A Sensitive Fluorescence Polarization Immunoassay for the Rapid Detection of Okadaic Acid in Environmental Waters

 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Synthesis of the Coating Antigens
2.3. ELISA of OA
2.4. Synthesis of Ethylenediamine Fluoresceinthiocarbamyl
2.5. Synthesis of OA–EDF and OA–AMF
2.6. FPIA of OA
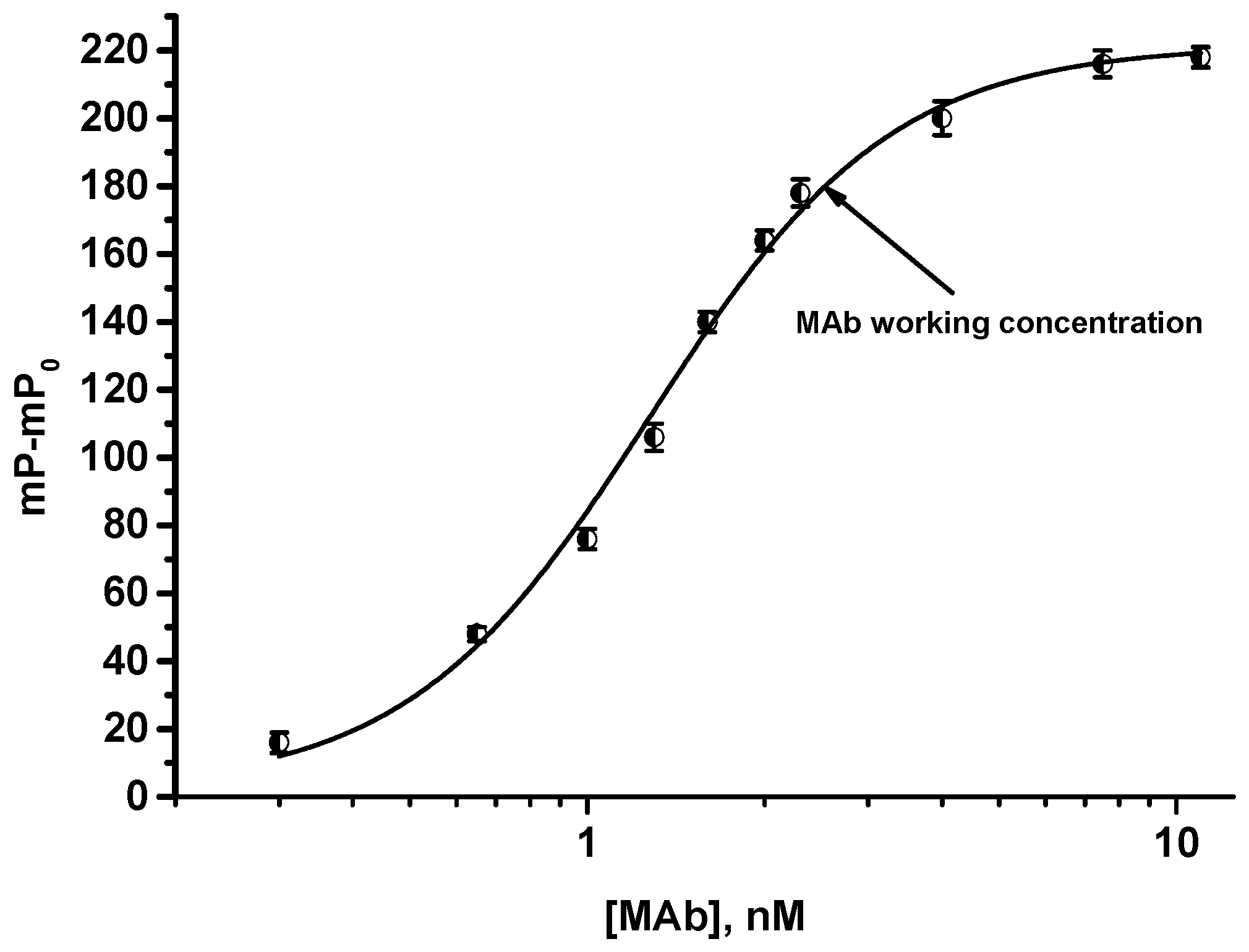
2.6.1. Choice of Tracer and MAbs Working Concentrations
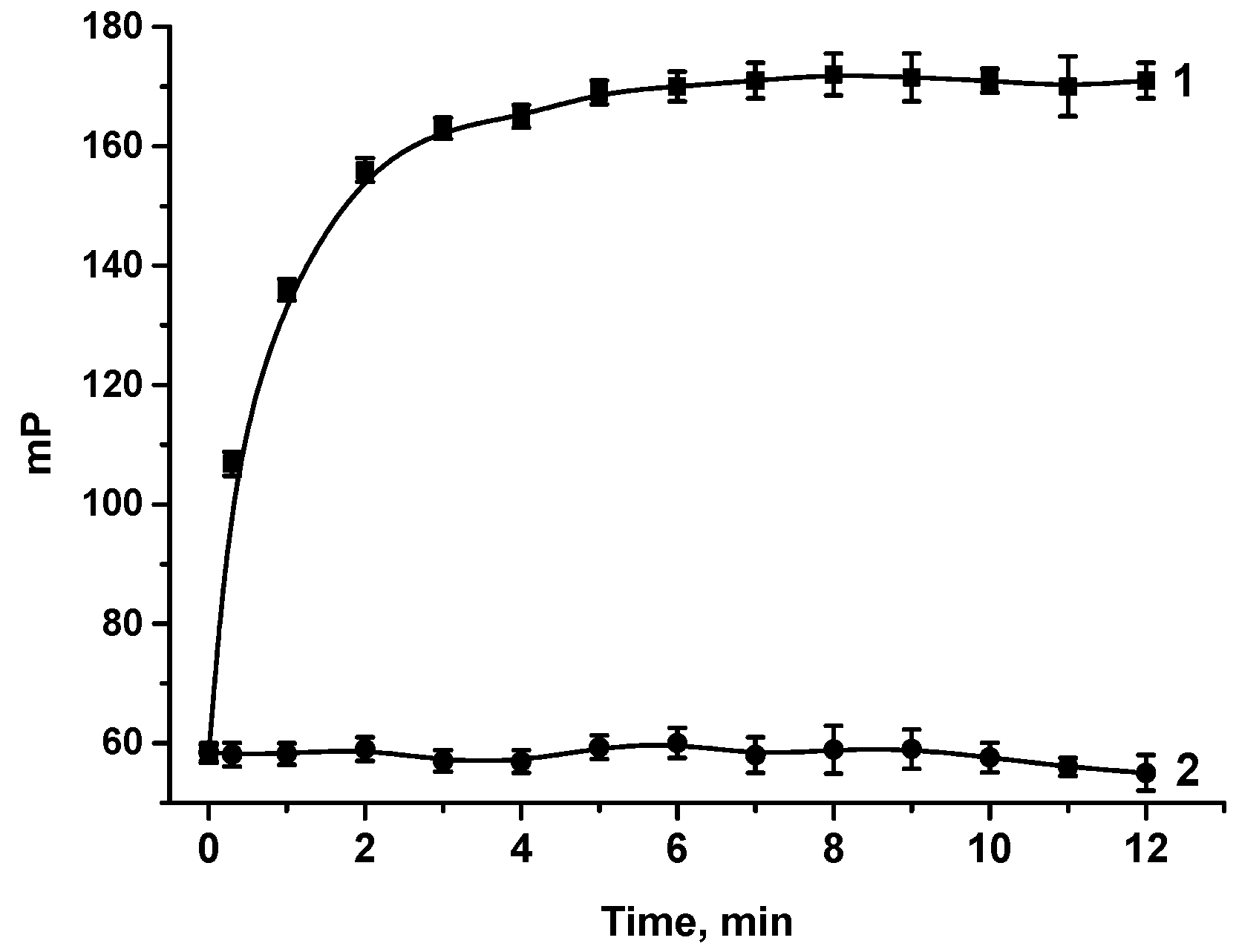
2.6.2. Kinetics of MAbs—Tracer Interaction
2.6.3. Competitive FPIA
2.7. Evaluation of the Assay Results
2.8. Pretreatment of Seawater Samples
3. Results and Discussion
3.1. Obtaining and Characterization of the Specific Reagents
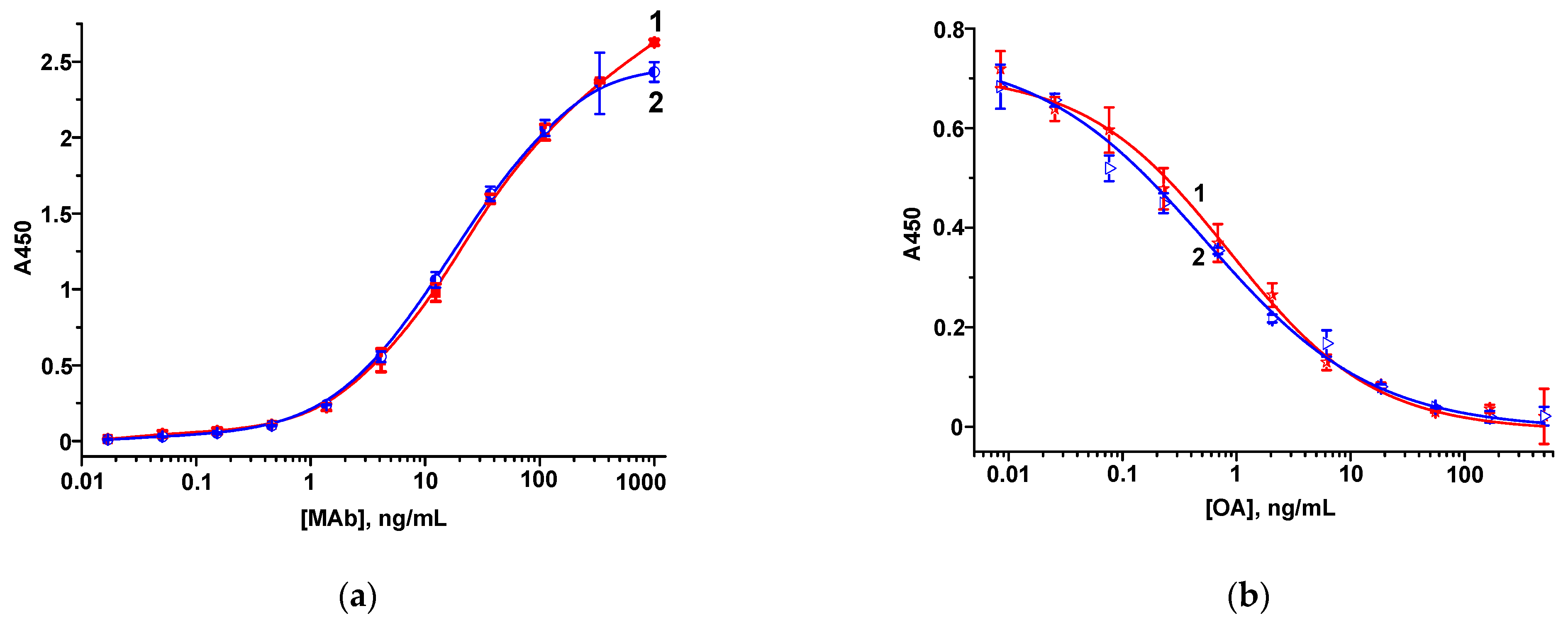
3.1.1. Hapten–Protein Conjugates and MAbs
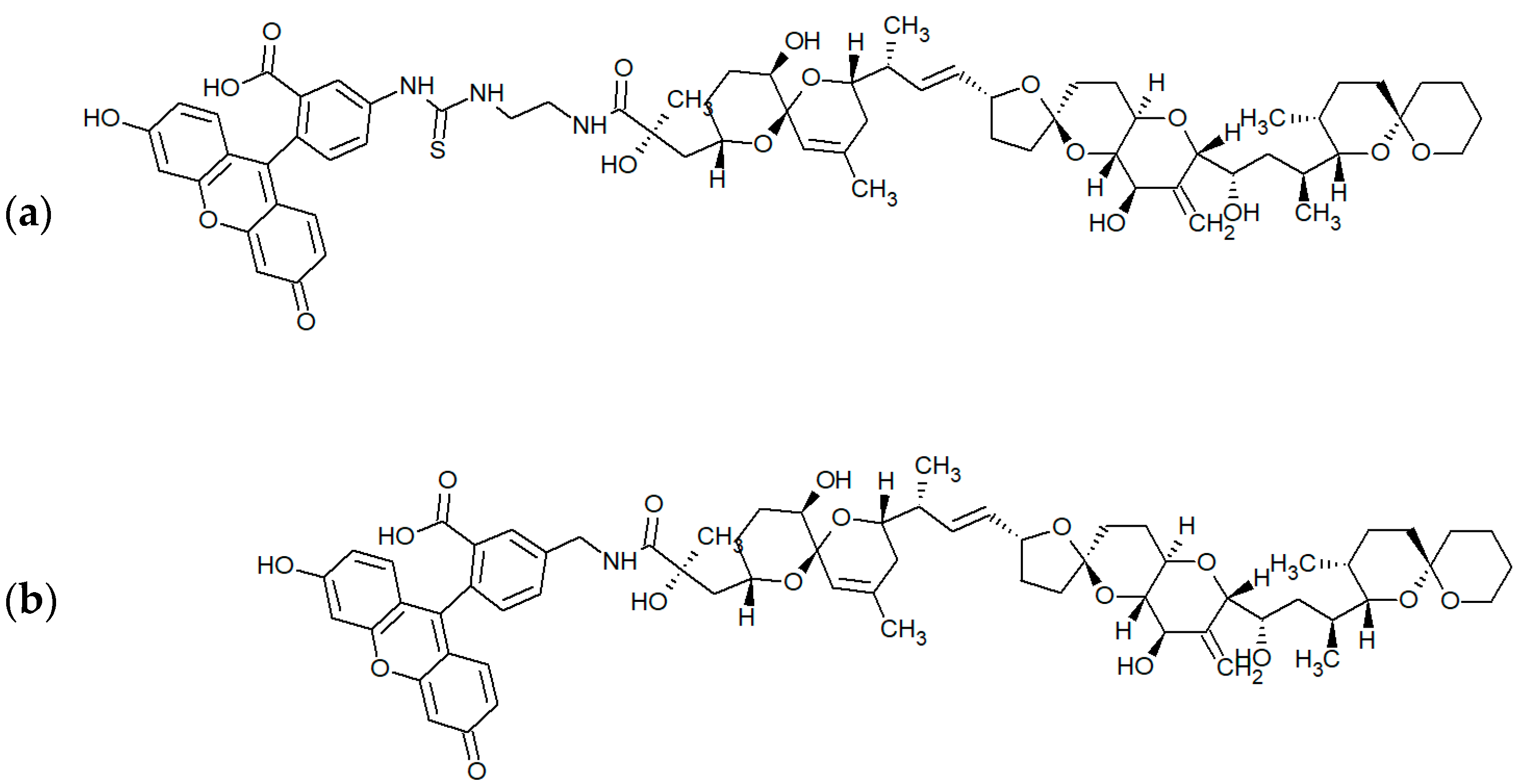
3.1.2. Fluorescent Tracers
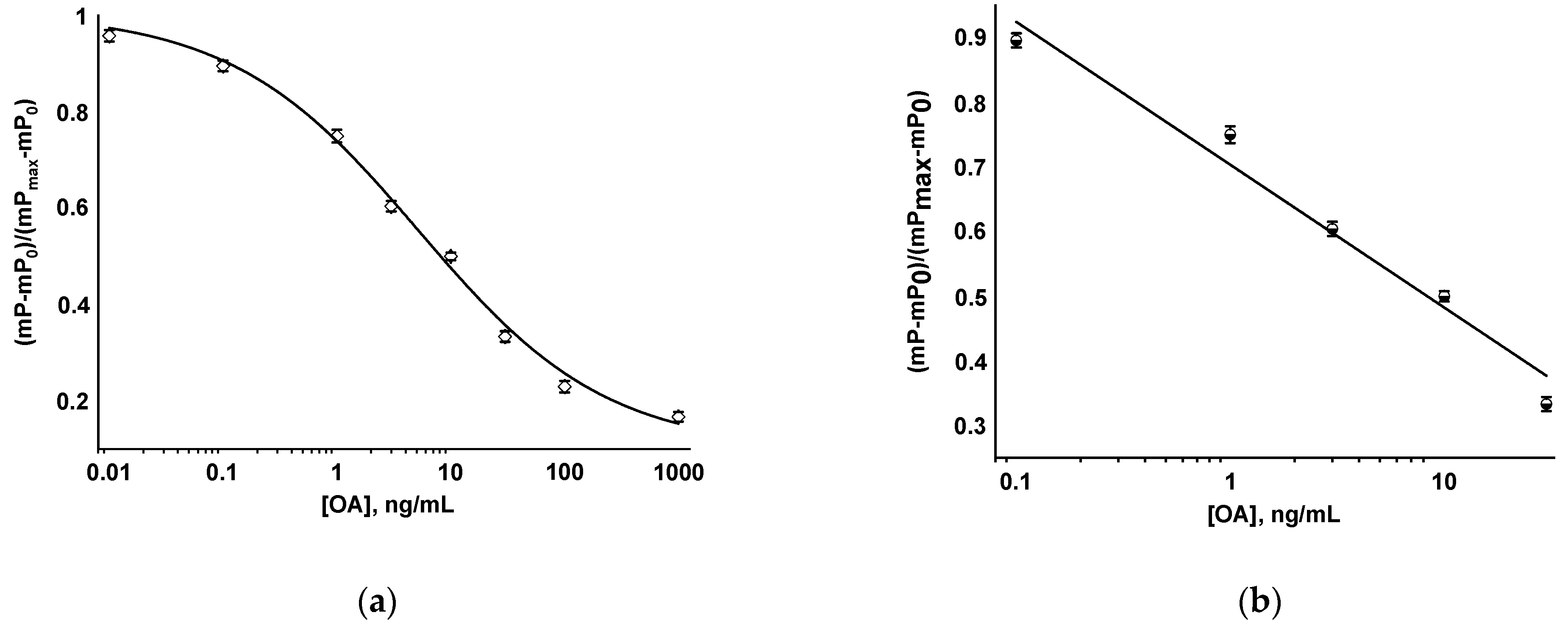
3.2. FPIA of OA
3.3. Study of the Assay Specificity
3.4. Determination of OA in Water
3.5. Comparison of the Obtained Results with Other Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ji, Y.; Gao, K. Effects of climate change factors on marine macroalgae: A review. Adv. Mar. Biol. 2021, 88, 91–136. [Google Scholar]
- Neves, R.A.F.; Nascimento, S.M.; Santos, L.N. Harmful algal blooms and shellfish in the marine environment: An overview of the main molluscan responses, toxin dynamics, and risks for human health. Environ. Sci. Pollut. Res. 2021, 28, 55846–55868. [Google Scholar] [CrossRef] [PubMed]
- Zepernick, B.N.; Wilhelm, S.W.; Bullerjahn, G.S.; Paerl, H.W. Climate change and the aquatic continuum: A cyanobacterial comeback story. Environ. Microbiol. Rep. 2023, 15, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Pires, E.; da Cunha Lana, P.; Laureno Mafra, L., Jr. Phycotoxins and marine annelids—A global review. Harmful Algae 2023, 122, 102373. [Google Scholar] [CrossRef] [PubMed]
- Louzao, M.C.; Vilariño, N.; Vale, C.; Costas, C.; Cao, A.; Raposo-Garcia, S.; Vieytes, M.R.; Botana, L.M. Current trends and new challenges in marine phycotoxins. Mar. Drugs 2022, 20, 198. [Google Scholar] [CrossRef]
