Fluholoscopy—Compact and Simple Platform Combining Fluorescence and Holographic Microscopy


Abstract
:1. Introduction
2. Materials and Methods
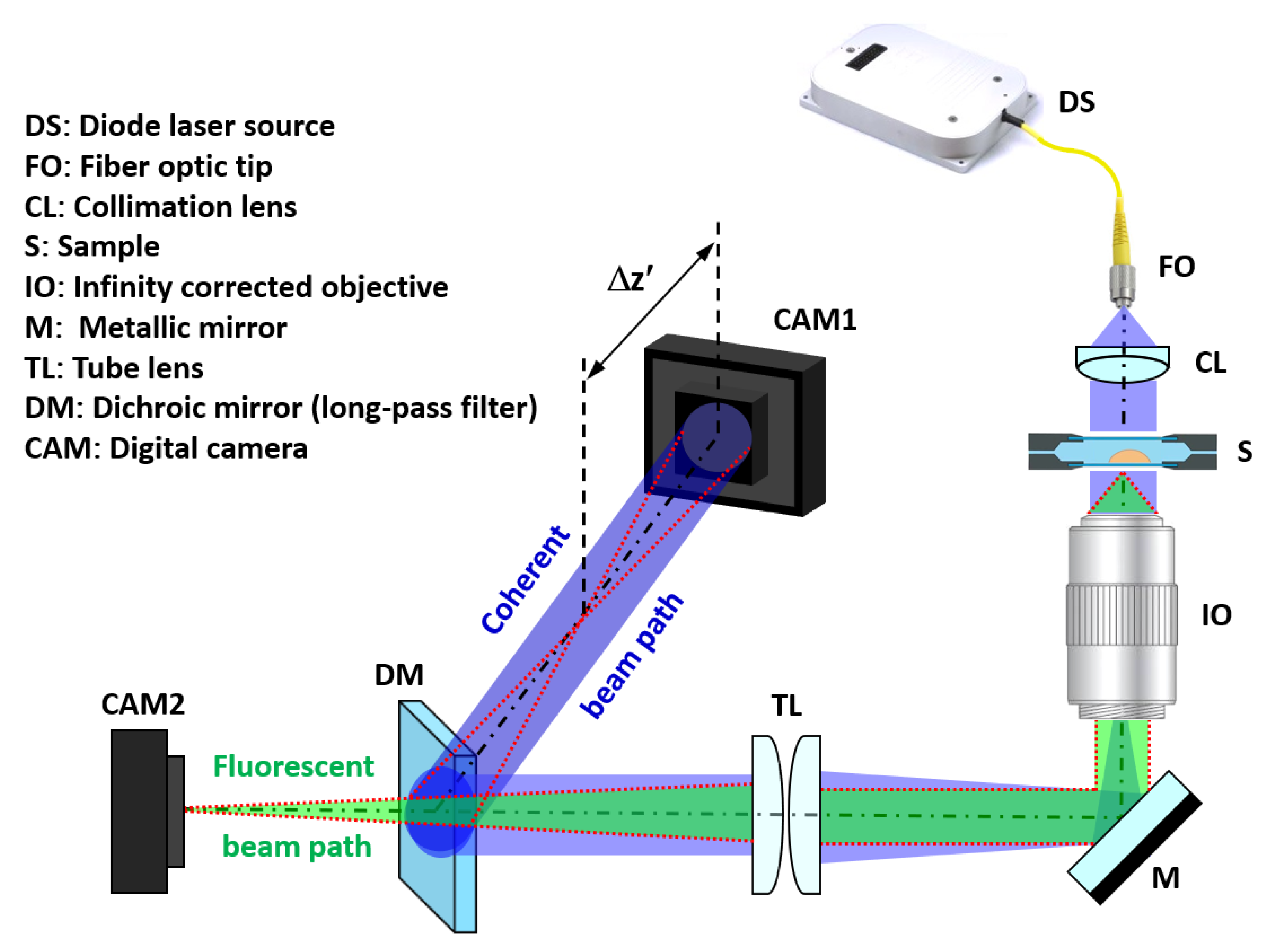
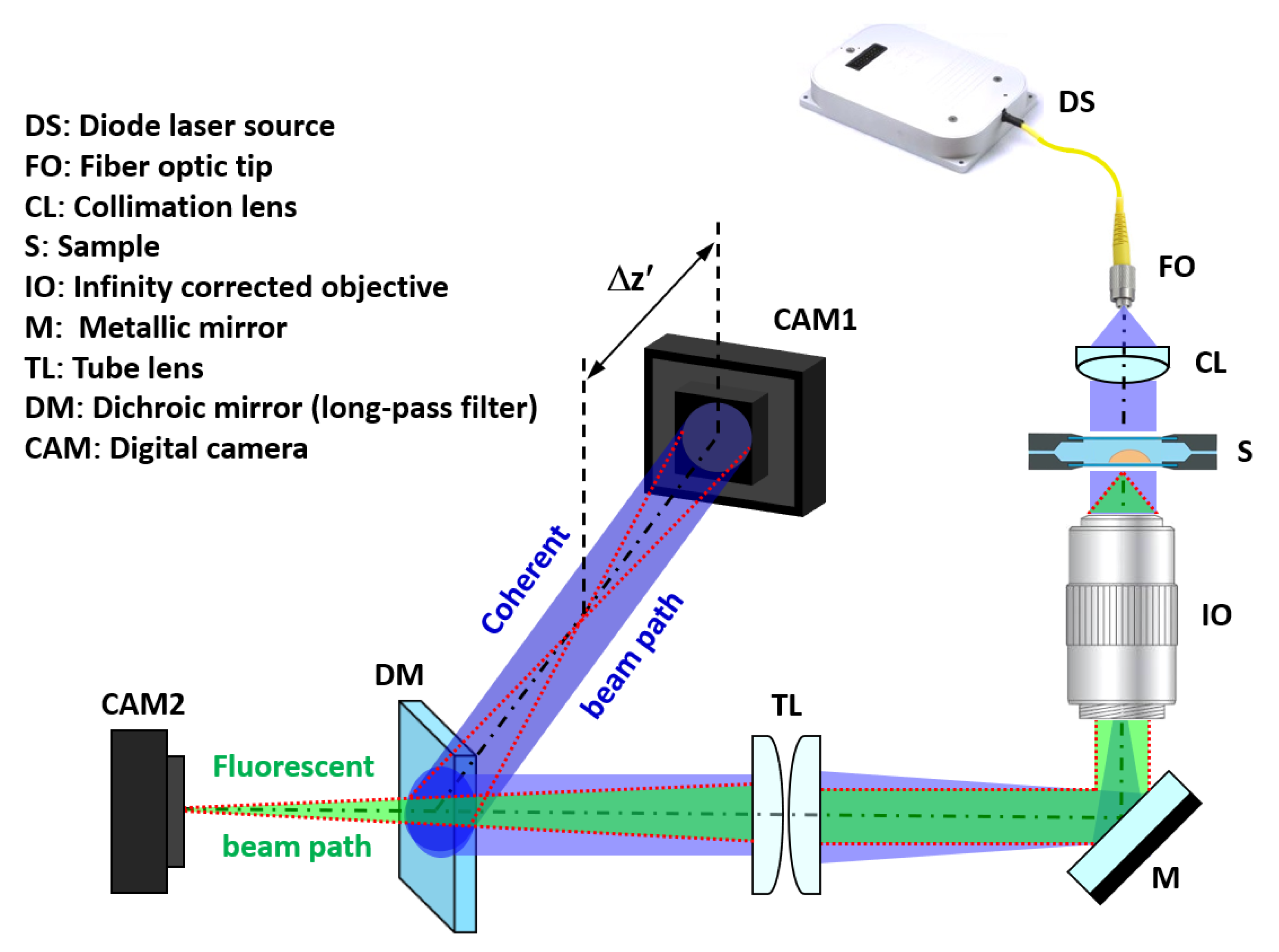
2.1. Experimental Setup and Numeric Propagation
2.2. Calibration Targets and Imaging of the Samples
2.3. Preparation of Fluorescence Micro-Beads and Living Samples
3. Results
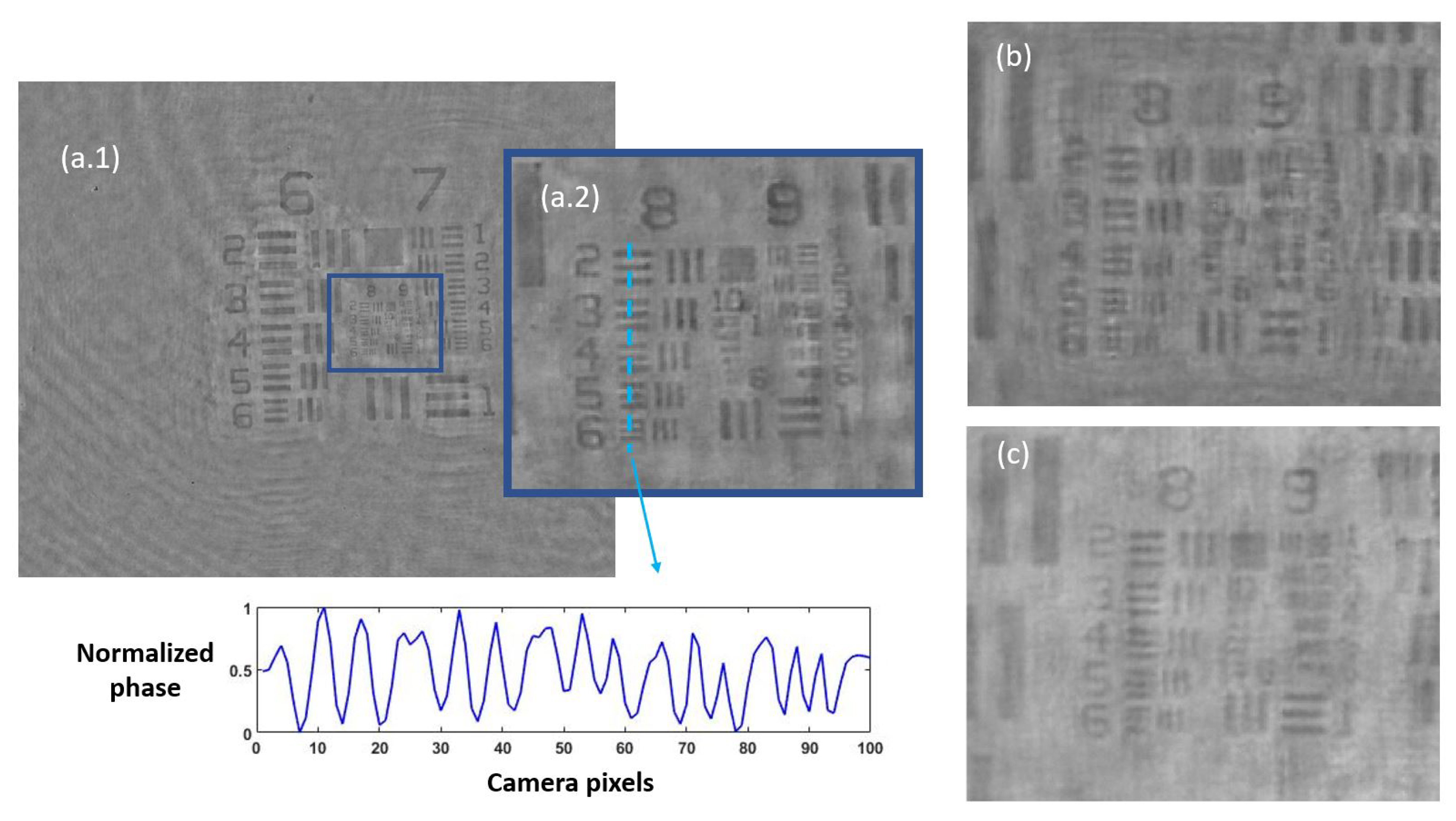
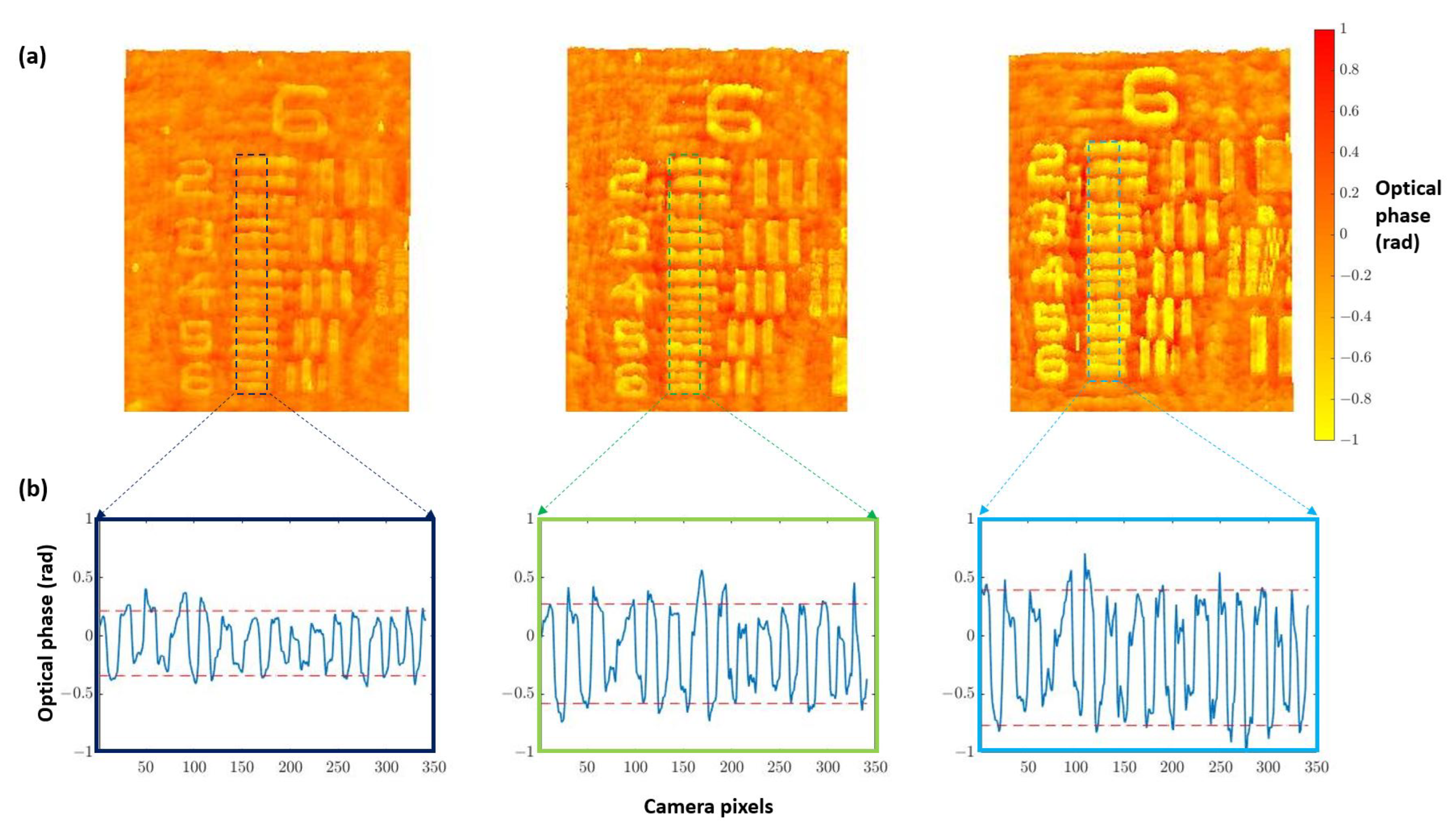
3.1. Phase Imaging Calibration
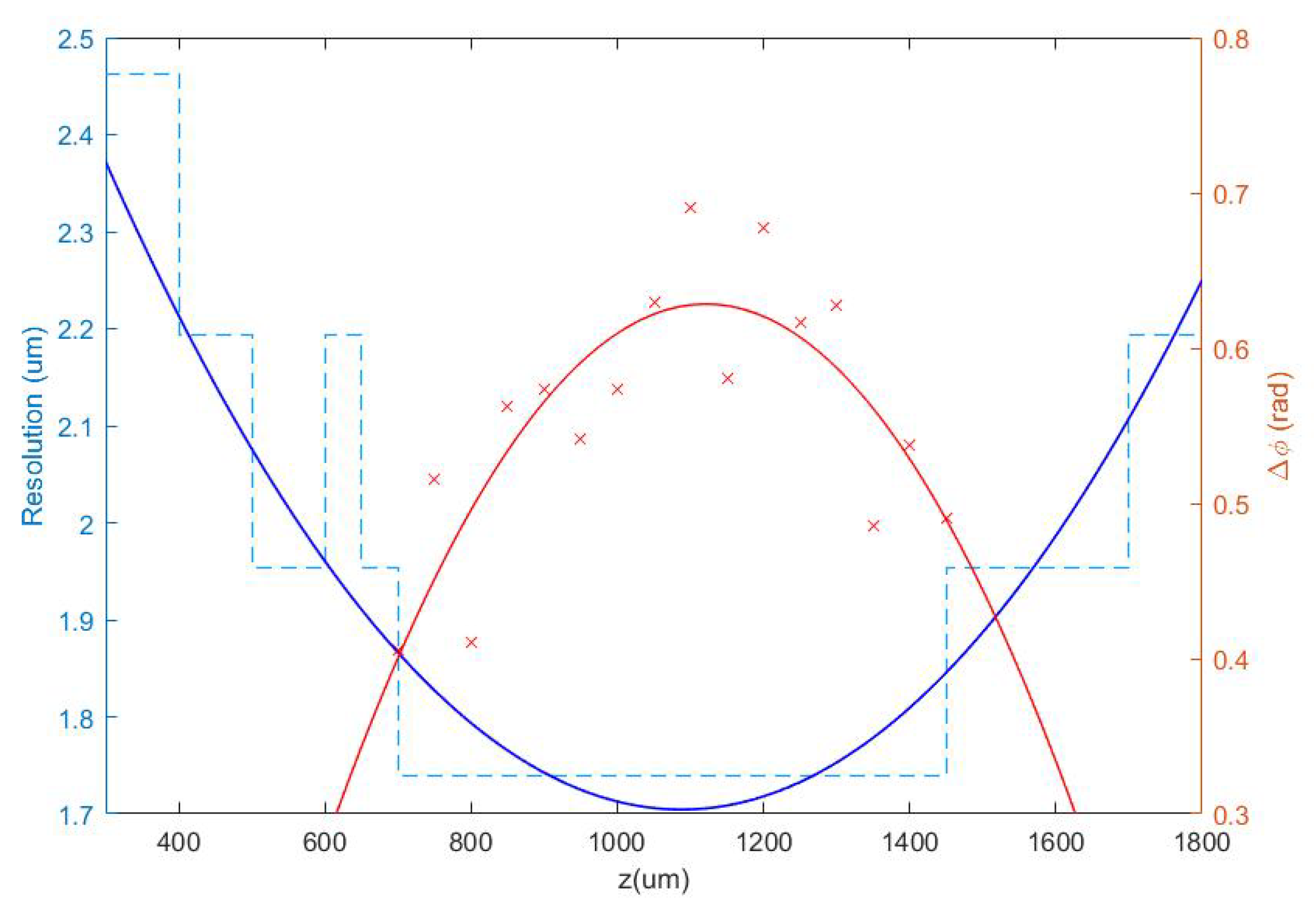
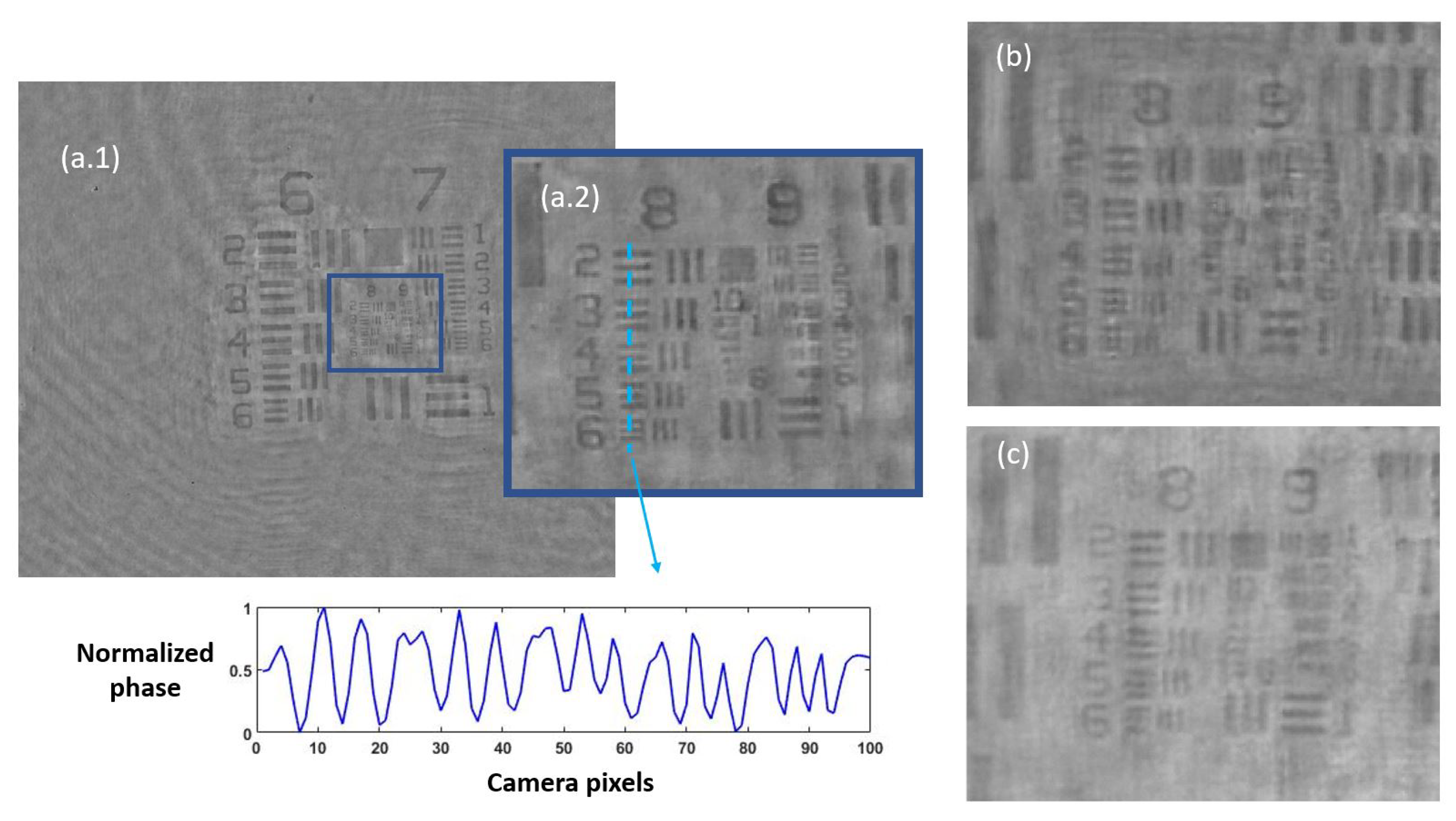
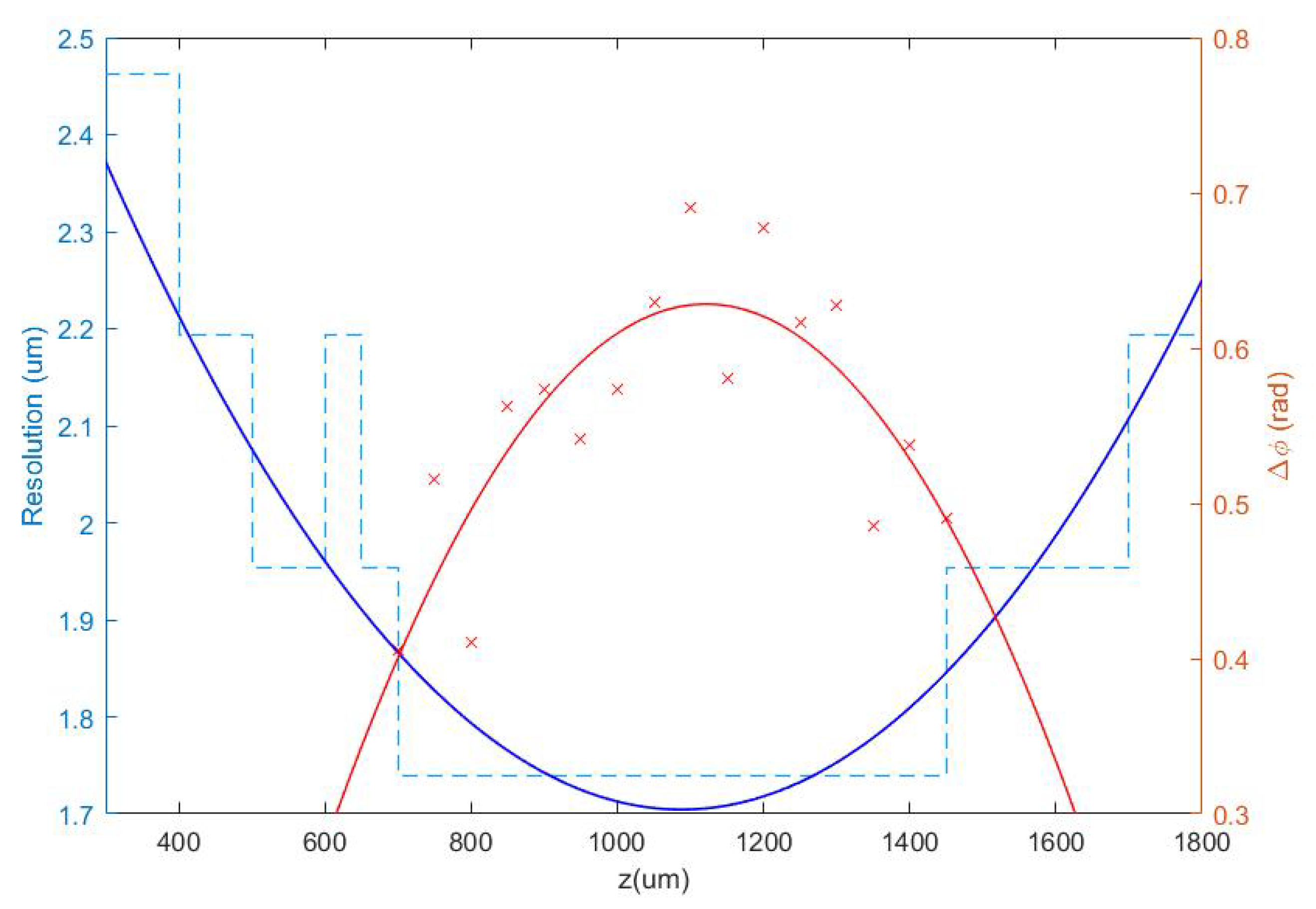
3.1.1. Defocus Distance
3.1.2. Quantitative Validation
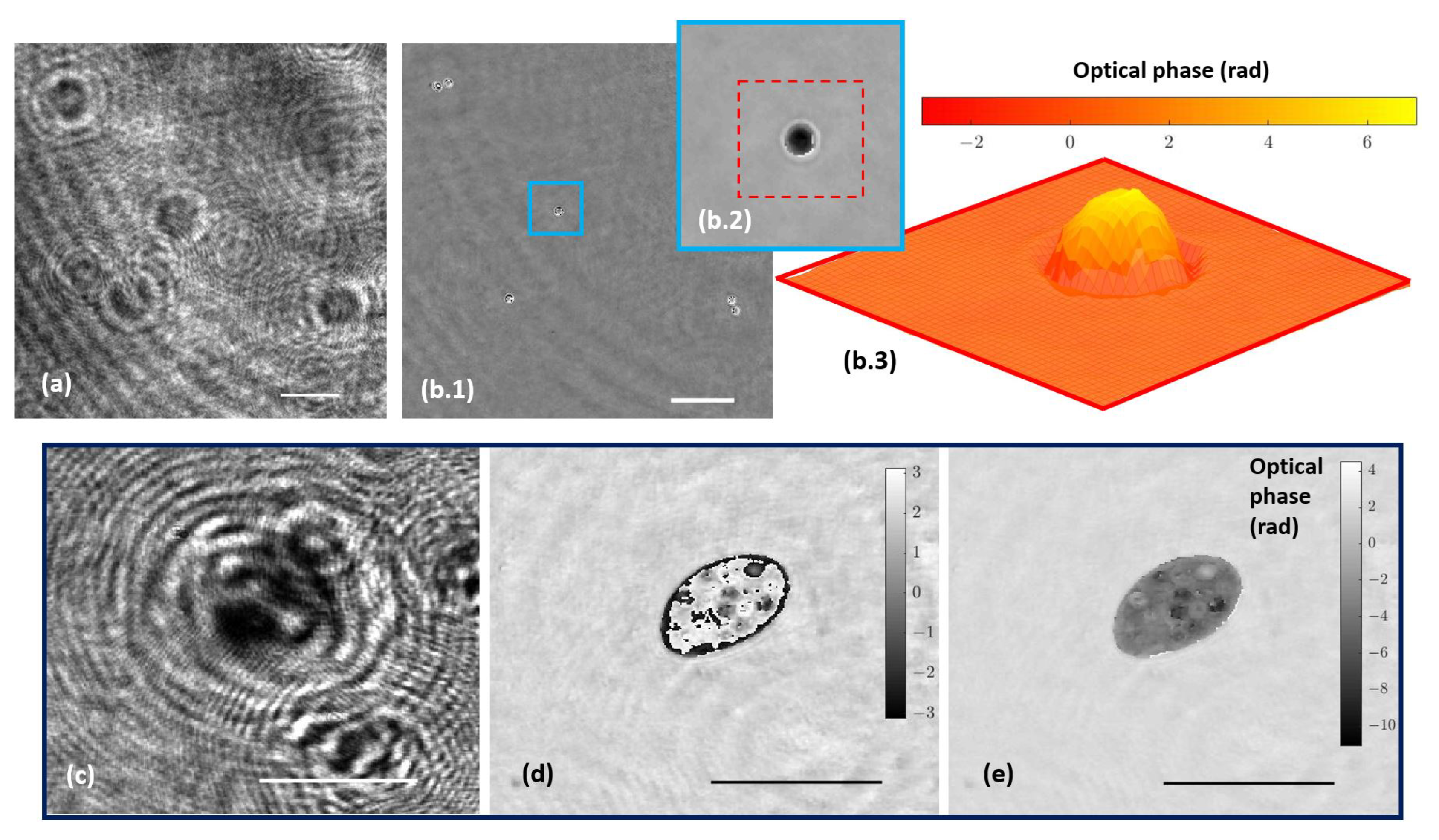
3.2. Fluorescence Imaging Calibration
3.3. Combined Station
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FI | Fluorescence imaging |
| QPI | Quantitative phase imaging |
| NA | Numerical aperture |
| FOV | Field of view |
| USAF | United State Air Force |
| CAM | Camera |
References
- Vogler, N.; Heuke, S.; Bocklitz, T.W.; Schmitt, M.; Popp, J. Multimodal imaging spectroscopy of tissue. Annu. Rev. Anal. Chem. 2015, 8, 359–387. [Google Scholar] [CrossRef] [PubMed]
