Enhancing Colorimetric Detection of Nucleic Acids on Nitrocellulose Membranes: Cutting-Edge Applications in Diagnostics and Forensics

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Oligonucleotides
2.3. EMSA
2.4. DNA Immobilization on Gold Nanoparticles
2.5. LFA Production and Assembly
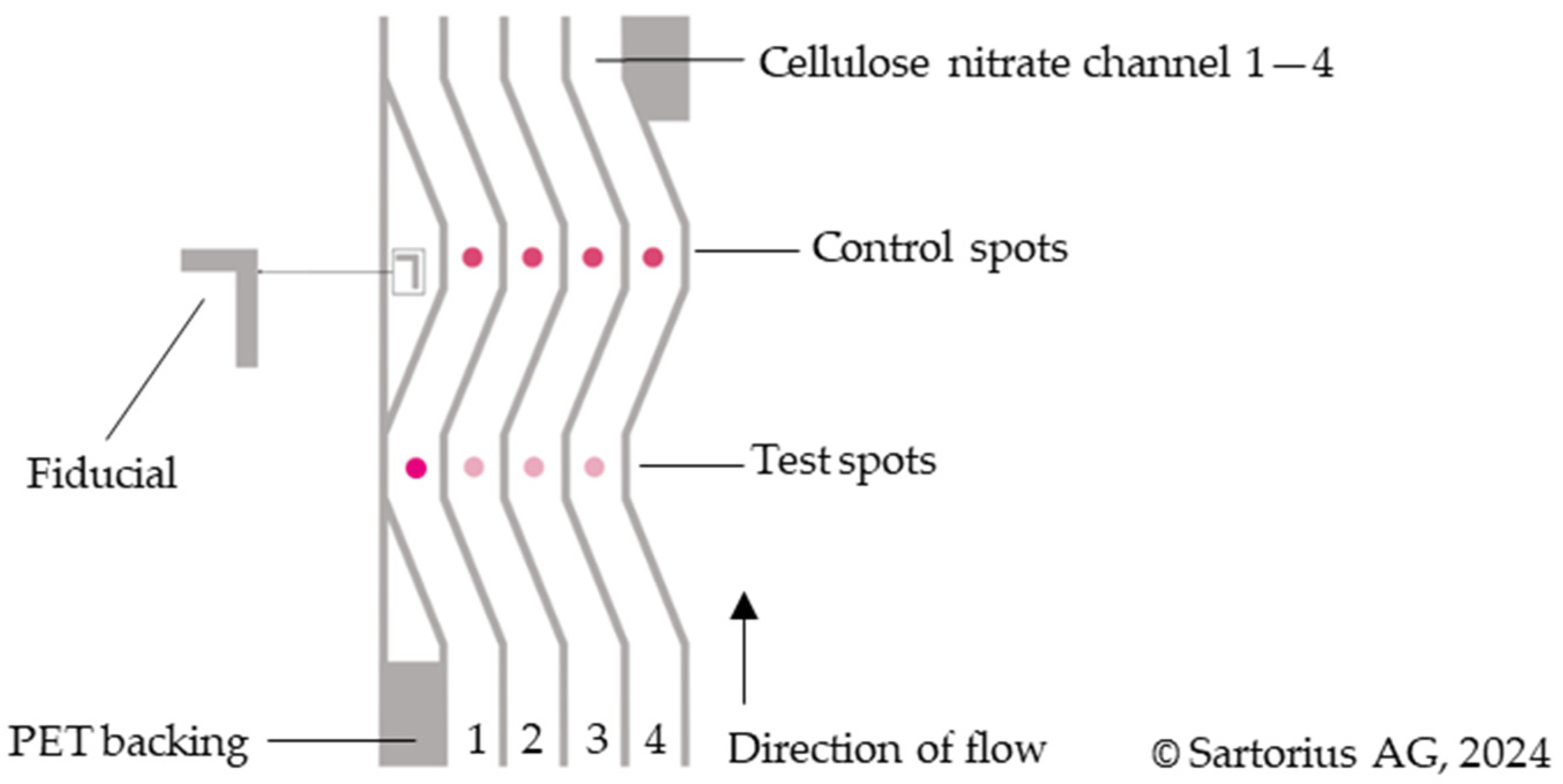
2.5.1. Lined LFA
2.5.2. Spotted LFA
3. Results
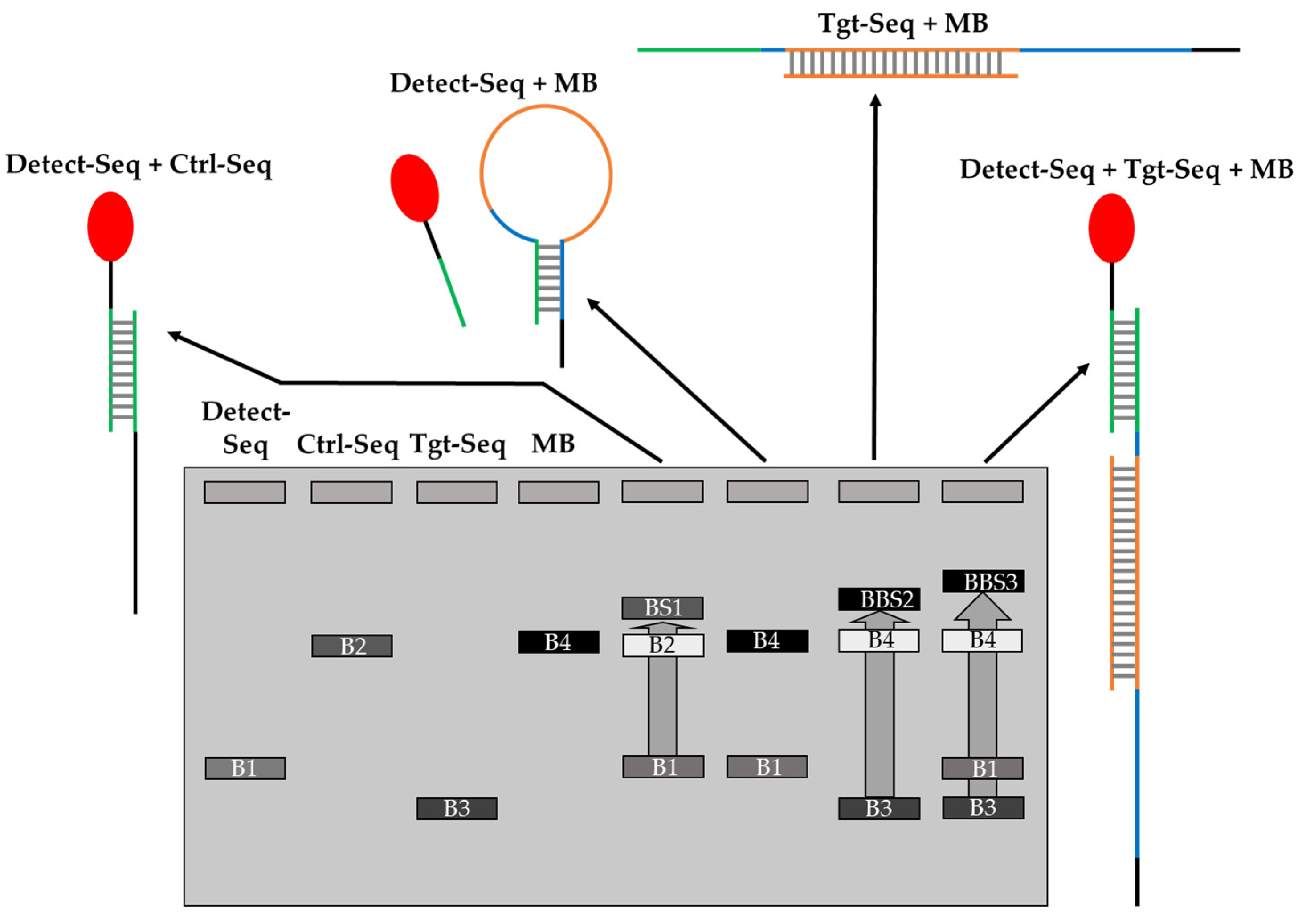
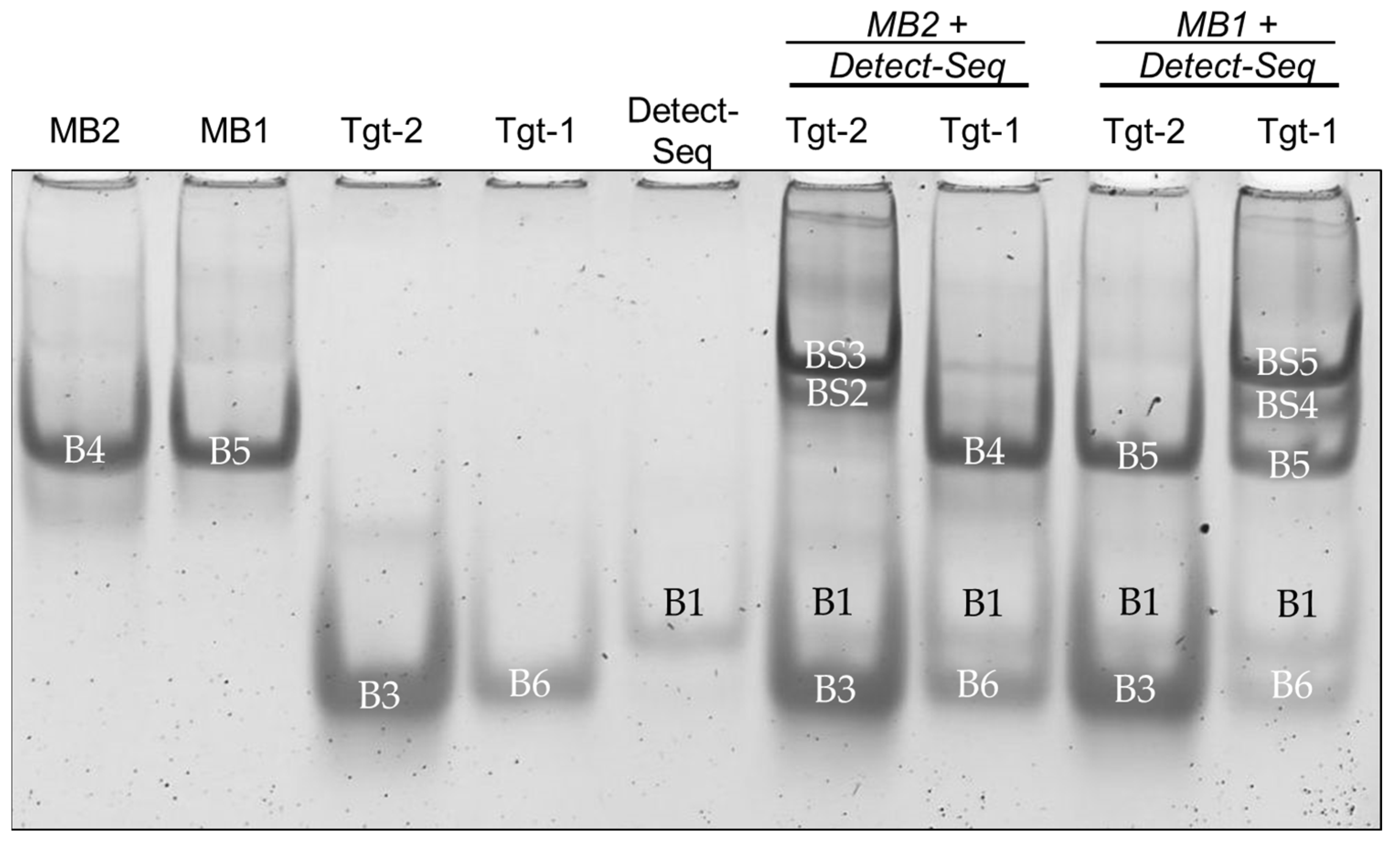
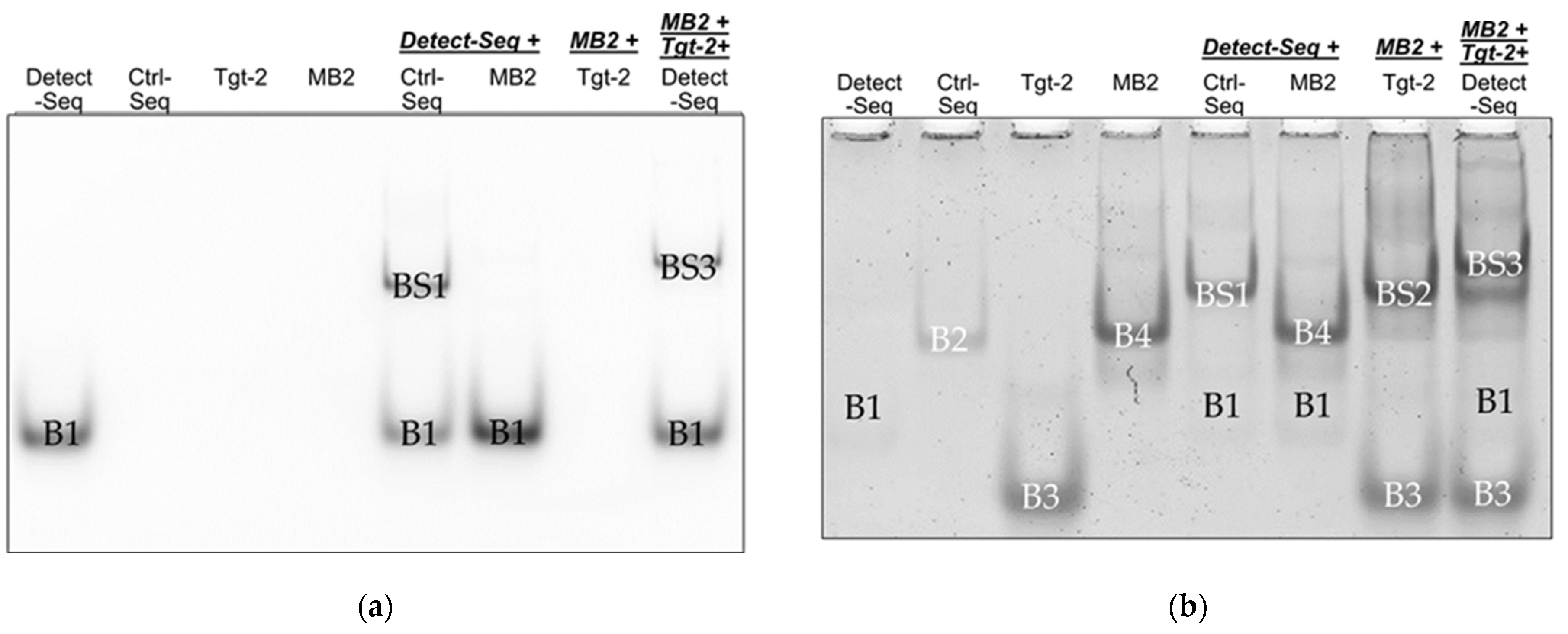
3.1. Verification of Oligonucleotide Hybridization with EMSA
3.1.1. Band Shift Analysis for Optimizing EMSA Parameters
3.1.2. Fluorescence Monitoring of Band Shifts in EMSA
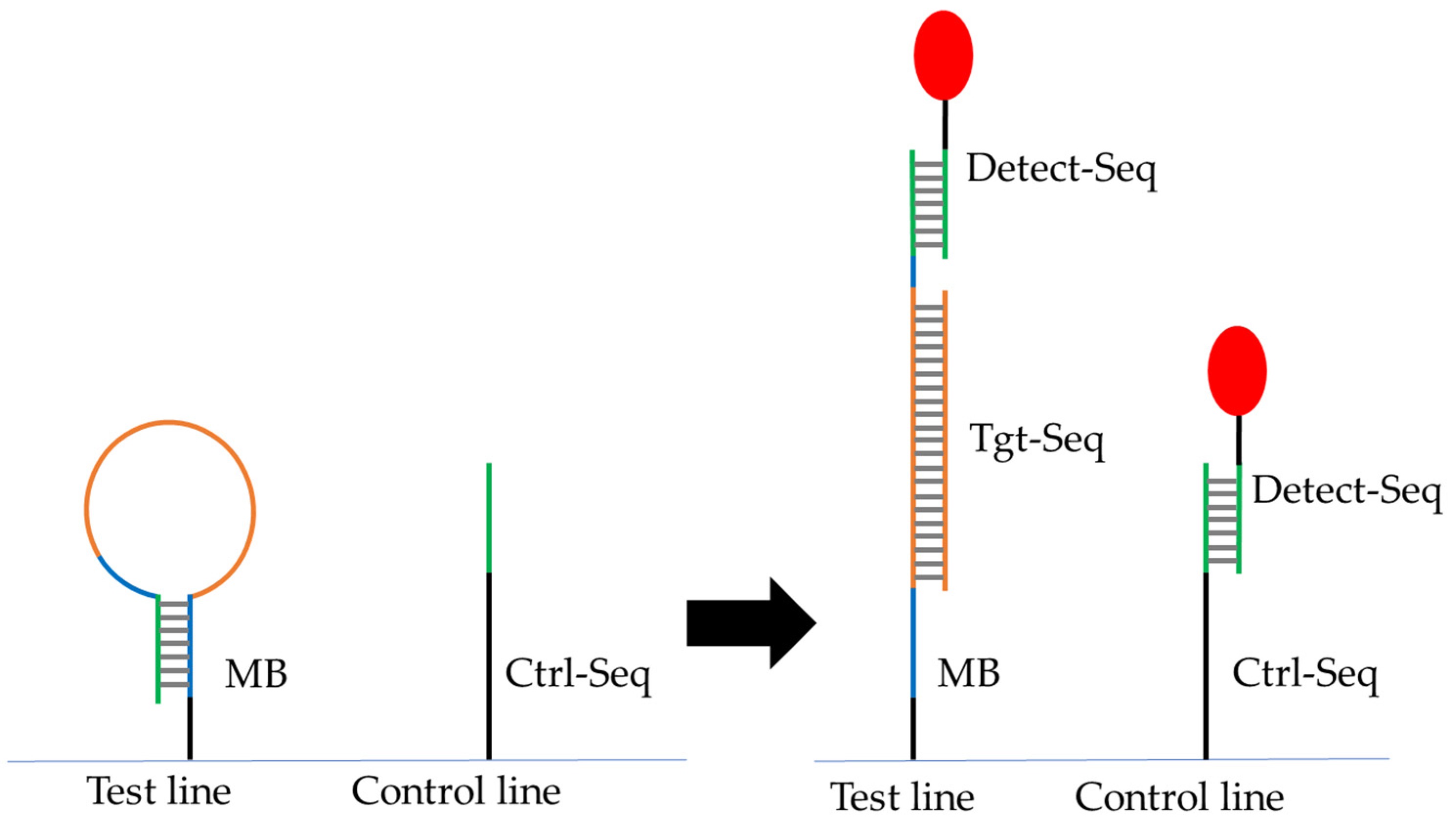
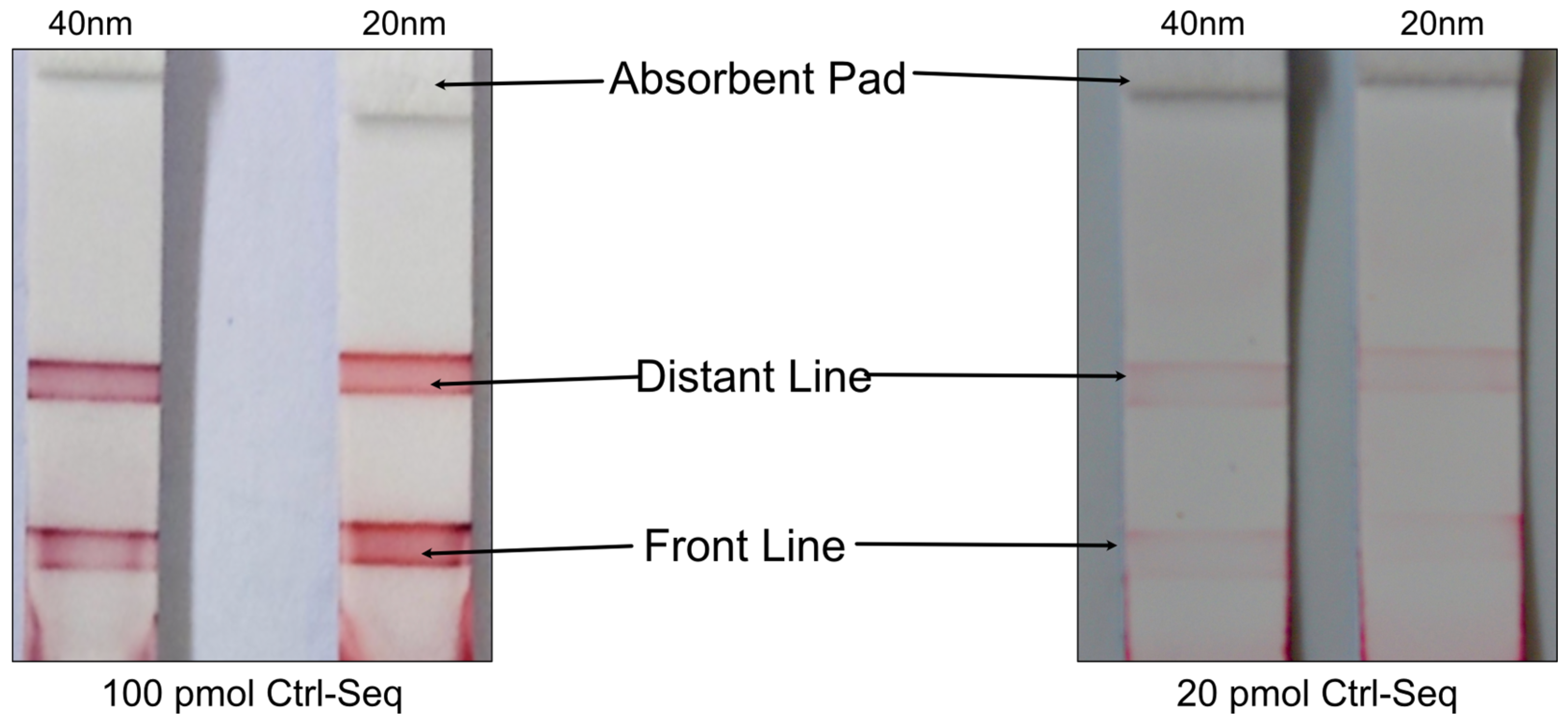
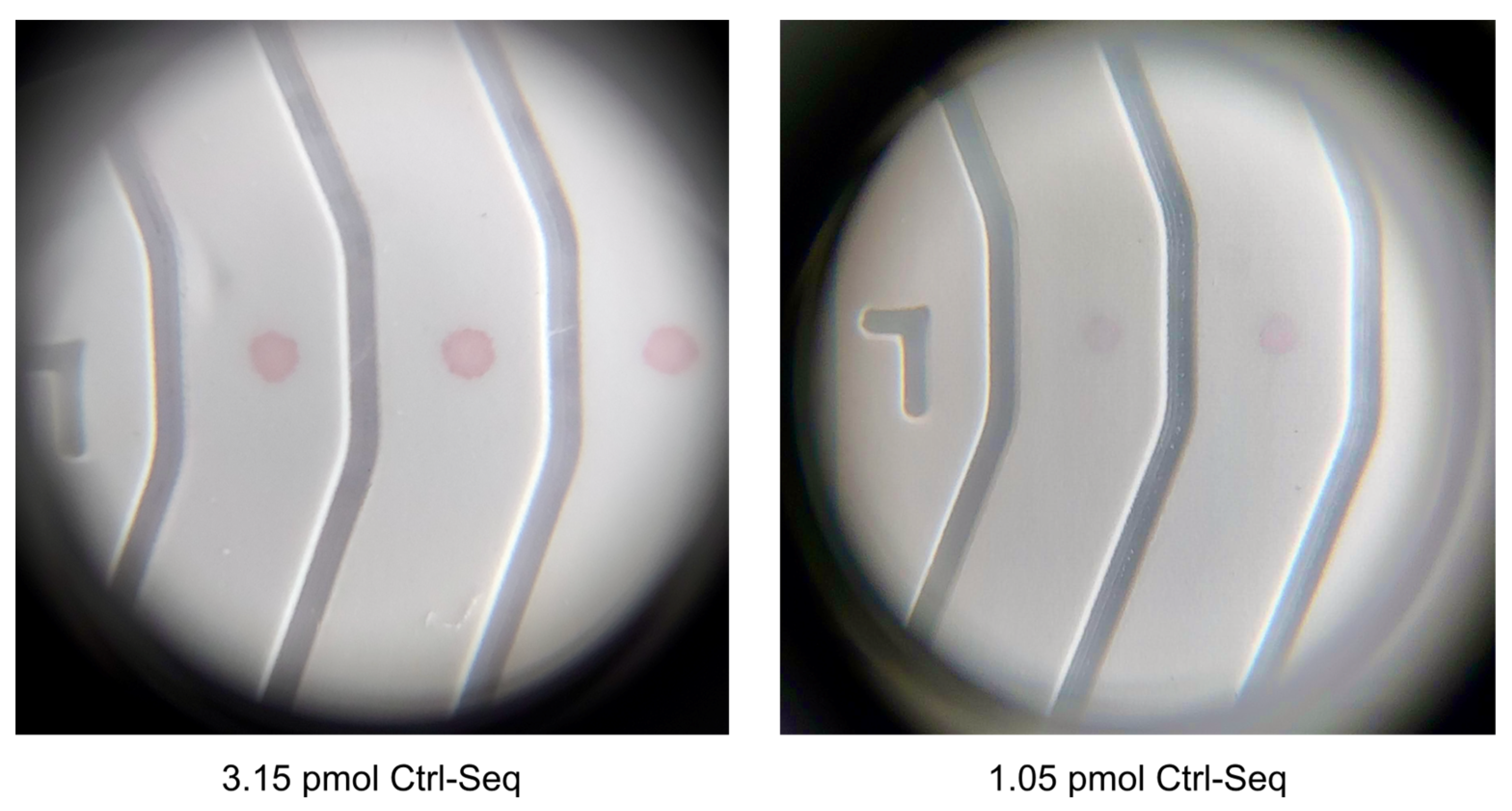
3.2. Protein-Free NALFA Test Performance and Assessment of Limit of Detection
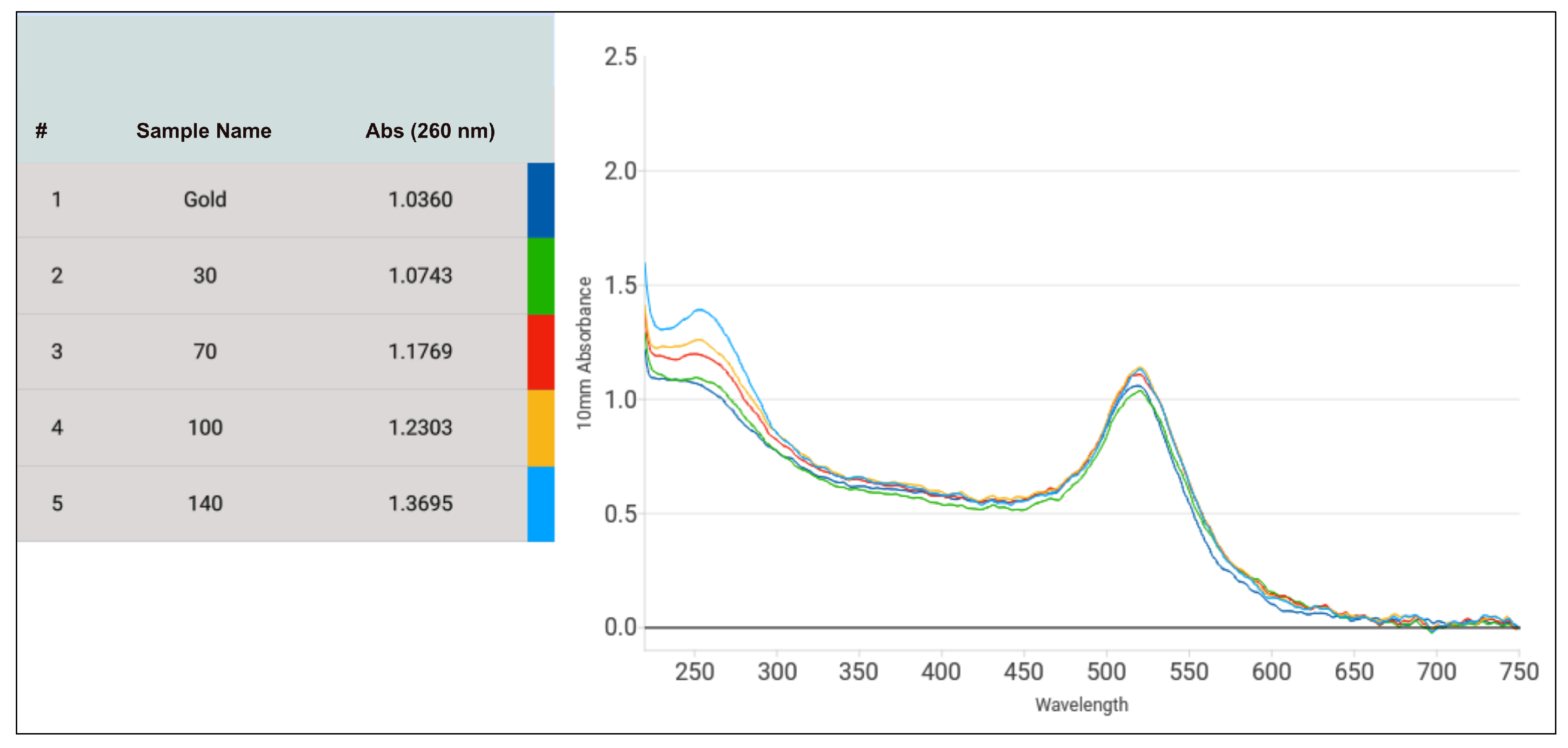
3.3. Optimization of the DNA-AuNP Conjugation Regarding the DNA to AuNP Ratio
3.4. Verification of Limit of Detection and Sensitivity
3.5. Specificity Experiments with Three Different Molecular Beacons and Their Target Sequences
3.6. Verification of Functionality with Human Body Fluids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H. The structure of DNA. Cold Spring Harb. Symp. Quant. Biol. 