A New Approach for Interferent-Free Amperometric Biosensor Production Based on All-Electrochemically Assisted Procedures

 ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Apparatus
2.3. Polymer Electrosynthesis Deposition Procedure
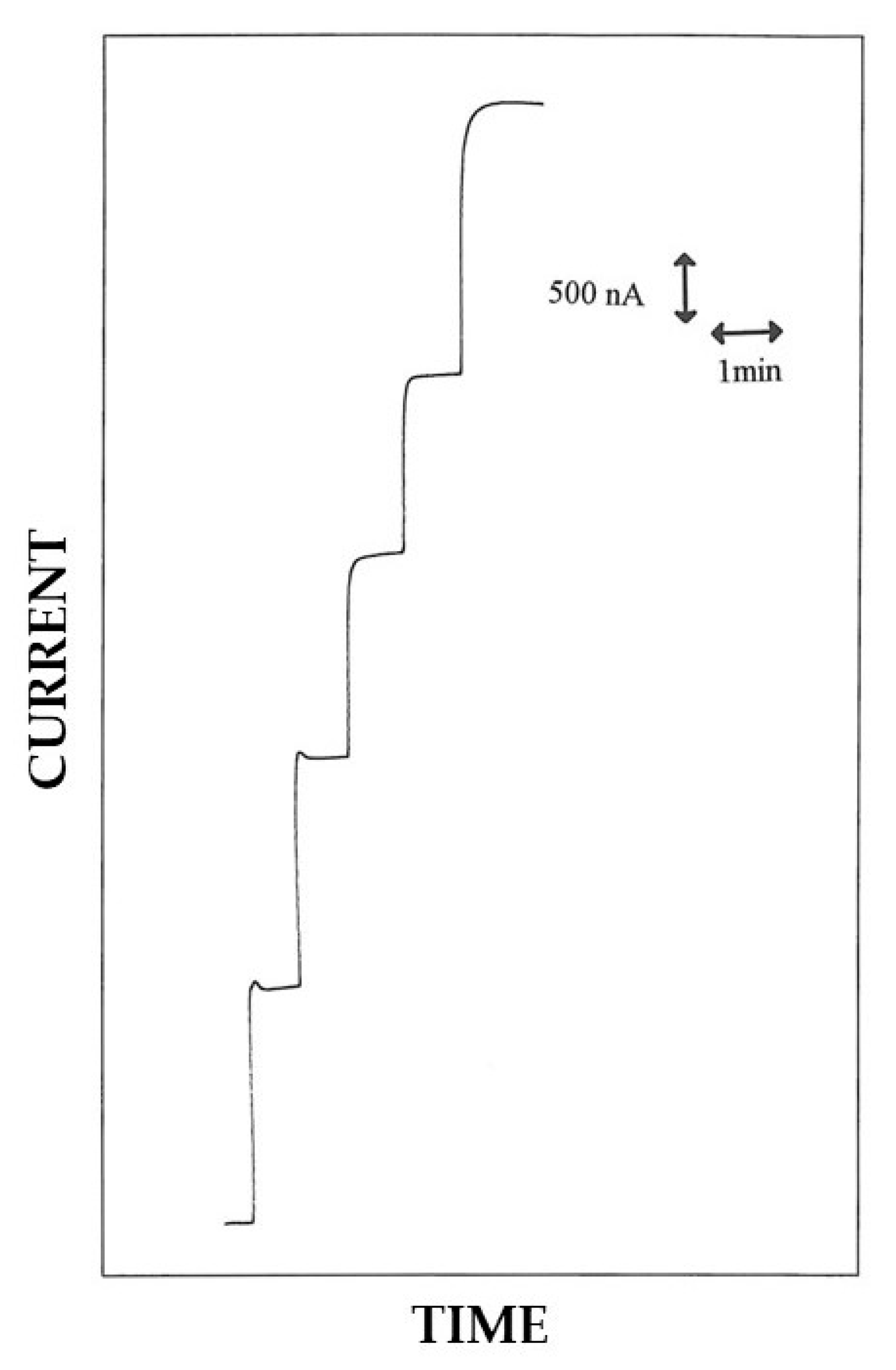
2.4. EPD Procedure for Enzyme Immobilization
2.5. Biosensor Fabrication and Characterization
3. Results and Discussion
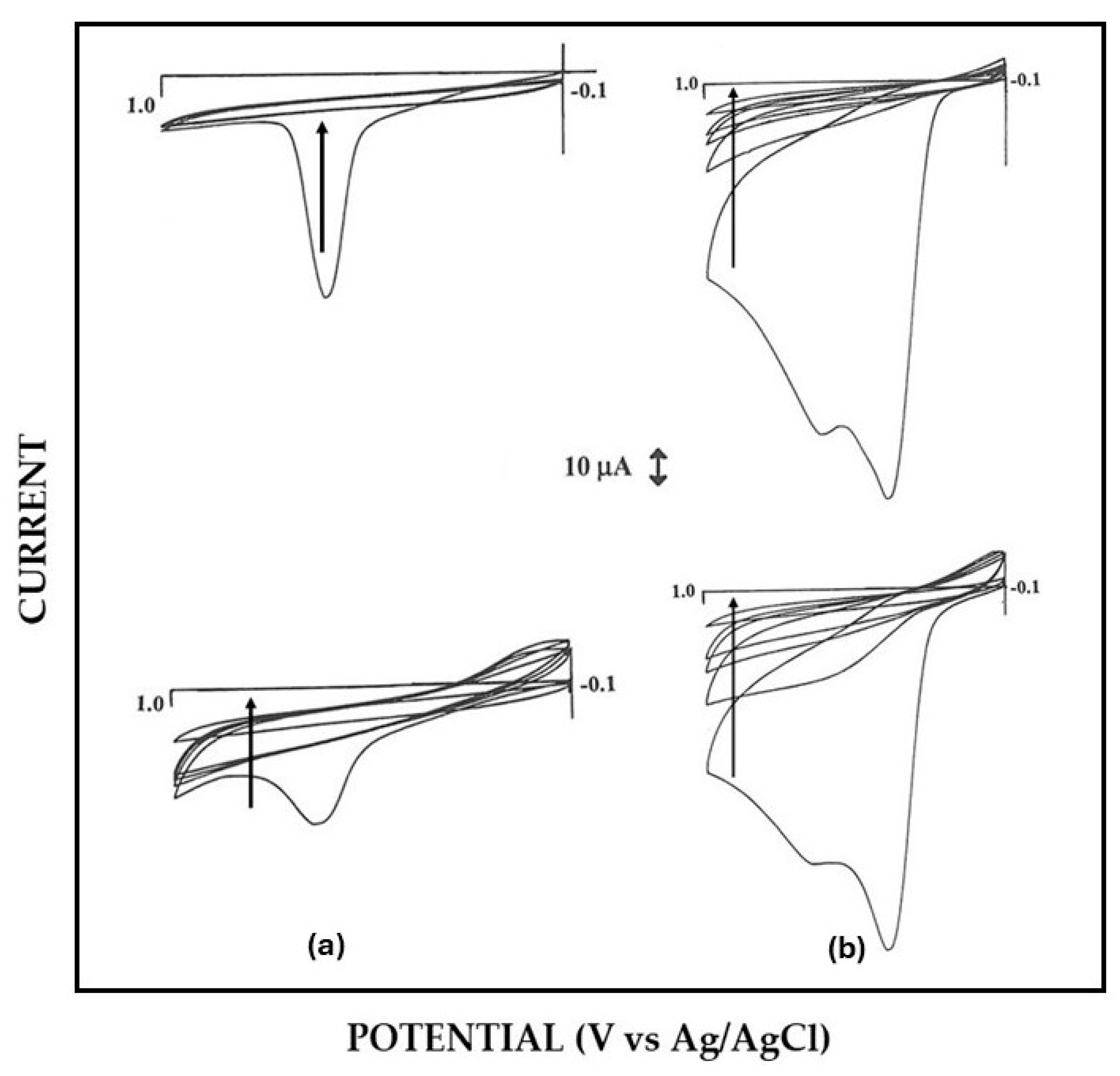
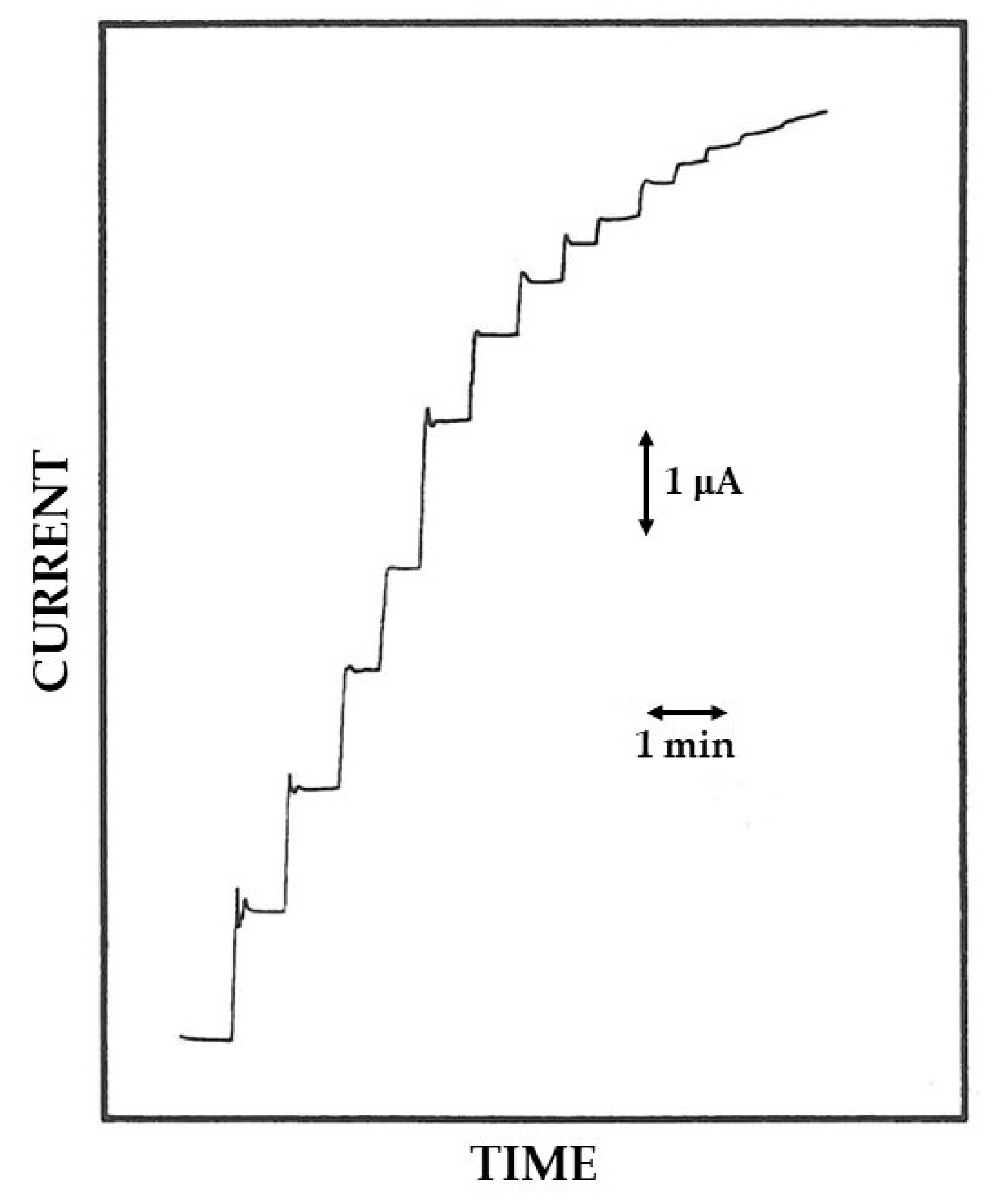
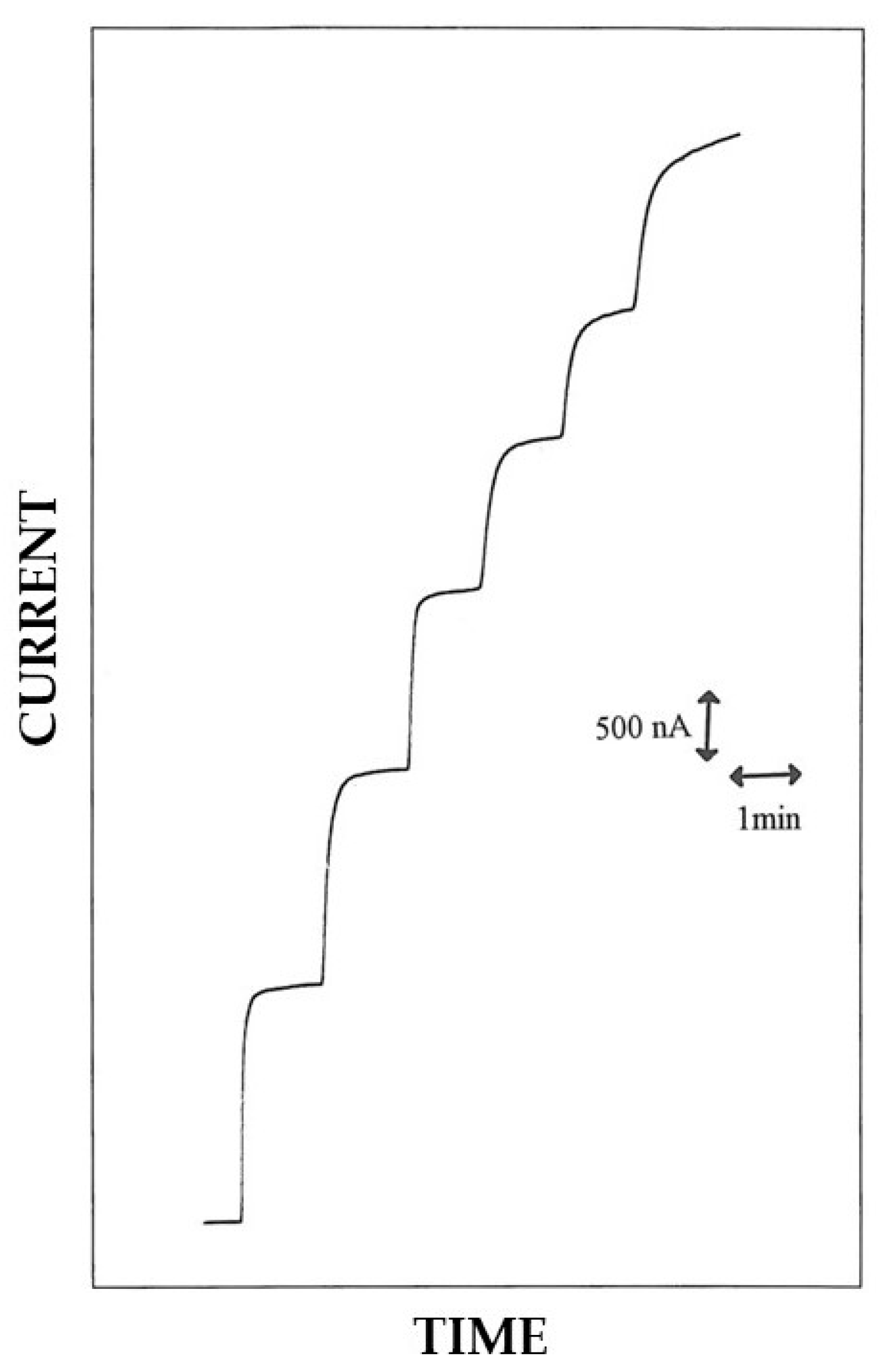
3.1. EPD and in Situ Co-Crosslinking of Glucose Oxidase and Bovine Serum Albumin onto the Electrode Surface
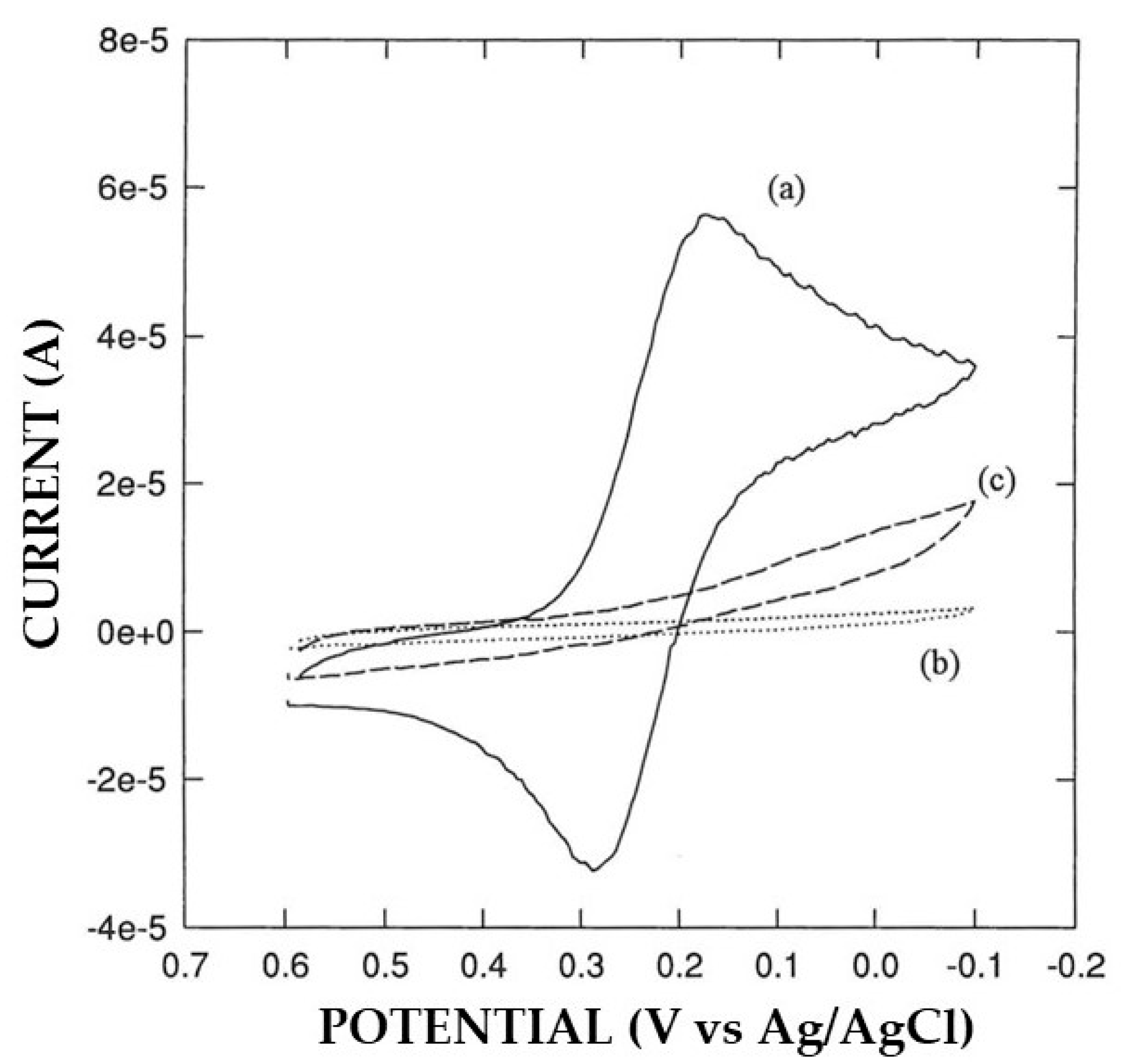
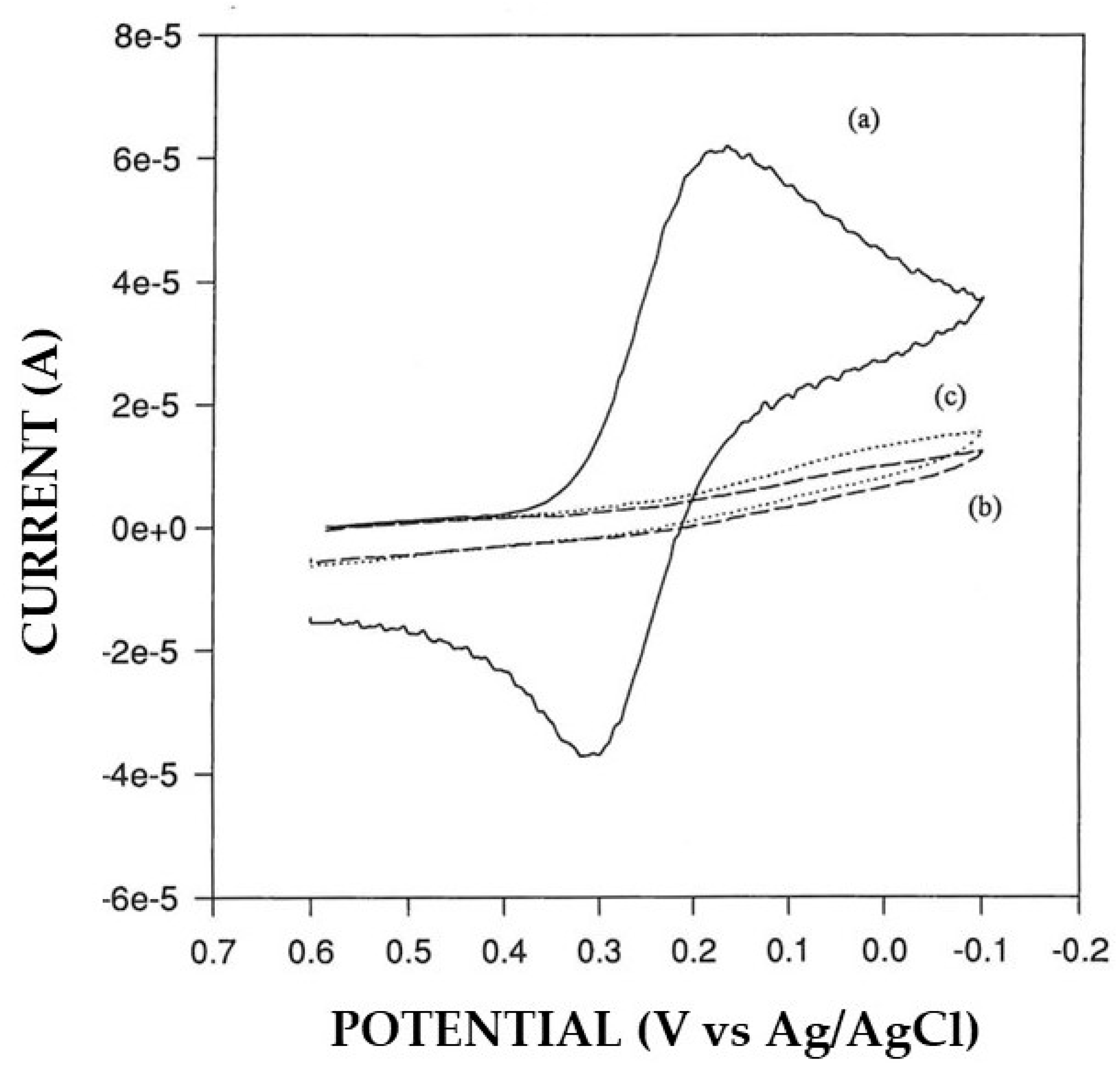
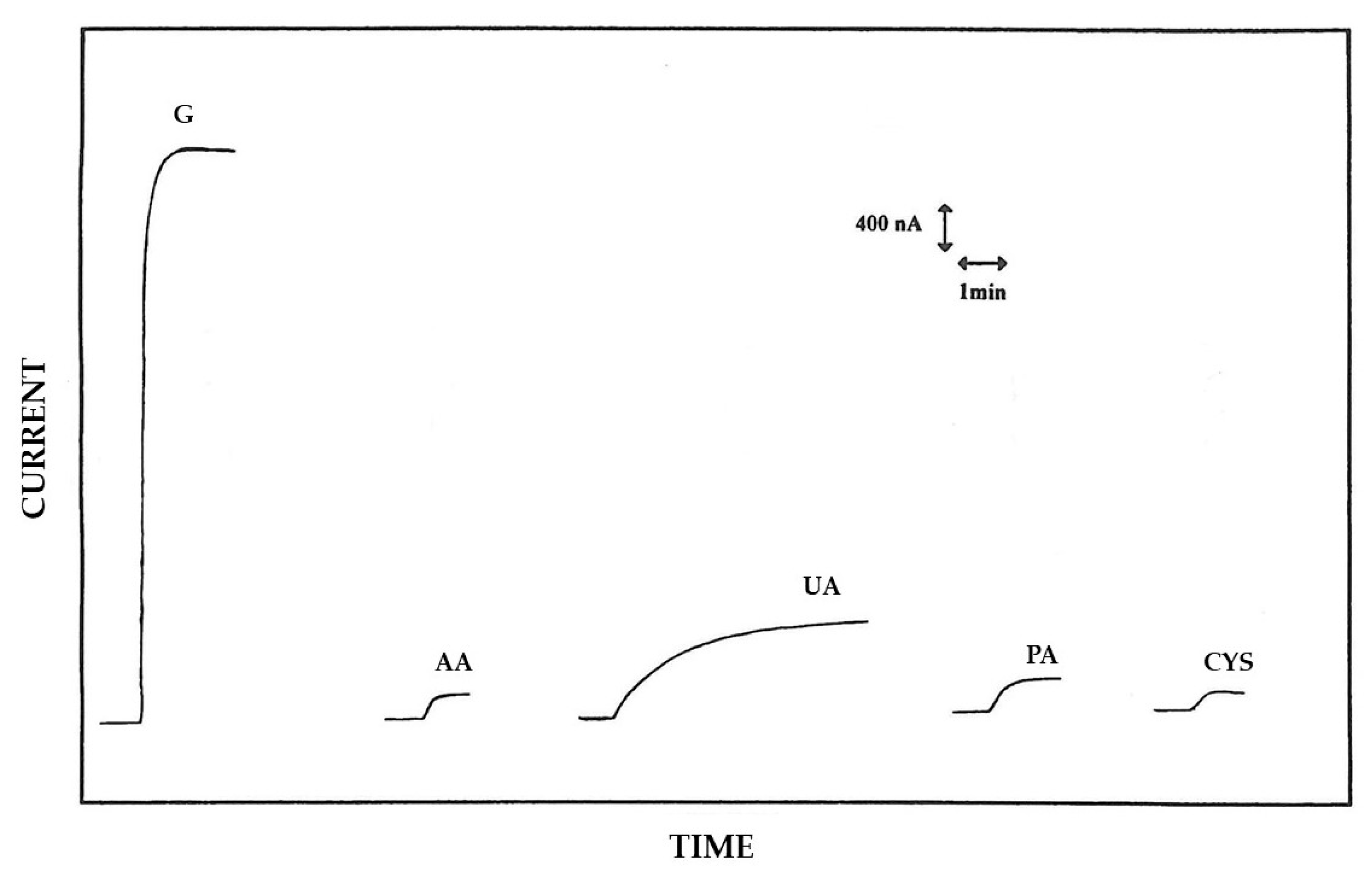
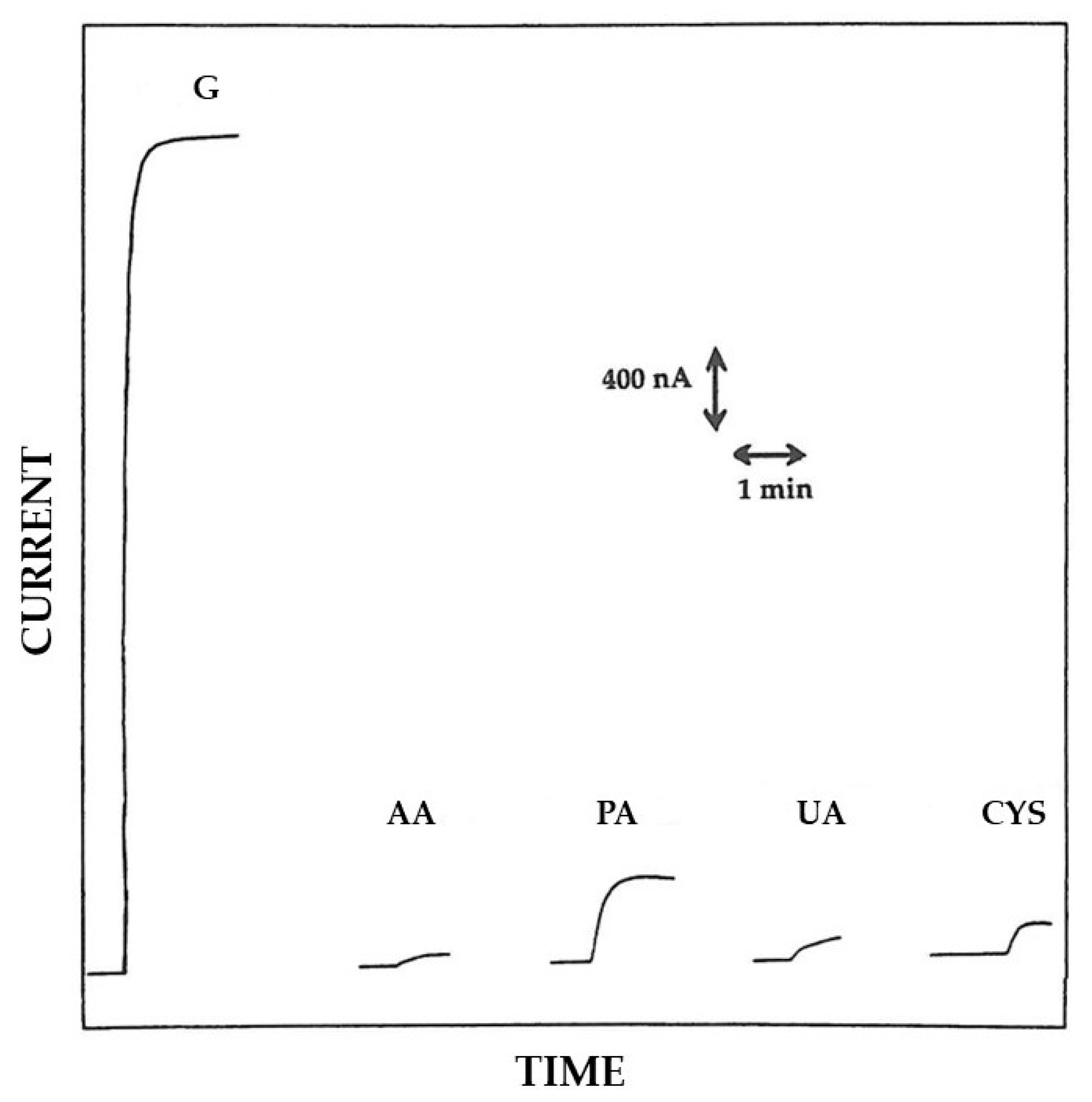
3.2. Permselective Behavior of the Enzyme Layer as Deposited by EPD
3.3. Study of the Coupling of the Electrosynthesis of Permselective, Non-Conducting Polymers with Enzyme Immobilization by EPD
3.4. Analytical Characterization of Pt/Polymer/GOD Biosensors
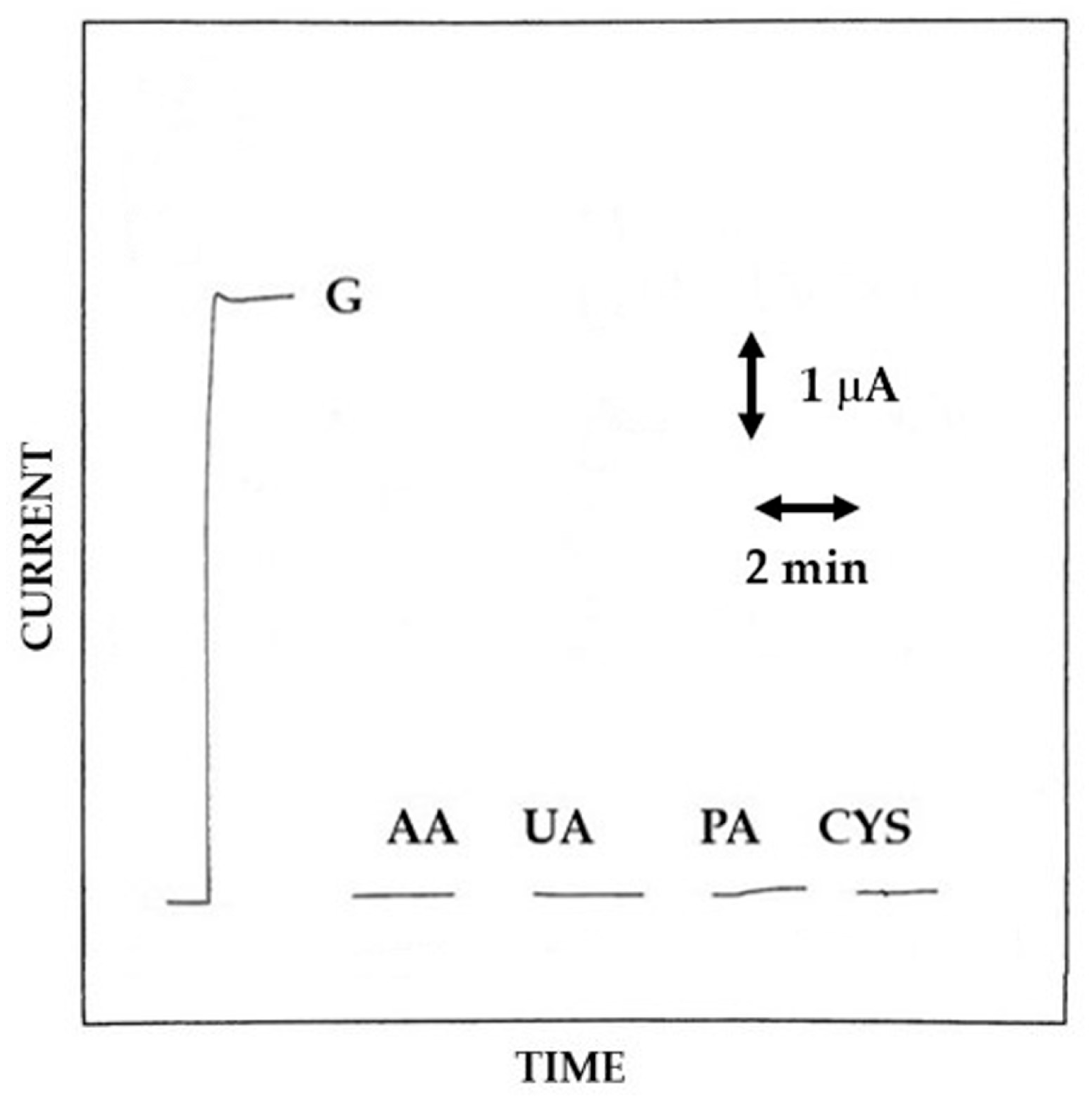