- Morabito, S.; Silvestro, S.; Faggio, C. How the marine biotoxins affect human health. Nat. Prod. Res. 2018, 32, 621–631. [Google Scholar] [CrossRef]
- Pradhan, B.; Kim, H.; Abassi, S.; Ki, J.S. Toxic effects and tumor promotion activity of marine phytoplankton toxins: A review. Toxins 2022, 14, 397. [Google Scholar] [CrossRef]
- Casas-Rodriguez, A.; Cameán, A.M.; Jos, A. Potential endocrine disruption of cyanobacterial toxins, microcystins and cylindrospermopsin: A review. Toxins 2022, 14, 882. [Google Scholar] [CrossRef]
- Zhang, S.; Du, X.; Liu, H.; Losiewic, M.D.; Chen, X.; Ma, Y.; Wang, R.; Tian, Z.; Shi, L.; Guo, H.; et al. The latest advances in the reproductive toxicity of microcystin-LR. Environ. Res. 2021, 192, 110254. [Google Scholar] [CrossRef]
- Kameneva, P.A.; Orlova, T.Y. Okadaic acid group toxins in hydrobionts of Russian seas: Producers, distribution, and safety regulation. Russ. J. Mar.Biol. 2017, 43, 331–341. [Google Scholar] [CrossRef]
- Valdiglesias, V.; Prego-Faraldo, M.V.; Pasaro, E.; Mendez, J.; Laffon, B. Okadaic acid: More than a diarrheic toxin. Mar. Drugs 2013, 11, 4328–4349. [Google Scholar] [CrossRef]
- Sassolas, A.; Hayat, A.; Catanante, G.; Marty, J.-L. Detection of the marine toxin okadaic acid: Assessing seafood safety. Talanta 2013, 105, 306–316. [Google Scholar] [CrossRef]
- Alexander, J.; Benford, D.; Boobis, A.; Ceccatelli, S.; Cravedi, J.-P.; Di Domenico, A.; Doerge, D.; Dogliotti, E.; Edler, L.; Farmer, P.; et al. Marine biotoxins in shellfish—Summary on regulated marine biotoxins. Scientific opinion of the panel on contaminants in the food chain in feed and food. EFSA J. 2009, 1306, 1–23. [Google Scholar]
- Prego-Faraldo, M.V.; Valdiglesias, V.; Méndez, J.; Eirín-López, J.M. Okadaic acid meet and greet: An insight into detection methods, response strategies and genotoxic effects in marine invertebrates. Mar. Drugs 2013, 11, 2829–2845. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-Y.; Zhou, Y.; Li, Y.-S.; Lin, C.; Meng, X.-M.; Yan, D.-M.; Li, Z.-H.; Yu, S.-Y.; Liu, Z.-S.; Ren, H.-L. Production of monoclonal antibody and application in indirect competitive ELISA for detecting okadaic acid and dinophytoxin-1 in seafood. Environ. Sci. Pollut. Res. Int. 2011, 19, 2619–2626. [Google Scholar] [CrossRef]