- Jena, B.P.; Taatjes, D.J. NanoCellBiology: Multimodal Imaging in Biology and Medicine, 1st ed.; Pan Stanford Publishing: Singapore, 2014. [Google Scholar]
- Zhou, Q.; Chen, Z. Multimodality Imaging: For Intravascular Application, 1st ed.; Springer: New York, NY, USA, 2020. [Google Scholar]
- Sen, H.N.; Read, R.W. Multimodal Imaging in Uveitis, 1st ed.; Springer: New York, NY, USA, 2018. [Google Scholar]
- Suresh, A.; Udendran, R.; Vimal, S. Deep Neural Networks for Multimodal Imaging and Biomedical Applications, 1st ed.; IGI Global: Pensilvania, PA, USA, 2020. [Google Scholar]
- Baz, A.E.; Suri, J.S. Big Data in Multimodal Medical Imaging, 1st ed.; Chapman & Hall: London, UK, 2020. [Google Scholar]
- Mazumder, N.; Balla, N.K.; Zhuo, G.-Y.; Kistenev, Y.V.; Kumar, R.; Kao, F.-J.; Brasselet, S.; Nikolaev, V.V.; Krivova, N.A. Label-Free Non-linear Multimodal Optical Microscopy—Basics, Development, and Applications. Front. Phys. 2019, 7, 170. [Google Scholar] [CrossRef]
- Yu, Z.G.; Spandana, K.U.; Sindhoora, K.M.; Kistenev, Y.V.; Kao, F.-J.; Nikolaev, V.V.; Zuhayri, H.; Krivova, A.N.; Mazumber, N. Label-free multimodal nonlinear optical microscopy for biomedical applications. J. Appl. Phys. 2021, 129, 214901. [Google Scholar]