3.5. Analytical Characterization of Pt/GOD/Polymer Biosensors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Owen, V.M.; Turner, A.P.F. Biosensors: A revolution in clinical analysis? Endeavour 1987, 11, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.P.F. Current trends in biosensor research and development. Sens. Actuators 1989, 17, 433–450. [Google Scholar] [CrossRef]
- Clark, L.C.; Lyons, C. ELECTRODE SYSTEMS FOR CONTINUOUS MONITORING IN CARDIOVASCULAR SURGERY. Ann. N. Y. Acad. Sci. 1962, 102, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P. Enzyme-based amperometric biosensors: 60 years later … Quo Vadis? Anal. Chim. Acta 2022, 1234, 340517. [Google Scholar] [CrossRef] [PubMed]
- Kalita, N.; Gogoi, S.; Minteer, S.D.; Goswami, P. Advances in Bioelectrode Design for Developing Electrochemical Biosensors. ACS Meas. Sci. Au 2023, 3, 404–433. [Google Scholar] [CrossRef] [PubMed]
- Cass, A.E.G. (Ed.) Biosensors. A Practical Approach; IRL Press at Oxford University Press: Oxford, UK, 1990; ISBN 9780199630479. [Google Scholar]
- Bartlett, P.N.; Cooper, J.M. A review of the immobilization of enzymes in electropolymerized films. J. Electroanal. Chem. 1993, 362, 1–12. [Google Scholar] [CrossRef]