- Pang, L.; Quan, H.; Sun, Y.; Wang, P.; Ma, D.; Mu, P.; Chai, T.; Zhang, Y.; Hammock, B.D. A rapid competitive ELISA assay of okadaic acid level based on epoxy-functionalized magnetic beads. Food Agric. Immunol. 2019, 30, 1286–1302. [Google Scholar] [CrossRef]
- Jawaid, W.; Meneely, J.P.; Campbell, K.; Melville, K.; Holmes, S.J.; Rice, J.; Elliott, C.T. Development and validation of a lateral flow immunoassay for the rapid screening of okadaic acid and all dinophysis toxins from shellfish extracts. J. Agric. Food Chem. 2015, 63, 8574–8583. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Li, X.; Zhang, D.; Wang, K.; Zhao, W.; Zhao, Q.; Wang, R.; Yuan, J.; Xin, S.; Wang, S. Detection of okadaic acid (OA) and tetrodotoxin (TTX) simultaneously in seafood samples using colloidal gold immunoassay. Toxicon 2019, 165, 103–109. [Google Scholar] [CrossRef]
- Hendrickson, O.D.; Zvereva, E.A.; Panferov, V.G.; Solopova, O.N.; Zherdev, A.V.; Sveshnikov, P.G.; Dzantiev, B.B. Application of Au@Pt Nanozyme as enhancing label for the sensitive lateral flow immunoassay of okadaic acid. Biosensors 2022, 12, 1137. [Google Scholar] [CrossRef]
- Hendrickson, O.D.; Zvereva, E.A.; Solopova, O.N.; Zherdev, A.V.; Sveshnikov, P.G.; Eremin, S.A.; Dzantiev, B.B. Double immunochromatographic test system for sensitive detection of phycotoxins domoic acid and okadaic acid in seawater and seafood. Micromachines 2022, 13, 1506. [Google Scholar] [CrossRef]
- Ueda, H.; Dong, J. From fluorescence polarization to Quenchbody: Recent progress in fluorescent reagentless biosensors based on antibody and other binding proteins. Biochim. Biophys. Acta 2014, 1844, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.S.; Eremin, S.A. Fluorescence polarization immunoassays and related methods for simple, high-throughput screening of small molecules. Anal. Bioanal. Chem. 2008, 394, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, O.D.; Taranova, N.A.; Zherdev, A.V.; Dzantiev, B.B.; Eremin, S.A. Fluorescence