1953, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Southern, E.M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 1975, 98, 503–517. [Google Scholar] [CrossRef]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Langer-Safer, P.R.; Levine, M.; Ward, D.C. Immunological method for mapping genes on Drosophila polytene chromosomes. Proc. Natl. Acad. Sci. USA 1982, 79, 4381–4385. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.; Pulham, C.R.; Morrison, C.A. Structure and properties of nitrocellulose: Approaching 200 years of research. RSC Adv. 2023, 13, 32321–32333. [Google Scholar] [CrossRef]
- Shields, M.J.; Siegel, J.N.; Clark, C.R.; Hines, K.K.; Potempa, L.A.; Gewurz, H.; Anderson, B. An appraisal of polystyrene-(ELISA) and nitrocellulose-based (ELIFA) enzyme immunoassay systems using monoclonal antibodies reactive toward antigenically distinct forms of human C-reactive protein. J. Immunol. Methods 1991, 141, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Posthuma-Trumpie, G.A.; Korf, J.; van Amerongen, A. Lateral flow (immuno)assay: Its strengths, weaknesses, opportunities and threats. A literature survey. Anal. Bioanal. Chem. 2009, 393, 569–582. [Google Scholar] [CrossRef]
- Nguyen, V.-T.; Song, S.; Park, S.; Joo, C. Recent advances in high-sensitivity detection methods for paper-based lateral-flow assay. Biosens. Bioelectron. 2020, 152, 112015. [Google Scholar] [CrossRef]
