Construction of Graphene Oxide Probes Loaded with Antisense Peptide Nucleic Acid and Doxorubicin for Regulating Telomerase Activity and Inducing Apoptosis of Cancer Cells


Abstract
1. Introduction
2. Materials and Methods
2.1. Apparatus
2.2. Reagents
2.3. Experimental Sections
2.3.1. Preparation of FA-PNA-GO Probe, PNA-GO Probe, and Dox-FA-PNA-GO Probe
2.3.2. Fluorescence Quenching of GO on Antisense-PNA and Fluorescence Recovery of FA-PNA-GO Probe Triggered by Target-DNA
2.3.3. Determination of Dox Encapsulation Efficiency and Loading Capacity
2.3.4. Cell Culture
2.3.5. Confocal Fluorescence Imaging
2.3.6. Determination of Intracellular hTERT Protein Expression Levels
2.3.7. RNA Extraction and Quantitation of hTERT mRNA by PCR
2.3.8. Cellular Viability Study
2.3.9. Flow Cytometric Analysis of Cell Apoptosis Induced by FA-PNA-GO Probes
3. Results and Discussion
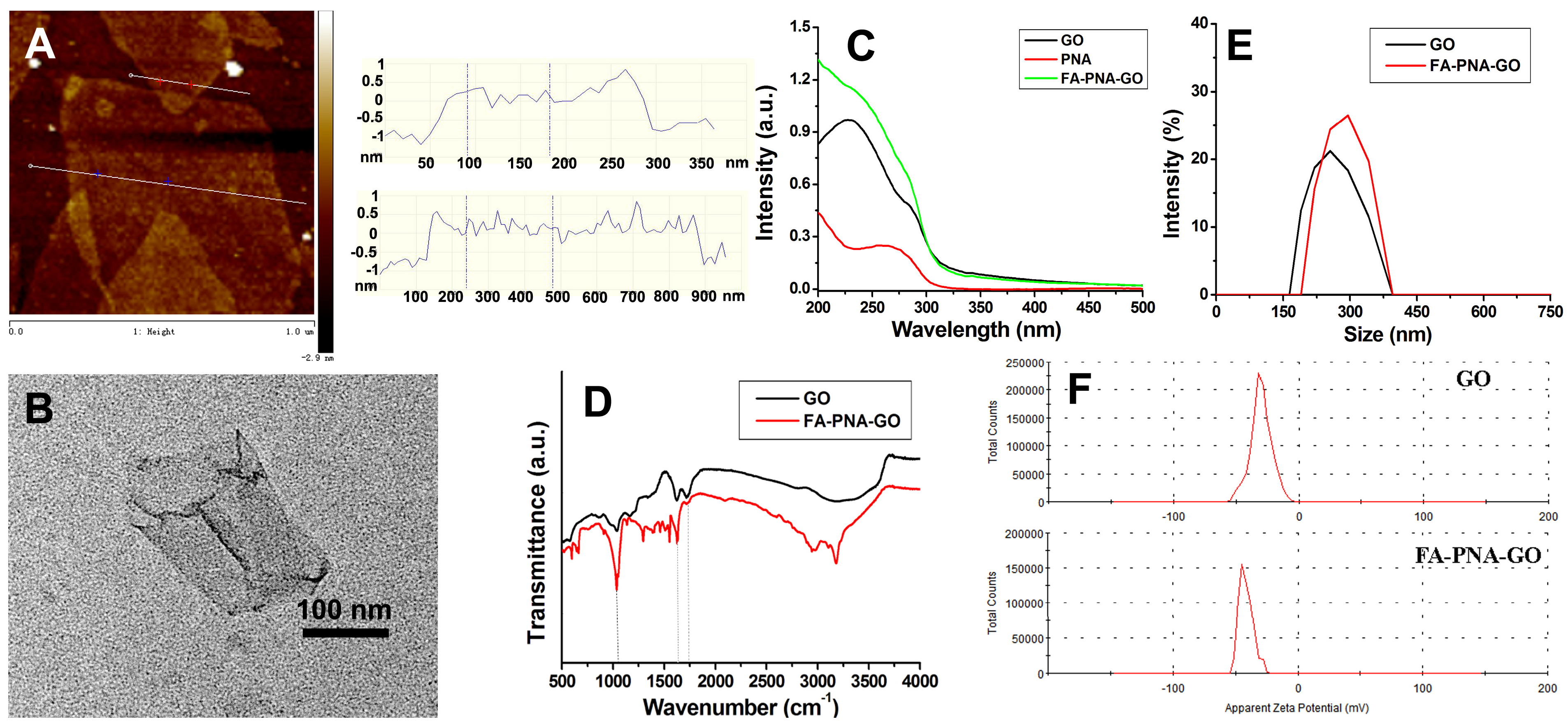
3.1. Characterization of GO and FA-PNA-GO Probe
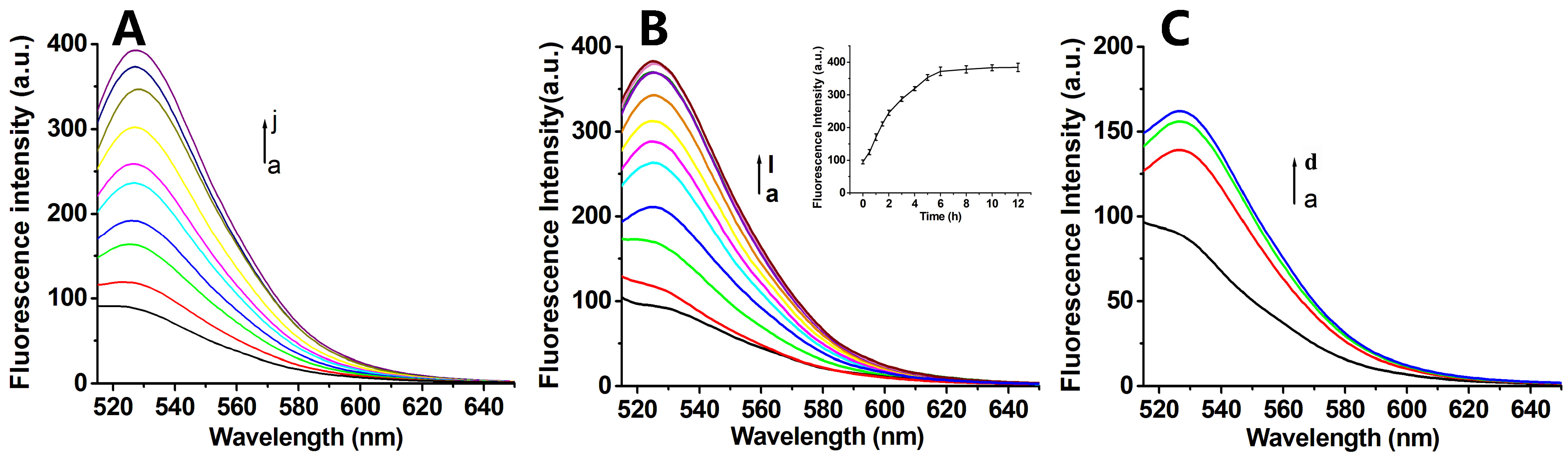
3.2. Fluorescence Spectroscopic Analysis of GO-Mediated Antisense-PNA Adsorption and Fluorophore Quenching
3.3. Fluorescence Spectroscopic Analysis of PNA-GO and FA-PNA-GO Probes in Response to Complementary Targets
3.4. In Situ Detection, Fluorescence Imaging, and Time Response of Intracellular hTERT mRNA Using the FA-PNA-GO Probes
3.5. Design and Characterization of the Dox-FA-PNA-GO Probe
3.6. Real-Time Tracking of Therapeutic Cargo Release from Dox-FA-PNA-GO Nanoplatform in Cancer Cells
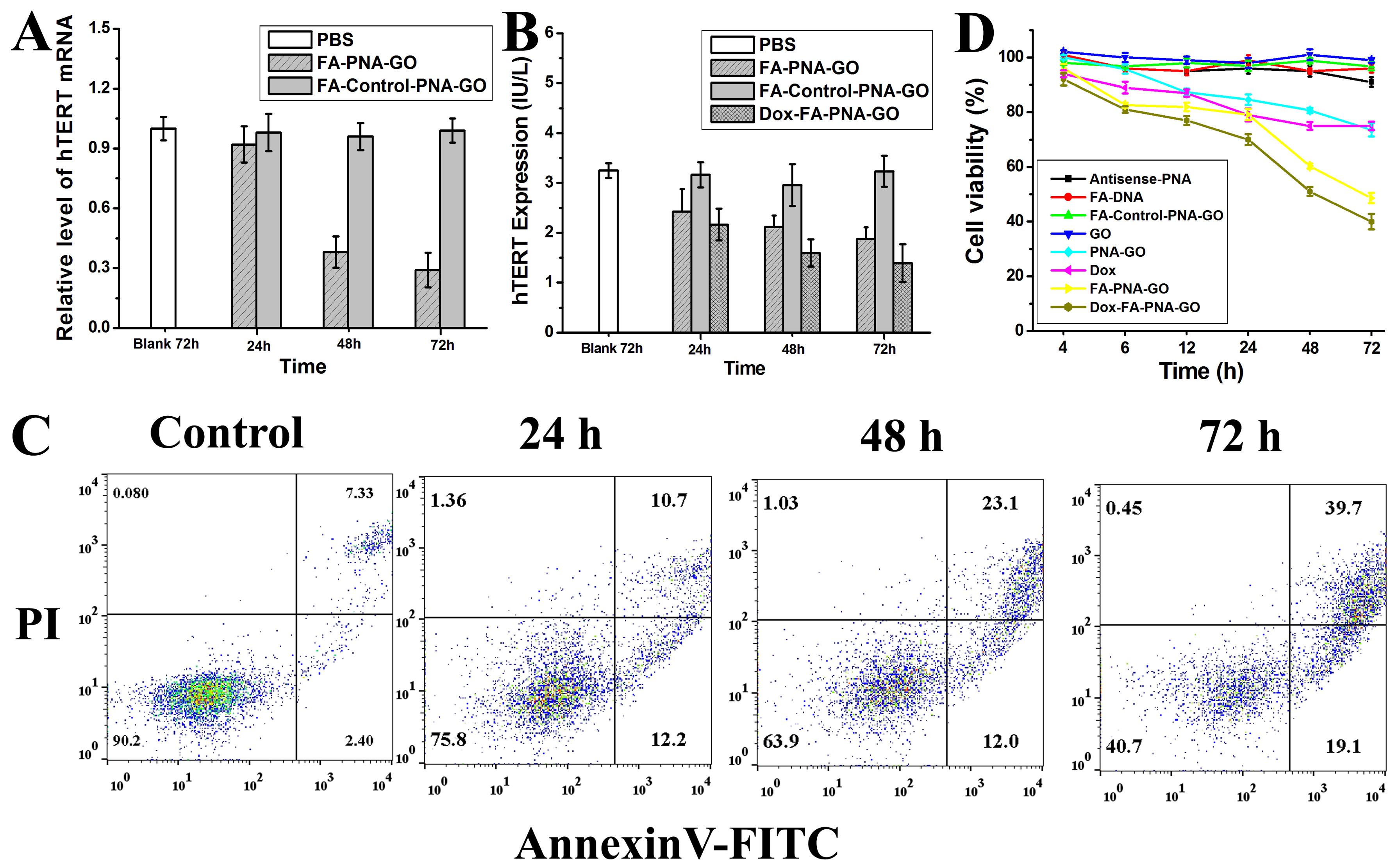
3.7. Investigating the FA-PNA-GO Probe’s Dual Functionality: hTERT mRNA/Protein Downregulation and Proapoptotic Activity in Cancer Cells
3.8. Synergistic Telomerase Suppression by Dox-FA-PNA-GO Nanoplatform
3.9. Assessing the Cytotoxic Efficacy of Different Nanosystems: Time-Dependent Modulation of Cellular Viability