polarization-based bioassays: New horizons. Sensors 2020, 20, 7132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, S.; De Ruyck, K.; Beloglazova, N.V.; Eremin, S.A.; De Saeger, S.; Zhang, S.; Shen, J.; Wang, Z. Fluorescence polarization assays for chemical contaminants in food and environmental analyses. TrAC Trends Anal. Chem. 2019, 114, 293–313. [Google Scholar] [CrossRef]
- Chen, J.; Liu, J.; Chen, X.; Qiu, H. Recent progress in nanomaterial-enhanced fluorescence polarization/anisotropy sensors. Chin. Chem. Lett. 2019, 30, 1575–1580. [Google Scholar] [CrossRef]
- Adunphatcharaphon, S.; Elliottm, C.T.; Sooksimuang, T.; Charlermroj, R.; Petchkongkaew, A.; Karoonuthaisiri, N. The evolution of multiplex detection of mycotoxins using immunoassay platform technologies. J. Hazard. Mater. 2022, 432, 128706. [Google Scholar] [CrossRef]
- Duan, C.; Zhang, Y.; Li, P.; Li, Q.; Yu, W.; Wen, K.; Eremin, S.A.; Shen, J.; Yu, X.; Wang, Z. Dual-wavelength fluorescence polarization immunoassay for simultaneous detection of sulfonamides and antibacterial synergists in milk. Biosensors 2022, 12, 1053. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, J.; Tao, Z.; Eremin, S.A.; Hua, X.; Wang, M. Development of fluorescence polarization immunoassay for imidacloprid in environmental and agricultural samples. Front. Chem. 2020, 8, 615594. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, S.; Beier, R.C.; Beloglazova, N.V.; Lei, H.; Sun, X.; Ke, Y.; Zhang, S.; Wang, Z. Simple, high efficiency detection of microcystins and Nodularin-R in water by fluorescence polarization immunoassay. Anal. Chim. Acta 2017, 992, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, O.D.; Zherdev, A.V.; Mukhametova, L.I.; Hibara, A.; Tokeshi, M.; Eremin, S.A. Fluorescence polarization immunoassay for microcystin-LR. Public Health Toxicol. 2022, 2, A5. [Google Scholar] [CrossRef]
- Pourfarzaneh, M.; White, G.W.; Landon, J.; Smith, D.S. Cortisol directly determined in serum by fluoroimmunoassay with magnetizable solid phase. Clin. Chem. 1980, 26, 730–733. [Google Scholar] [CrossRef] [PubMed]