- Soh, J.H.; Chan, H.-M.; Ying, J.Y. Strategies for developing sensitive and specific nanoparticle-based lateral flow assays as point-of-care diagnostic device. Nano Today 2020, 30, 100831. [Google Scholar] [CrossRef]
- He, X.; Liu, Z.; Yang, Y.; Li, L.; Wang, L.; Li, A.; Qu, Z.; Xu, F. Sensitivity Enhancement of Nucleic Acid Lateral Flow Assays through a Physical-Chemical Coupling Method: Dissoluble Saline Barriers. ACS Sens. 2019, 4, 1691–1700. [Google Scholar] [CrossRef]
- Li, X.-M.; Fu, P.-Y.; Liu, J.-M.; Zhang, S.-S. Biosensor for multiplex detection of two DNA target sequences using enzyme-functionalized Au nanoparticles as signal amplification. Anal. Chim. Acta 2010, 673, 133–138. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Broughton, J.P.; Deng, X.; Yu, G.; Fasching, C.L.; Servellita, V.; Singh, J.; Miao, X.; Streithorst, J.A.; Granados, A.; Sotomayor-Gonzalez, A.; et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020, 38, 870–874. [Google Scholar] [CrossRef] [PubMed]
- van den Hurk, R.; Evoy, S. A Review of Membrane-Based Biosensors for Pathogen Detection. Sensors 2015, 15, 14045–14078. [Google Scholar] [CrossRef] [PubMed]
- Koczula, K.M.; Gallotta, A. Lateral flow assays. Essays Biochem. 2016, 60, 111–120. [Google Scholar] [CrossRef]