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, J.; Wang, Y.; Susac, L.; Chan, H.; Basu, R.; Zhou, Z.H.; Feigon, J. Structure of Telomerase with Telomeric DNA. Cell 2018, 173, 1179–1190. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Cui, Y.; Yin, H.; Scheid, A.; Hendricks, W.P.; Schmidt, J.; Sekulic, A.; Kong, D.; Trent, J.M.; Gokhale, V.; et al. A Pharmacological Chaperone Molecule Induces Cancer Cell Death by Restoring Tertiary DNA Structures in Mutant hTERT Promoters. J. Am. Chem. Soc. 2016, 138, 13673–13692. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, K.; Fuessel, S.; Schmidt, U.; Kotzsch, M.; Schwenzzer, B.; Wirth, M.P.; Meye, A. Antisense-mediated hTERT inhibition specifically reduces the growth of human bladder cancer cells. Clin. Cancer Res. 2003, 9, 3794–3800. [Google Scholar]
- Qi, Z.; Mi, R. Inhibition of human telomerase reverse transcriptase in vivo and in vitro for retroviral vector-based antisense oligonucleotide therapy in ovarian cancer. Cancer Gene Ther. 2016, 23, 36–42. [Google Scholar] [CrossRef]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- Wang, F.; Liu, L.S.; Lau, C.H.; Chang, T.J.H.; Tam, D.Y.; Leung, H.M.; Tin, C.; Lo, P.K. Synthetic α-l-Threose Nucleic Acids Targeting BcL-2 Show Gene Silencing and in Vivo Antitumor Activity for Cancer Therapy. ACS Appl. Mater. Interfaces 2019, 11, 38510–38518. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015, 518, 107–111. [Google Scholar] [CrossRef]
- Shen, H.; Liu, M.; He, H.X.; Zhang, L.M.; Huang, J.; Chong, Y.; Dai, J.W.; Zhang, Z.J. PEGylated Graphene Oxide-Mediated Protein Delivery for Cell Function Regulation. ACS Appl. Mater. Interfaces 2012, 4, 6317–6323. [Google Scholar] [CrossRef]