- DeRose, P.C.; Kramer, G.W. Bias in the absorption coefficient determination of a fluorescent dye, standard reference material 1932 fluorescein solution. J. Lumin. 2005, 113, 314–320. [Google Scholar] [CrossRef]
- Hendrickson, O.D.; Zvereva, E.A.; Shanin, I.A.; Zherdev, A.V.; Dzantiev, B.B. Development of a multicomponent immunochromatographic test system for the detection of fluoroquinolone and amphenicol antibiotics in dairy products. J. Sci. Food Agric. 2019, 99, 3834–3842. [Google Scholar] [CrossRef]
- Uhrovcik, J. Strategy for determination of LOD and LOQ values—Some basic aspects. Talanta 2014, 119, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Rodbard, D. Statistical estimation of the minimal detectable concentration (“sensitivity”) for radioligand assays. Anal. Biochem. 1978, 90, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, H.; Yang, Q.; Feng, L.; Hua, X.; Wang, M. A fluorescence polarization immunoassay for detection of thiacloprid in environmental and agricultural samples. RSC Adv. 2019, 9, 36825–36830. [Google Scholar] [CrossRef]
- Zhao, Q.; Bai, Y.; Wang, H. Directing a rational design of aptamer-based fluorescence anisotropy assay for sensitive detection of immunoglobulin E by site-specific binding study. Talanta 2020, 217, 121018. [Google Scholar] [CrossRef]
- Hayat, A.; Barthelmebs, L.; Marty, J.-L. Enzyme-linked immunosensor based on super paramagnetic nanobeads for easy and rapid detection of okadaic acid. Anal. Chim. Acta 2011, 690, 248–252. [Google Scholar] [CrossRef]
- Li, X.; Cheng, X.; Xu, R.; Zhang, Z.; Qi, X.; Chen, L.; Zhu, M. A smartphone-assisted microarray immunosensor coupled with GO-based multi-stage signal amplification strategy for high-sensitivity detection of okadaic acid. Talanta 2022, 247, 123567. [Google Scholar] [CrossRef]
- Liang, C.; Ji, Y.; Ma, J.; Zhang, C.; Zhao, H. Development of a highly sensitive and specific monoclonal antibody-based immunoassay for detection of okadaic acid in oysters and green mussels. Food Agric. Immunol. 2022, 33, 346–359. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Wang, L.; Du, X.; Wei, D. Production of a soluble single-chain variable fragment antibody against okadaic acid and exploration of its specific binding. Anal. Biochem. 