A Multipurpose and Multilayered Microneedle Sensor for Redox Potential Monitoring in Diverse Food Analysis




Abstract
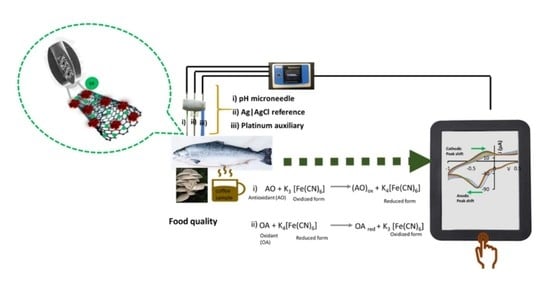
1. Introduction
2. Experimental Section
2.1. Materials and Reagents
2.2. Fabrication of the MN Redox Sensor
2.3. Instrumentation
2.4. Electrochemical Characterization of the MN Redox Sensor
2.5. Voltammetric Quantification of Oxidant and Antioxidant Compounds Using the MN Redox Sensor
2.6. Antioxidant and ORP Quantification and Fish Spoilage Analysis Using the MN Redox Sensor
3. Results and Discussion
3.1. Morphological and Electrochemical Characterization of MN Redox Sensor

3.2. Electrochemical Analysis of Oxidant, Antioxidant, and Polyamine Compounds Using MN Redox Sensor


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | PANI@CNT/CNC MN Capacitive Sensitivity (µF·mL/ng) | MN Redox Sensor Capacitive Sensitivity (µF·mL/ng) | PANI@CNT/CNC MN ORP Sensitivity (V·mL/ng) | MN Redox Sensor ORP Sensitivity (V·mL/ng) |
|---|---|---|---|---|
| Ascorbic Acid | 3.98 × 10−4 | 9.66 × 10−4 | −2.89 × 10−5 | −7.12 × 10−5 |
| H2O2 | 1.85 × 10−3 | 2.87 × 10−3 | −1.76 × 10−4 | −3.12 × 10−4 |
| Putrescine | 5.81 × 10−4 | 2.87 × 10−3 | −6.19 × 10−5 | 2.01 × 10−4 |
| Sensing Method | Analyte | Concentration Range (ng/mL) | LOD (ng/mL) | Reference |
|---|---|---|---|---|
| 3-D nitrogen-doped graphene-modified GCE | Ascorbic Acid | 3.52 × 103–1.76 × 106 | 689 | [33] |
| K4Fe(CN)6-doped Ppy-modified platinum electrode | Ascorbic Acid | 176–1.76 × 104 | 44.0 | [34] |
| K3[Fe(CN)6]/K4[Fe(CN)6]-infused PANI@CNT/CNC MN electrode | Ascorbic Acid | 180–1.78 × 103 (Capacitive) 180–1.43 × 103 ascorbic acid equivalents (ORP) | 49.9 (Capacitive) 2. 75 × 103 ascorbic acid equivalents (ORP) | This work |
| Screen-printed carbon electrode modified with a carboxylated triazole copper complex | H2O2 | 340–1.78 × 104 | 19.4 | [35] |
| Copper oxide/graphitic carbon nitride-modified GCE | H2O2 | 17.0–1.70 × 103 | 10.5 | [36] |
| K3[Fe(CN)6]/K4[Fe(CN)6]-infused PANI@CNT/CNC MN electrode | H2O2 | 49.8–476 (Capacitive) 49.8–431 H2O2 equivalents (ORP) | 14.3 (Capacitive) 55.4 H2O2 equivalents (ORP) | This work |
| K4Fe(CN)6-doped Ppy-modified screen-printed carbon electrode | Putrescine | 88.2–8.82 × 103 | 30.0 | [37] |
| Diamine oxidase, Prussian blue, and indium tin oxide nanoparticle-modified screen-printed carbon electrode | Putrescine | 696–2.64 × 105 | 688 | [38] |
| K3[Fe(CN)6]/K4[Fe(CN)6]-infused PANI@CNT/CNC MN electrode | Putrescine | 88.0–872 (Capacitive) 88.0–872 putrescine equivalents (ORP) | 263 (Capacitive) 25.8 putrescine equivalents (ORP) | This work |
| Nano-LC-ESI-MS and HPLC-UV-Vis | Anthocyanins | 6.00 × 103–5.00 × 104 (Nano-LC-ESI-MS) 100–5.00 × 104 (HPLC-UV-Vis) | 6.00 × 103 (Nano-LC-ESI-MS) 30.0 (HPLC-UV-Vis) | [15] |
| HPLC-DAD | Tea antioxidants | Several | 60.0–910 | [13] |
| Modified GC-MS/MS method | Phenolic antioxidants | 0.100–1.00 | 0.00814–0.0255 | [16] |
3.3. Determination of Antioxidant Content in King Mushroom and Brewed Coffee Samples
3.4. Electrochemical Monitoring of Fish Spoilage
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pottosin, I.; Velarde-Buendía, A.M.; Bose, J.; Zepeda-Jazo, I.; Shabala, S.; Dobrovinskaya, O. Cross-Talk between Reactive Oxygen Species and Polyamines in Regulation of Ion Transport across the Plasma Membrane: Implications for Plant Adaptive Responses. J. Exp. Bot. 2014, 65, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Aran, K.; Parades, J.; Rafi, M.; Yau, J.F.; Acharya, A.P.; Zibinsky, M.; Liepmann, D.; Murthy, N. Stimuli-Responsive Electrodes Detect Oxidative Stress and Liver Injury. Adv. Mater. 2015, 27, 1433–1436. [Google Scholar] [CrossRef]