- Song, S.; Liang, Z.; Zhang, J.; Wang, L.; Li, G.; Fan, C. Gold-nanoparticle-based multicolor nanobeacons for sequence-specific DNA analysis. Angew. Chem. Int. Ed. Engl. 2009, 48, 8670–8674. [Google Scholar] [CrossRef]
- Qing, T.; He, D.; He, X.; Wang, K.; Xu, F.; Wen, L.; Shangguan, J.; Mao, Z.; Lei, Y. Nucleic acid tool enzymes-aided signal amplification strategy for biochemical analysis: Status and challenges. Anal. Bioanal. Chem. 2016, 408, 2793–2811. [Google Scholar] [CrossRef]
- Du, Y.; Dong, S. Nucleic Acid Biosensors: Recent Advances and Perspectives. Anal. Chem. 2017, 89, 189–215. [Google Scholar] [CrossRef]
- Javani, A.; Javadi-Zarnaghi, F.; Rasaee, M.J. A multiplex protein-free lateral flow assay for detection of microRNAs based on unmodified molecular beacons. Anal. Biochem. 2017, 537, 99–105. [Google Scholar] [CrossRef]
- Kor, K.; Turner, A.P.; Zarei, K.; Atabati, M.; Beni, V.; Mak, W.C. Structurally responsive oligonucleotide-based single-probe lateral-flow test for detection of miRNA-21 mimics. Anal. Bioanal. Chem. 2016, 408, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Yershov, G.; Barsky, V.; Belgovskiy, A.; Kirillov, E.; Kreindlin, E.; Ivanov, I.; Parinov, S.; Guschin, D.; Drobishev, A.; Dubiley, S.; et al. DNA analysis and diagnostics on oligonucleotide microchips. Proc. Natl. Acad. Sci. USA 1996, 93, 4913–4918. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Gibson, U.E.; Heid, C.A.; Williams, P.M. A novel method for real time quantitative RT-PCR. Genome Res. 1996, 6, 995–1001. [Google Scholar] [CrossRef]