- Campbell, M.A.; Wengel, J. Locked vs. unlocked nucleic acids (LNA vs. UNA): Contrasting structures work towards common therapeutic goals. Chem. Soc. Rev. 2011, 40, 5680–5689. [Google Scholar] [CrossRef]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M.; Buchardt, O. Peptide nucleic acid (PNA). A DNA mimic with a peptide backbone. Bioconjug. Chem. 1994, 5, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.C.; Piatyszek, M.A.; Wright, W.E.; Shay, J.W.; Corey, D.R. Inhibition of human telomerase activity by peptide nucleic acids. Nat. Biotechnol. 1996, 4, 615–619. [Google Scholar] [CrossRef]
- Cutts, S.M.; Nudelman, A.; Rephaeli, A.; Phillips, D.R.S.; Cutts, A.; Nudelman, A.; Phillips, D.R. The Power and Potential of Doxorubicin-DNA Adducts. IUBMB Life 2005, 57, 73–81. [Google Scholar] [CrossRef]
- Mollaei, M.; Hassan, Z.M.; Khorshidi, F.; Langroudi, L. Chemotherapeutic drugs: Cell death- and resistance-related signalingpathways. Are they really as smart as the tumor cells? Transl. Oncol. 2021, 14, 101056. [Google Scholar] [CrossRef]
- Zhu, X.; Kumar, R.; Mandal, M.; Sharma, N.; Sharma, H.W.; Dhingra, U.; Sokoloski, J.A.; Hsiao, R.; Narayanan, R. Cell cycle-dependent modulation of telomerase activity in tumor cells. Proc. Natl. Acad. Sci. USA 1996, 93, 6091–6095. [Google Scholar] [CrossRef]
- Feng, T.T.; Feng, D.; Shi, W.; Li, X.; Ma, H. A graphene oxide-peptide fluorescence sensor for proteolytically active prostate-specific antigen. Mol. BioSyst. 2012, 8, 1441–1445. [Google Scholar] [CrossRef]
- Liao, X.J.; Wang, Q.B.; Ju, H.X. A peptide nucleic acid-functionalized carbon nitride nanosheet as a probe for in situ monitoring of intracellular microRNA. Analyst 2015, 140, 4245–4252. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Zhang, X.Y.; Liu, Z.F.; Ma, Y.F.; Huang, Y.; Chen, Y.S. High-Efficiency Loading and Controlled Release of Doxorubicin Hydrochloride on Graphene Oxide. J. Phys. Chem. C 2008, 112, 17554–17558. [Google Scholar] [CrossRef]