- Peres, C.; Nardin, C.; Yang, G.; Mammano, F. Commercially derived versatile optical architecture for two-photon STED, wavelength mixing and label-free microscopy. Biomed. Opt. Express 2022, 13, 1410–1429. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.D.; Clemente, P.; Tajahuerce, E.; Lancis, J. Dual-mode optical microscope based on single-pixel imaging. Opt. Lasers. Eng. 2016, 82, 87–94. [Google Scholar] [CrossRef]
- Picazo-Bueno, J.A.; Cojoc, D.; Iseppon, F.; Torre, V.; Micó, V. Single-shot, dual-mode, water-immersion microscopy platform for biological applications. Appl. Opt. 2018, 57, A242–A249. [Google Scholar] [CrossRef]
- Yeh, L.-H.; Chowdhury, S.; Repina, N.A.; Waller, L. Speckle-structured illumination for 3D phase and fluorescence computational microscopy. Opt. Express. 2019, 10, 3635–3653. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, B.; Choi, S. On-Chip Cell Staining and Counting Platform for the Rapid Detection of Blood Cells in Cerebrospinal Fluid. Sensors 2018, 18, 1124. [Google Scholar] [CrossRef]
- Pavillon, N.; Fujita, K.; Smith, N.I. Multimodal label-free microscopy. J. Innov. Opt. Health Sci. 2014, 7, 1330009. [Google Scholar] [CrossRef]