- Mackay, I.M. Real-time PCR in the microbiology laboratory. Clin. Microbiol. Infect. 2004, 10, 190–212. [Google Scholar] [CrossRef]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006, 1, 1559–1582. [Google Scholar] [CrossRef]
- Henderson, W.A.; Xiang, L.; Fourie, N.H.; Abey, S.K.; Ferguson, E.G.; Diallo, A.F.; Kenea, N.D.; Kim, C.H. Simple lateral flow assays for microbial detection in stool. Anal. Methods 2018, 10, 5358–5363. [Google Scholar] [CrossRef]
- Moon, Y.; Moon, H.; Chang, J.; Kim, H.D.; Lee, J.H.; Lee, J. Development of a highly sensitive lateral flow strip device for nucleic acid detection using molecular beacons. Front. Sens. 2022, 3, 1012775. [Google Scholar] [CrossRef]
- Zheng, J.; Yang, R.; Shi, M.; Wu, C.; Fang, X.; Li, Y.; Li, J.; Tan, W. Rationally designed molecular beacons for bioanalytical and biomedical applications. Chem. Soc. Rev. 2015, 44, 3036–3055. [Google Scholar] [CrossRef]
- Garner, M.M.; Revzin, A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: Application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 1981, 9, 3047–3060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Servos, M.R.; Liu, J. Instantaneous and quantitative functionalization of gold nanoparticles with thiolated DNA using a pH-assisted and surfactant-free route. J. Am. Chem. Soc. 2012, 134, 7266–7269. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, B.; Dave, N.; Servos, M.R.; Liu, J. Instantaneous attachment of an ultrahigh density of nonthiolated DNA to gold nanoparticles and its applications. Langmuir 2012, 28, 17053–17060. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, Y. Preparation of aptamer-linked gold nanoparticle purple aggregates for colorimetric sensing of analytes. Nat. Protoc. 2006, 1, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Zadehkafi, A.; Siavashi, M.; Asiaei, S.; Bidgoli, M.R. Simple geometrical modifications for substantial color intensity and detection limit enhancements in lateral-flow immunochromatographic assays. J. Chromatogr. B 2019, 1110–1111, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sartorius. Unisart StructSure® Membranes—The Next Generation of Lateral Flow Assays; Sartorius: Göttingen, Germany, 2023. [Google Scholar]
- Andersson, K.; Björkelund, H.; Malmqvist, M. Antibody-antigen interactions: What is the required time to equilibrium? Nat. Preced. 2010. [Google Scholar] [CrossRef]
- Ndao, M. Diagnosis of parasitic diseases: Old and new approaches. Interdiscip. Perspect. Infect. Dis. 2009, 2009, 278246. [Google Scholar] [CrossRef]
- Smith, S.; Korvink, J.G.; Mager, D.; Land, K. The potential of paper-based diagnostics to meet the ASSURED criteria. RSC Adv. 2018, 8, 34012–34034. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Number of Drops Deposited per Spot | Total Volume Deposited per Spot, Vtot [nL] | Amount of Ctrl-DNA Deposited [pmol] |
|---|---|---|---|
| 1 | 1 | 0.42 | 0.105 |
| 2 | 10 | 4.2 | 1.05 |
| 3 | 20 | 8.4 | 2.10 |
| 4 | 30 | 12.6 | 3.15 |
| 5 | 40 | 16.8 | 4.20 |
| 6 | 50 | 21.0 | 5.25 |
| Feature | Nucleic Acid-Based LFA | Antibody Based LFA | PCR (Polymerase Chain Reaction) | qPCR (Quantitative PCR) | CRISPR-Based Assays | Microarrays |
|---|---|---|---|---|---|---|
| Sensitivity | High (pM) | Moderate (ng/mL) | Very High (aM) | Very High (aM) | High (pM) | Moderate (ng/mL) |
| Specificity | Very High | High | Very High | Very High | High | High |
| Time to Result | 10–20 min | 20–30 min | 1–3 h | 1–3 h | 30–60 min | 4–8 h |
| Ease of Use | Simple, user-friendly | Simple, user-friendly | Requires specialized equipment | Requires specialized equipment | Simple, user-friendly | Complex, requires specialized equipment |
| Cost | Low to Moderate | Moderate | High | High | Moderate to High | High |
| Equipment Required | Minimal (LFA device) | Minimal (LFA device) | PCR Machine | qPCR Machine | Minimal (LFA device) | Microarray Scanner |
| Sample Preparation | Minimal | Minimal | Moderate to High | Moderate to High | Minimal | High |