- Sen, C.K.; Roy, S. Redox Signals in Wound Healing. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Brainina, K.Z.; Galperin, L.G.; Gerasimova, E.L.; Khodos, M.Y. Noninvasive Potentiometric Method of Determination of Skin Oxidant/Antioxidant Activity. IEEE Sens. J. 2012, 12, 527–532. [Google Scholar] [CrossRef]
- Brainina, K.Z.; Zaharov, A.S.; Vidrevich, M.B. Potentiometry for the Determination of Oxidant Activity. Anal. Methods 2016, 8, 5667–5675. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free Radicals, Antioxidants and Functional Foods: Impact on Human Health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Flint, R.J.; Fanous, M.; Delahunt, B.; Kilmartin, P.A.; Cooper, G.J.S.; Windsor, J.A.; Phillips, A.R.J. Redox Status of Acute Pancreatitis as Measured by Cyclic Voltammetry: Initial Rodent Studies to Assess Disease Severity. Crit. Care Med. 2008, 36, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.A.; Crapo, J.D. Biology of Disease. Free Radicals and Tissue Injury. Lab. Investig. 1982, 47, 412–426. [Google Scholar]
- Haslam, E.; Cai, Y. Plant Polyphenols (Vegetable Tannins): Gallic Acid Metabolism. Nat. Prod. Rep. 1994, 11, 41–66. [Google Scholar] [CrossRef]
- Muñoz-Esparza, N.C.; Latorre-Moratalla, M.L.; Comas-Basté, O.; Toro-Funes, N.; Veciana-Nogués, M.T.; Vidal-Carou, M.C. Polyamines in Food. Front. Nutr. 2019, 6, 108. [Google Scholar] [CrossRef]
- Draisci, R.; Volpe, G.; Lucentini, L.; Cecilia, A.; Federico, R.; Palleschi, G. Determination of Biogenic Amines with an Electrochemical Biosensor and Its Application to Salted Anchovies. Food Chem. 1998, 62, 225–232. [Google Scholar] [CrossRef]
- Yang, Y.; Ge, L. Sensor Coating Employed to Preliminarily Evaluate the Banana Ripeness. Colloids Surf. A Physicochem. Eng. Asp. 2021, 616, 126057. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Q.; Xing, H.; Lu, X.; Zhao, L.; Qu, K.; Bi, K. Evaluation of Antioxidant Activity of Ten Compounds in Different Tea Samples by Means of an On-Line HPLC–DPPH Assay. Food Res. Int. 2013, 53, 847–856. [Google Scholar] [CrossRef]
- Brainina, K.; Tarasov, A.; Khamzina, E.; Stozhko, N.; Vidrevich, M. Contact Hybrid Potentiometric Method for On-Site and in Situ Estimation of the Antioxidant Activity of Fruits and Vegetables. Food Chem. 2020, 309, 125703. [Google Scholar] [CrossRef] [PubMed]
- Fanali, C.; Dugo, L.; D’Orazio, G.; Lirangi, M.; Dachà, M.; Dugo, P.; Mondello, L. Analysis of Anthocyanins in Commercial Fruit Juices by Using Nano-Liquid Chromatography-Electrospray-Mass Spectrometry and High-Performance Liquid Chromatography with UV-Vis Detector. J. Sep. Sci. 2011, 34, 150–159. [Google Scholar] [CrossRef]
- Gupta, M.K.; Anand, A.; Asati, A.; Thati, R.; Katragunta, K.; Agarwal, R.; Mudiam, M.K.R. Quantitative Determination of Phenolic Antioxidants in Fruit Juices by GC-MS/MS Using Automated Injector Port Silylation after QuEChERS Extraction. Microchem. J. 2021, 160, 105705. [Google Scholar] [CrossRef]
- Di Fusco, M.; Federico, R.; Boffi, A.; MacOne, A.; Favero, G.; Mazzei, F. Characterization and Application of a Diamine Oxidase from Lathyrus Sativus as Component of an Electrochemical Biosensor for the Determination of Biogenic Amines in Wine and Beer. Anal. Bioanal. Chem. 2011, 401, 707–716. [Google Scholar] [CrossRef]
- Huang, T.H.; Salter, G.; Kahn, S.L.; Gindt, Y.M. Redox Titration of Ferricyanide to Ferrocyanide with Ascorbic Acid: Illustrating the Nernst Equation and Beer-Lambert Law. J. Chem. Educ. 2007, 84, 1461. [Google Scholar] [CrossRef]
- Mugo, S.M.; Alberkant, J. Flexible Molecularly Imprinted Electrochemical Sensor for Cortisol Monitoring in Sweat. Anal. Bioanal. Chem. 2020, 412, 1825–1833. [Google Scholar] [CrossRef]