- Malitesta, C.; Palmisano, F.; Torsi, L.; Zambonin, P.G. Glucose fast-response amperometric sensor based on glucose oxidase immobilized in an electropolymerized poly(o-phenylenediamine) film. Anal. Chem. 1990, 62, 2735–2740. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, P.N.; Tebbutt, P.; Tyrrell, C.H. Electrochemical immobilization of enzymes. 3. Immobilization of glucose oxidase in thin films of electrochemically polymerized phenols. Anal. Chem. 1992, 64, 138–142. [Google Scholar] [CrossRef]
- Ciriello, R.; Cataldi, T.R.I.; Centonze, D.; Guerrieri, A. Permselective Behavior of an Electrosynthesized, Nonconducting Thin Film of Poly(2-naphthol) and Its Application to Enzyme Immobilization. Electroanalysis 2000, 12, 825–830. [Google Scholar] [CrossRef]
- Guerrieri, A.; Ciriello, R.; Centonze, D. Permselective and enzyme-entrapping behaviours of an electropolymerized, non-conducting, poly(o-aminophenol) thin film-modified electrode: A critical study. Biosens. Bioelectron. 2009, 24, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, F.; Centonze, D.; Guerrieri, A.; Zambonin, P.G. An interference-free biosensor based on glucose oxidase electrochemically immobilized in a non-conducting poly(pyrrole) film for continuous subcutaneous monitoring of glucose through microdialysis sampling. Biosens. Bioelectron. 1993, 8, 393–399. [Google Scholar] [CrossRef]