2016, 503, 21–27. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.-C.; Li, J.; Jiang, W.; Chen, Y.-J.; Song, N.-N. Development of a chemiluminescent ELISA for determining okadaic acid in shellfish. J. Food Qual. 2012, 35, 76–82. [Google Scholar] [CrossRef]
- Tian, Y.; Yuan, L.; Zhang, M.; He, Y.; Lin, X. Sensitive detection of the okadaic acid marine toxin in shellfish by Au@Pt NPs/horseradish peroxidase dual catalysis immunoassay. Anal. Methods 2022, 14, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Laycock, M.V.; Jellett, J.F.; Easy, D.J.; Donovan, M.A. First report of a new rapid assay for diarrhetic shellfish poisoning toxins. Harm. Algae 2006, 5, 74–78. [Google Scholar] [CrossRef]
- Liu, B.-H.; Hung, C.-T.; Lu, C.-C.; Chou, H.-N.; Yu, F.-Y. Production of monoclonal antibody for okadaic acid and its utilization in ultrasensitive enzyme-linked immunosorbent assay and one-step immunochromatographic strip. J. Agric. Food Chem. 2014, 62, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zeng, L.; Yang, H.; Zhong, Y.; Wang, J.; Ling, S.; Saeed, A.f.; Yuan, J.; Wang, S. Detection of Okadaic Acid (OA) using ELISA and colloidal gold immunoassay based on monoclonal antibody. J. Haz. Mater. 2017, 339, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Hongli, Q.; Miao, L.; Liao, G.; Yan, F. Rapid enzyme-linked immunosorbent assay and colloidal gold immunoassay for assessing okadaic acid and its derivatives in shellfish. Food Agric. Immunol. 2022, 33, 677–691. [Google Scholar] [CrossRef]
- Hu, L.; Liu, J.; Wang, Q.; Zhang, Y.; Jia, R.; Cai, C.; Wu, W.; Chen, S.-F.; He, P. Development of an immunochromatographic strip test for the rapid detection of okadaic acid in shellfish sample. Appl. Phycol. 2013, 25, 1091–1099. [Google Scholar] [CrossRef]
- Lu, S.-Y.; Lin, C.; Li, Y.-S.; Zhou, Y.; Meng, X.-M.; Yu, S.-Y.; Li, Z.-H.; Li, L.; Ren, H.-L.; Liu, Z.-S. A screening lateral flow immunochromatographic assay for on-site detection of okadaic acid in shellfish products. Anal. Biochem. 2012, 422, 59–65. [Google Scholar] [CrossRef]