- Krafft, C.; Schmitt, M.; Schie, I.W.; Cialla, D.-M.; Matthaus, C.; Bocklitz, T. Label-Free Molecular Imaging of Biological Cells and Tissues by Linear and Nonlinear Raman Spectroscopic Approaches. Angew. Chem. Int. Ed. 2017, 56, 4392–4430. [Google Scholar] [CrossRef]
- Ryu, J.; Kang, U.; Song, J.W.; Kim, J.; Kim, J.M.; Yoo, H.; Gweon, B. Multimodal microscopy for the simultaneous visualization of five different imaging modalities using a single light source. Biomed. Opt. Express 2021, 12, 5452–5469. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Popescu, G.; Badizadegan, K.; Dasari, R.R.; Feld, M.S. Diffraction phase and fluorescence microscopy. Opt. Express 2006, 14, 8263–8268. [Google Scholar] [CrossRef] [PubMed]
- Yelleswarapu, C.S.; Tipping, M.; Kothapalli, S.R.; Veraksa, A.; Rao, D.V.G.L.N. Common-path multimodal optical microscopy. Opt. Lett. 2009, 34, 1243–1245. [Google Scholar] [CrossRef]
- Quan, X.; Nitta, K.; Matoba, O.; Xia, P.; Awatsuji, Y. Phase and fluorescence imaging by combination of digital holographic microscopy and fluorescence microscopy. Opt. Lett. 2015, 22, 349–353. [Google Scholar] [CrossRef]