| Field Applicability | High | Moderate | Low (lab-based) | Low (lab-based) | Moderate | Low (lab-based) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subhashini, N.; Kerler, Y.; Menger, M.M.; Böhm, O.; Witte, J.; Stadler, C.; Griberman, A. Enhancing Colorimetric Detection of Nucleic Acids on Nitrocellulose Membranes: Cutting-Edge Applications in Diagnostics and Forensics. Biosensors 2024, 14, 430. https://doi.org/10.3390/bios14090430
Subhashini N, Kerler Y, Menger MM, Böhm O, Witte J, Stadler C, Griberman A. Enhancing Colorimetric Detection of Nucleic Acids on Nitrocellulose Membranes: Cutting-Edge Applications in Diagnostics and Forensics. Biosensors. 2024; 14(9):430. https://doi.org/10.3390/bios14090430
Chicago/Turabian StyleSubhashini, Nidhi, Yannick Kerler, Marcus M. Menger, Olga Böhm, Judith Witte, Christian Stadler, and Alexander Griberman. 2024. "Enhancing Colorimetric Detection of Nucleic Acids on Nitrocellulose Membranes: Cutting-Edge Applications in Diagnostics and Forensics" Biosensors 14, no. 9: 430. https://doi.org/10.3390/bios14090430
APA StyleSubhashini, N., Kerler, Y., Menger, M. M., Böhm, O., Witte, J., Stadler, C., & Griberman, A. (2024). Enhancing Colorimetric Detection of Nucleic Acids on Nitrocellulose Membranes: Cutting-Edge Applications in Diagnostics and Forensics. Biosensors, 14(9), 430. https://doi.org/10.3390/bios14090430