- Pantano, P.; Kuhr, W.G. Enzyme-modified microelectrodes for in vivo neurochemical measurements. Electroanalysis 1995, 7, 405–416. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Whitaker, R.G. Electrochemical immobilisation of enzymes: Part II. Glucose oxidase immobilised in poly-N-methylpyrrole. J. Electroanal. Chem. Interfacial Electrochem. 1987, 224, 37–48. [Google Scholar] [CrossRef]
- De Benedetto, G.E.; Malitesta, C.; Zambonin, C.G. Electroanalytical/X-ray photoelectron spectroscopy investigation on glucose oxidase adsorbed on platinum. J. Chem. Soc. Faraday Trans. 1994, 90, 1495. [Google Scholar] [CrossRef]
- Sasso, S.V.; Pierce, R.J.; Walla, R.; Yacynych, A.M. Electropolymerized 1, 2-Diaminobenzene as a Means To Prevent Interferences and Fouling and To Stabilize Immobilized Enzyme in Electrochemical Biosensors. Anal. Chem. 1990, 62, 1111–1117. [Google Scholar] [CrossRef]
- Geise, R.J.; Adams, J.M.; Barone, N.J.; Yacynych, A.M. Electropolymerized films to prevent interferences and electrode fouling in biosensors. Biosens. Bioelectron. 1991, 6, 151–160. [Google Scholar] [CrossRef]