- Chowdhury, S.; Eldridge, W.J.; Wax, A.; Izatt, J.A. Spatial frequency-domain multiplexed microscopy for simultaneous, single-camera, one-shot, fluorescent, and quantitative-phase imaging. Opt. Lett. 2015, 40, 4839–4842. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Eldridge, W.J.; Wax, A.; Izatt, J.A. Structured illumination multimodal 3D-resolved quantitative phase and fluorescence sub-diffraction microscopy. Biomed. Opt. Express 2017, 8, 2496–2518. [Google Scholar] [CrossRef]
- Shen, Z.; He, Y.; Zhang, G.; He, Q.; Li, D.; Ji, Y. Dual-wavelength digital holographic phase and fluorescence microscopy for an optical thickness encoded suspension array. Opt. Lett. 2018, 43, 739–742. [Google Scholar] [CrossRef]
- Dubey, V.; Ahmad, A.; Singh, R.; Wolfson, D.L.; Basnet, P.; Acharya, G.; Mehta, D.S.; Ahluwalia, B.S. Multi-modal chip-based fluorescence and quantitative phase microscopy for studying inflammation in macrophages. Opt. Express 2018, 26, 19864–19876. [Google Scholar] [CrossRef]
- Nygate, Y.N.; Singh, G.; Barnea, I.; Shaked, N.T. Simultaneous off-axis multiplexed holography and regular fluorescence microscopy of biological cells. Opt. Lett. 2018, 43, 2587–2590. [Google Scholar] [CrossRef]
- Kernier, I.D.; Cherif, A.A.; Rongeat, N.; Cioni, O.; Morales, S.; Savatier, J.; Monneret, S.; Blandin, P. Large field-of-view phase and fluorescence mesoscope with microscopic resolution. J. Biomed. Opt. 2019, 24, 036501. [Google Scholar] [CrossRef]