- Zvereva, E.A.; Hendrickson, O.D.; Solopova, O.N.; Zherdev, A.V.; Sveshnikov, P.G.; Dzantiev, B.B. Triple immunochromatographic test system for detection of priority aquatic toxins in water and fish. Anal. Bioanal. Chem. 2022, 414, 7553–7563. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, O.D.; Zvereva, E.A.; Zherdev, A.V.; Dzantiev, B.B. Cascade-enhanced lateral flow immunoassay for sensitive detection of phycotoxin okadaic acid in seawater, fish, and seafood. Foods 2022, 11, 1691. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Water Sample | Sampling Location | Added OA Concentration, ng/mL | Detected OA Concentration, ng/mL | Recovery, % |
|---|---|---|---|---|
| 1 | The Volkhov River | 1 | 0.9 ± 0.1 | 90.0 ± 2.0 |
| 3 | 3.2 ± 0.2 | 107.0 ± 1.5 | ||
| 20 | 21.2 ± 0.2 | 106.0 ± 1.6 | ||
| 2 | The Barents Sea | 1 | 1.1 ± 0.1 | 110.0 ± 2.0 |
| 3 | 3.1 ± 0.2 | 103.0 ± 3.1 | ||
| 20 | 19.5 ± 0.2 | 97.5 ± 1.2 | ||
| 3 | The Mediterranean Sea | 1 | 1.1 ± 0.1 | 112.0 ± 2.0 |
| 3 | 3.1 ± 0.2 | 103.0 ± 1.5 | ||
| 20 | 22.2 ± 0.2 | 111.0 ± 1.6 | ||
| 4 | The Aegean Sea | 1 | 1.2 ± 0.1 | 115.0 ± 2.0 |
| 3 | 2.8 ± 0.2 | 93.3 ± 1.2 | ||
| 20 | 21.6 ± 0.2 | 108.0 ± 1.0 |
| N | Assay Format | Assay Performance | Assay Duration, min | Matrix | Reference |
|---|---|---|---|---|---|
| 1 | ELISA | IC50 = 4.4 ng/mL | n.i. * | Mollusks | [15] |
| 2 | ELISA with magnetic beads | LOD = 0.35 ng/mL | n.i. | Shellfish | [16] |
| 3 | Enzyme-linked immunosensor based on super-paramagnetic nanobeads | LOD = 0.38 ng/mL | 60 | Mussels | [38] |
| 4 | Smartphone-assisted microarray immunosensor based on ELISA | LOD = 0.02 ng/mL | n.i. | Shellfish | [39] |
| 5 | ELISA | IC50 = 0.15 ng/mL | n.i. | Oysters and green mussels | [40] |
| 6 | Chemiluminescent ELISA | LOD = 0.012 ng/mL | n.i. | Buffer | [41] |
| 7 | Chemiluminescent ELISA | LOD = 0.175 ng/g | n.i. | Shellfish | [42] |
| 8 | Enhanced ELISA with nanozymes | LOD = 0.04 ng/mL | n.i. | Oysters, mussels, and clams | [43] |
| 9 | ELISA/ICA | IC50 = 6.4/2.4 ng/mL | n.i./30 | Shellfish | [44] |
| 10 | ELISA/ICA | IC50 = 0.077 ng/mL/ LOD = 5 ng/mL | n.i./10 | Clams, scallops, mussels, and oysters | [45] |