- Kennedy, J.F.; White, C.A. Principles of immobilization of enzymes. In Handbook of Enzyme Biotechnology; Wiseman, A., Ed.; Ellis Horwood Lim., John Wiley & Sons: Hoboken, NJ, USA, 1985; pp. 147–207. ISBN 0133829200. [Google Scholar]
- Guerrieri, A.; De Benedetto, G.E.; Palmisano, F.; Zambonin, P.G. Electrosynthesized non-conducting polymers as permselective membranes in amperometric enzyme electrodes: A glucose biosensor based on a co-crosslinked glucose oxidase/overoxidized polypyrrole bilayer. Biosens. Bioelectron. 1998, 13, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Guerrieri, A.; Lattanzio, V.; Palmisano, F.; Zambonin, P.G. Electrosynthesized poly(pyrrole)/poly(2-naphthol) bilayer membrane as an effective anti-interference layer for simultaneous determination of acethylcholine and choline by a dual electrode amperometric biosensor. Biosens. Bioelectron. 2006, 21, 1710–1718. [Google Scholar] [CrossRef] [PubMed]
- Guerrieri, A.; Ciriello, R.; Cataldi, T.R.I. A novel amperometric biosensor based on a co-crosslinked l-lysine-α-oxidase/overoxidized polypyrrole bilayer for the highly selective determination of l-lysine. Anal. Chim. Acta 2013, 795, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, R.; Guerrieri, A. A Crosstalk- and Interferent-Free Dual Electrode Amperometric Biosensor for the Simultaneous Determination of Choline and Phosphocholine. Sensors 2021, 21, 3545. [Google Scholar] [CrossRef] [PubMed]
- Guerrieri, A.; Ciriello, R.; Acquavia, M.A.; Bianco, G.; Di Capua, A. Electrophoretic Protein Deposition as a Tool for In Situ Co-Crosslinking Enzyme Immobilization: An Electrochemical/Quartz Crystal Microbalance Study. Appl. Sci. 2023, 14, 212. [Google Scholar] [CrossRef]
- Lacefield, W.R. Current Status of Ceramic Coatings for Dental Implants. Implant Dent. 1998, 7, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Boccaccini, A.R.; Keim, S.; Ma, R.; Li, Y.; Zhitomirsky, I. Electrophoretic deposition of biomaterials. J. R. Soc. Interface 2010, 7, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Atanasov, P.; Wilkins, E. Development of needle-type glucose sensor with high selectivity. Sensors Actuators B Chem. 1998, 46, 249–256. [Google Scholar] [CrossRef]
- Zaborsky, O. Immobilized Enzymes; CRC Press: Cleveland, OH, USA, 1973; ISBN 9780878190164. [Google Scholar]