- Wen, H.Y.; Dong, C.Y.; Dong, H.Q.; Shen, A.J.; Xia, W.J.; Cai, X.J.; Song, Y.Y.; Li, X.Q.; Li, Y.Y.; Shi, D.L. Engineered Redox-Responsive PEG Detachment Mechanism in PEGylated Nano-Graphene Oxide for Intracellular Drug Delivery. Small 2012, 8, 760–769. [Google Scholar] [CrossRef]
- Liu, Z.; Robinson, J.T.; Sun, X.M.; Dai, H.J. PEGylated Nanographene Oxide for Delivery of Water-Insoluble Cancer Drugs. J. Am. Chem. Soc. 2008, 130, 10876–10877. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.B.; Wei, Z.H.; Zhao, Z.P.; Miao, Y.L.; Qiu, Y.D.; Yang, W.J.; Jia, X.; Liu, Z.Y.; Hou, H.W. Design and Development of Graphene Oxide Nanoparticle/Chitosan Hybrids Showing pH-Sensitive Surface Charge-Reversible Ability for Efficient Intracellular Doxorubicin Delivery. ACS Appl. Mater. Interfaces 2018, 10, 6608–6617. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, S.R.; Lee, J.; Yeo, J.; Na, H.K.; Kim, Y.K.; Jang, H.; Lee, J.H.; Han, S.W.; Lee, Y.; Kim, V.N.; et al. Quantitative and Multiplexed MicroRNA Sensing in Living Cells Based on Peptide Nucleic Acid and Nano Graphene Oxide (PANGO). ACS Nano 2013, 7, 5882–5891. [Google Scholar] [CrossRef] [PubMed]
- Stobniecka, M.; Dworakowska, B.; Jakiela, S.; Lukasik, A.; Chalupa, A.; Zembrzycki, K. Sensing of survivin mRNA in malignant astrocytes using graphene oxide nanocarrier-supported oligonucleotide molecular beacons. Sens. Actuators B Chem. 2016, 235, 136–145. [Google Scholar]
- Baek, A.; Baek, Y.M.; Kim, H.; Jun, B.; Kim, D. Polyethylene Glycol-Engrafted Graphene Oxide as Biocompatible Materials for Peptide Nucleic Acid Delivery into Cells. Bioconjug. Chem. 2018, 29, 528–537. [Google Scholar] [CrossRef]
- Das, M.; Mohanty, C.; Sahoo, S.K. Ligand-based targeted therapy for cancer tissue. Expert Opin. Drug Deliv. 2009, 6, 285–304. [Google Scholar] [CrossRef]