- Mandula, O.; Kleman, J.P.; Lacroix, F.; Allier, C.; Fiole, D.; Hervé, L.; Blandin, P.; Kraemer, D.C.; Morales, S. Phase and fluorescence imaging with a surprisingly simple microscope based on chromatic aberration. Opt. Express 2020, 28, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Quan, X.; Awatsuji, Y.; Tamada, Y.; Matoba, O. Digital holographic multimodal cross-sectional fluorescence and quantitative phase imaging system. Sci. Rep. 2020, 10, 7580. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.; Gao, Z.; Fang, X.; Liu, M.; Zheng, J.; Ma, M.; Zalevsky, Z.; Gao, P. Structured illumination microscopy with partially coherent illumination for phase and fluorescent imaging. Opt. Express 2021, 29, 33679–33693. [Google Scholar] [CrossRef] [PubMed]
- Quan, X.; Kumar, M.; Rajput, S.K.; Tamada, Y.; Awatsuji, Y.; Matoba, O. Multimodal microscopy: Fast acquisition of quantitative phase and fluorescence imaging in 3D space. IEEE J. Sel. Topics Quantum. Electron. 2021, 27, 6800911. [Google Scholar] [CrossRef]
- Liu, P.Y.; Chin, L.K.; Ser, W.; Chen, H.F.; Hsieh, C.M.; Lee, C.H.; Sung, K.B.; Ayi, T.C.; Yap, P.H.; Liedberg, B.; et al. Cell refractive index for cell biology and disease diagnosis: Past, present and future. Lab Chip 2016, 16, 634–644. [Google Scholar] [CrossRef]
- Lichtman, J.W.; Conchello, J.-A. Fluorescence microscopy. Nat. Methods 2005, 2, 910–919. [Google Scholar] [CrossRef]
- Micó, V.; Trindade, T.; Picazo-Bueno, J.A. Phase imaging microscopy under the Gabor regime in a minimally modified regular bright-field microscope. Opt. Express 2021, 29, 42738–42750. [Google Scholar] [CrossRef]
- Trusiak, M.; Cywińska, M.; Micó, V.; Picazo-Bueno, J.Á.; Zuo, C.; Zdańkowski, P.; Patorski, K. Variational Hilbert quantitative phase imaging. Sci. Rep. 2020, 10, 13955. [Google Scholar] [CrossRef]
- Zhai, J.; Shi, R.; Kong, L. Improving signal-to-background ratio by orders of magnitude in high-speed volumetric imaging in vivo by robust Fourier light field microscopy. Photon. Res. 2022, 10, 1255–1263. [Google Scholar] [CrossRef]
- Perucho, B.; Micó, V. Wavefront holoscopy: Application of digital in-line holography for the inspection of engraved marks in progressive addition lenses. J. Biomed. Opt. 2014, 19, 016017. [Google Scholar] [CrossRef]