- Gough, D.A.; Leypoldt, J.K. Membrane-covered, rotated disk electrode. Anal. Chem. 1979, 51, 439–444. [Google Scholar] [CrossRef]
- Levich, V.G. Physicochemical Hydrodynamics; Prentice-Hall: New York, NY, USA, 1962; ISBN 9780136744405. [Google Scholar]
- Allen, J.; Bard, L.R.F. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000; ISBN 978-0-471-04372-0. [Google Scholar]
- Sadana, A.; Sadana, N. Fabrication of Biosensors. In Handbook of Biosensors and Biosensor Kinetics; Sadana, A., Sadana, N., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; pp. 35–60. ISBN 978-0-444-53262-6. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biosensor | Sensitivity (µA/mM mm2) | Imax 2 (µA/mm2) | LD/LQ 3 (µM) | Linear Range (mM) | K′m 4 (mM) |
|---|---|---|---|---|---|
| Pt/PoAP/GOD | 0.60 | 10.8 | 55/180 | 0.1–8 | 13.4 |
| Pt/GOD/PoAP | 0.37 | 4.29 | 96/312 | 0.2–4 | 6.33 |
| Pt/GOD/P2NAP | 0.33 | 3.31 | 48/156 | 0.1–3 | 4.45 |
| Biosensor | Bias (%) 1 | |||
|---|---|---|---|---|
| AA 2 (0.1 mM) | UA 2 (0.5 mM) | PA 2 (0.2 mM) | CYS 2 (0.1 mM) | |
| Pt/PoAP/GOD | 3.85 | 16.9 | 5.38 | 3.46 |
| Pt/GOD/PoAP | 1.42 | 2.48 | 10.3 | 3.55 |