- Sabharanjak, S.; Mayor, S. Folate receptor endocytosis and trafficking. Adv. Drug Deliv. Rev. 2004, 56, 1099–1109. [Google Scholar] [CrossRef]
- Zhou, X.Z.; Huang, X.; Qi, X.Y.; Wu, S.X.; Xue, C.; Boey, F.Y.C.; Yan, Q.Y.; Chen, P.; Zhang, H. In Situ Synthesis of Metal Nanoparticles on Single-Layer Graphene Oxide and Reduced Graphene Oxide Surfaces. J. Phys. Chem. C 2009, 113, 10842–10846. [Google Scholar] [CrossRef]
- Hong, M.; Sun, H.X.; Yang, Q.Q.; Cheng, S.; Yu, S.X.; Fan, S.H.; Li, C.; Cui, C.; Tan, W.H. A microRNA-21-responsive doxorubicin-releasing sticky-flare for synergistic anticancer with silencing of microRNA and chemotherapy. Sci. China Chem. 2021, 64, 1009–1019. [Google Scholar] [CrossRef]
- Hong, M.; Xu, L.; Xue, Q.; Li, L.; Tang, B. Fluorescence Imaging of Intracellular Telomerase Activity Using Enzyme-Free Signal Amplification. Anal. Chem. 2016, 88, 12177–12182. [Google Scholar] [CrossRef]
- Qian, R.C.; Ding, L.; Yan, L.W.; Lin, M.F.; Ju, H.X. A Robust Probe for Lighting Up Intracellular Telomerase via Primer Extension To Open a Nicked Molecular Beacon. J. Am. Chem. Soc. 2014, 136, 8205–8208. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Gu, W.L.; Gong, M.Z.; Wang, J.; Li, D.Q. shRNA-mediated silencing of hTERT suppresses proliferation and promotes apoptosis in osteosarcoma cells. Cancer Gene Ther. 2017, 24, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Kao, F.H.; Akhtar, N.; Chen, C.C.; Chen, H.Y.; Thakur, M.K.; Chen, Y.Y.; Chen, C.L. In Vivo and in Vitro Demonstration of Gold Nanorod Aided Photothermal Presoftening of B16F10 Melanoma for Efficient Chemotherapy Using Doxorubicin Loaded Graphene Oxide. ACS Appl. Bio Mater. 2019, 2, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Nguyen, H.T.; Pham, T.T.; Choi, J.Y.; Choi, H.G.; Yong, C.S.; Kim, J.O. Development of a Graphene Oxide Nanocarrier for Dual-Drug Chemo-phototherapy to Overcome Drug Resistance in Cancer. ACS Appl. Mater. Interfaces 2015, 7, 28647–28655. [Google Scholar] [CrossRef]