| 11 | ELISA/ICA | LOD = 0.012/0.1 ng/mL | n.i./5 | Shellfish | [46] |
| 12 | ELISA/ICA | LOD = 0.023/5 ng/mL | n.i./10 | Mussels | [47] |
| 13 | ICA | LOD = 0.45 ng/mL | 40 | Shellfish | [48] |
| 14 | ICA | LOD = 50 ng/mL | 10 | Shellfish | [49] |
| 15 | ICA | LOD = 25 µg/kg | 20 | Shellfish | [17] |
| 16 | Double ICA of OA and tetrodotoxin | LOD = 0.75 ng/mL | 10 | Clams | [18] |
| 17 | Double ICA of OA and DA | LOD/cutoff = 0.1/2.5 ng/mL | 18 | Seawater, octopuses, mussels, tiger shrimps, crabs, whelks, and scallops | [20] |
| 18 | Triple ICA of OA, DA, and MC-LR | 0.1/2.0 ng/mL | 18 | Seawater, river water, and fish | [50] |
| 19 | Enhanced ICA with nanozyme | LOD/cutoff = 0.5/10 ng/mL | 20 | Seawater, river water, and fish | [19] |
| 20 | Enhanced with cascade amplification | LOD/cutoff = 0.03/1 ng/mL | 43 | Seawater, fish, tiger shrimps, and scallops | [51] |
| 21 | FPIA | LOD = 0.08 ng/mL | 5 | Environmental waters | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrickson, O.D.; Mukhametova, L.I.; Zvereva, E.A.; Zherdev, A.V.; Eremin, S.A. A Sensitive Fluorescence Polarization Immunoassay for the Rapid Detection of Okadaic Acid in Environmental Waters. Biosensors 2023, 13, 477. https://doi.org/10.3390/bios13040477
Hendrickson OD, Mukhametova LI, Zvereva EA, Zherdev AV, Eremin SA. A Sensitive Fluorescence Polarization Immunoassay for the Rapid Detection of Okadaic Acid in Environmental Waters. Biosensors. 2023; 13(4):477. https://doi.org/10.3390/bios13040477
Chicago/Turabian StyleHendrickson, Olga D., Liliya I. Mukhametova, Elena A. Zvereva, Anatoly V. Zherdev, and Sergei A. Eremin. 2023. "A Sensitive Fluorescence Polarization Immunoassay for the Rapid Detection of Okadaic Acid in Environmental Waters" Biosensors 13, no. 4: 477. https://doi.org/10.3390/bios13040477
APA StyleHendrickson, O. D., Mukhametova, L. I., Zvereva, E. A., Zherdev, A. V., & Eremin, S. A. (2023). A Sensitive Fluorescence Polarization Immunoassay for the Rapid Detection of Okadaic Acid in Environmental Waters. Biosensors, 13(4), 477. https://doi.org/10.3390/bios13040477