- Sanz, M.; Picazo-Bueno, J.A.; García, J.; Micó, V. Dual mode holographic microscopy imaging platform. Lab Chip 2018, 18, 1105–1112. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nominal Values (nm) | Measured Values (nm) | Retrieved Phase Steps (Rad) | Retrieved Thickness Values (nm) |
|---|---|---|---|
| 50 | 59.1 | 0.56 ± 0.11 | 77 ± 15 |
| 100 | 114.4 | 0.84 ± 0.17 | 120 ± 20 |
| 150 | 173.3 | 1.16 ± 0.16 | 160 ± 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso, D.; Garcia, J.; Micó, V. Fluholoscopy—Compact and Simple Platform Combining Fluorescence and Holographic Microscopy. Biosensors 2023, 13, 253. https://doi.org/10.3390/bios13020253
Alonso D, Garcia J, Micó V. Fluholoscopy—Compact and Simple Platform Combining Fluorescence and Holographic Microscopy. Biosensors. 2023; 13(2):253. https://doi.org/10.3390/bios13020253
Chicago/Turabian StyleAlonso, David, Javier Garcia, and Vicente Micó. 2023. "Fluholoscopy—Compact and Simple Platform Combining Fluorescence and Holographic Microscopy" Biosensors 13, no. 2: 253. https://doi.org/10.3390/bios13020253
APA StyleAlonso, D., Garcia, J., & Micó, V. (2023). Fluholoscopy—Compact and Simple Platform Combining Fluorescence and Holographic Microscopy. Biosensors, 13(2), 253. https://doi.org/10.3390/bios13020253