- Sun, H.X.; Hong, M.; Yang, Q.Q.; Li, C.; Zhang, G.Z.; Yue, Q.L.; Ma, Y.H.; Li, X.; Li, C.Z. Visualizing the down-regulation of hTERT mRNA expression using gold-nanoflare probes and verifying the correlation with cancer cell apoptosis. Analyst 2019, 144, 2994–3004. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Names | Sequences (5′→3′) |
|---|---|
| Antisense-PNA | FAM-OO-CCAGCCGCCAGCCCT |
| Control-PNA | FAM-OO-GATGCCGTAAGATCT |
| Target-DNA | AGGGCTGGCGGCTGG |
| 10T-Target-DNA | TTTTTTTTTTAGGGCTGGCGGCTGGTTTTTTTTTT |
| NH2-Poly A | NH2-AAAAAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Ji, Q.; Hong, M. Construction of Graphene Oxide Probes Loaded with Antisense Peptide Nucleic Acid and Doxorubicin for Regulating Telomerase Activity and Inducing Apoptosis of Cancer Cells. Biosensors 2025, 15, 337. https://doi.org/10.3390/bios15060337
Zhu Y, Ji Q, Hong M. Construction of Graphene Oxide Probes Loaded with Antisense Peptide Nucleic Acid and Doxorubicin for Regulating Telomerase Activity and Inducing Apoptosis of Cancer Cells. Biosensors. 2025; 15(6):337. https://doi.org/10.3390/bios15060337
Chicago/Turabian StyleZhu, Yanyan, Qinghong Ji, and Min Hong. 2025. "Construction of Graphene Oxide Probes Loaded with Antisense Peptide Nucleic Acid and Doxorubicin for Regulating Telomerase Activity and Inducing Apoptosis of Cancer Cells" Biosensors 15, no. 6: 337. https://doi.org/10.3390/bios15060337
APA StyleZhu, Y., Ji, Q., & Hong, M. (2025). Construction of Graphene Oxide Probes Loaded with Antisense Peptide Nucleic Acid and Doxorubicin for Regulating Telomerase Activity and Inducing Apoptosis of Cancer Cells. Biosensors, 15(6), 337. https://doi.org/10.3390/bios15060337