D.; Cheng, J.X. Label-free quantitative analysis of lipid metabolism in living caenorhabditis elegans. J. Lipid Res. 2010, 51, 672–677. [Google Scholar] [CrossRef]
- Samek, O.; Jonas, A.; Pilat, Z.; Zemanek, P.; Nedbal, L.; Triska, J.; Kotas, P.; Trtilek, M. Raman microspectroscopy of individual algal cells: Sensing unsaturation of storage lipids in vivo. Sensors 2010, 10, 8635–8651. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levchenko, S.M.; Qu, J. Biomolecular Component Analysis of Phospholipids Composition in Live HeLa Cells. Biosensors 2018, 8, 123. https://doi.org/10.3390/bios8040123
Levchenko SM, Qu J. Biomolecular Component Analysis of Phospholipids Composition in Live HeLa Cells. Biosensors. 2018; 8(4):123. https://doi.org/10.3390/bios8040123
Chicago/Turabian StyleLevchenko, Svitlana M., and Junle Qu. 2018. "Biomolecular Component Analysis of Phospholipids Composition in Live HeLa Cells" Biosensors 8, no. 4: 123. https://doi.org/10.3390/bios8040123
APA StyleLevchenko, S. M., & Qu, J. (2018). Biomolecular Component Analysis of Phospholipids Composition in Live HeLa Cells. Biosensors, 8(4), 123. https://doi.org/10.3390/bios8040123