| Pt/GOD/P2NAP | NV 3 | NV 3 | NV 3 | NV 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciriello, R.; Acquavia, M.A.; Bianco, G.; Di Capua, A.; Guerrieri, A. A New Approach for Interferent-Free Amperometric Biosensor Production Based on All-Electrochemically Assisted Procedures. Biosensors 2025, 15, 470. https://doi.org/10.3390/bios15080470
Ciriello R, Acquavia MA, Bianco G, Di Capua A, Guerrieri A. A New Approach for Interferent-Free Amperometric Biosensor Production Based on All-Electrochemically Assisted Procedures. Biosensors. 2025; 15(8):470. https://doi.org/10.3390/bios15080470
Chicago/Turabian StyleCiriello, Rosanna, Maria Assunta Acquavia, Giuliana Bianco, Angela Di Capua, and Antonio Guerrieri. 2025. "A New Approach for Interferent-Free Amperometric Biosensor Production Based on All-Electrochemically Assisted Procedures" Biosensors 15, no. 8: 470. https://doi.org/10.3390/bios15080470
APA StyleCiriello, R., Acquavia, M. A., Bianco, G., Di Capua, A., & Guerrieri, A. (2025). A New Approach for Interferent-Free Amperometric Biosensor Production Based on All-Electrochemically Assisted Procedures. Biosensors, 15(8), 470. https://doi.org/10.3390/bios15080470