- Dhanjai; Mugo, S.M.; Lu, W. Modified Stainless Steel Microneedle Electrode for Polyphenolics Detection. Anal. Bioanal. Chem. 2020, 412, 7063–7072. [Google Scholar] [CrossRef]
- Singh, B.P.; Samal, S.; Nayak, S.; Majhi, S.M.; Besra, L.; Bhattacharjee, S. The Production of a Multi-Walled Carbon Nanotube/Hexamethylene Diisocyanate Nanocomposite Coating on Copper by Electrophoretic Deposition. Surf. Coat. Technol. 2011, 206, 1319–1326. [Google Scholar] [CrossRef]
- Cochet, M.; Louarn, G.; Quillard, S.; Buisson, J.P.; Lefrant, S. Theoretical and Experimental Vibrational Study of Emeraldine in Salt Form. Part II. J. Raman Spectrosc. 2000, 31, 1041–1049. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Dresselhaus, G.; Saito, R.; Jorio, A. Raman Spectroscopy of Carbon Nanotubes. Phys. Rep. 2005, 409, 47–99. [Google Scholar] [CrossRef]
- Kar, P.; Choudhury, A. Carboxylic Acid Functionalized Multi-Walled Carbon Nanotube Doped Polyaniline for Chloroform Sensors. Sens. Actuators B Chem. 2013, 183, 25–33. [Google Scholar] [CrossRef]
- Martín-Yerga, D.; Pérez-Junquera, A.; González-García, M.B.; Perales-Rondon, J.V.; Heras, A.; Colina, A.; Hernández-Santos, D.; Fanjul-Bolado, P. Quantitative Raman Spectroelectrochemistry Using Silver Screen-Printed Electrodes. Electrochim. Acta 2018, 264, 183–190. [Google Scholar] [CrossRef]
- Habibi, B.; Jahanbakhshi, M. A Novel Nonenzymatic Hydrogen Peroxide Sensor Based on the Synthesized Mesoporous Carbon and Silver Nanoparticles Nanohybrid. Sens. Actuators B Chem. 2014, 203, 919–925. [Google Scholar] [CrossRef]
- Mugo, S.M.; Lu, W.; Wood, M.; Lemieux, S. Wearable Microneedle Dual Electrochemical Sensor for Simultaneous PH and Cortisol Detection in Sweat. Electrochem. Sci. Adv. 2021, 2, e2100039. [Google Scholar] [CrossRef]
- Chmielewski, M.; Heimbürger, O.; Stenvinkel, P.; Lindholm, B. Chapter 4—Uremic Toxicity. In Nutritional Management of Renal Disease; Kopple, J., Massry, S., Kalantar-Zadeh, K., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 49–77. [Google Scholar]
- Yano, Y.; Yokoyama, K.; Karube, I. Evaluation of Meat Spoilage Using a Chemiluminescence—Flow Injection Analysis System Based on Immobilized Putrescine Oxidase and a Photodiode. LWT—Food Sci. Technol. 1996, 29, 498–502. [Google Scholar] [CrossRef]
- Shanmugam, S.; Thandavan, K.; Gandhi, S.; Sethuraman, S.; Balaguru Rayappan, J.B.; Krishnan, U.M. Development and Evaluation of a Highly Sensitive Rapid Response Enzymatic Nanointerfaced Biosensor for Detection of Putrescine. Analyst 2011, 136, 5234–5240. [Google Scholar] [CrossRef]
- Equi, A.M.; Brown, A.M.; Cooper, A.; Her, S.K.; Watson, A.B.; Robins, D.J. Oxidation of Putrescine and Cadaverine Derivatives by Diamine Oxidases. Tetrahedron 1991, 47, 507–518. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Cimpeanu, C.; Predoi, G. Antioxidant Capacity Determination in Plants and Plant-Derived Products: A Review. Oxid. Med. Cell. Longev. 2016, 2016, 9130976. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ding, D.; Wang, J.; Lin, X.; Diao, G. Three-Dimensional Nitrogen-Doped Graphene-Based Metal-Free Electrochemical Sensors for Simultaneous Determination of Ascorbic Acid, Dopamine, Uric Acid, and Acetaminophen. Analyst 2021, 146, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Dinu, A.; Apetrei, C. Determination of Ascorbic Acid in Pharmaceuticals and Food Supplements with the New Potassium Ferrocyanide-Doped Polypyrrole-Modified Platinum Electrode Sensor. Chemosensors 2022, 10, 180. [Google Scholar] [CrossRef]
- Quezada, V.; Martinez, T.; Nelson, R.; Pérez-Fehrmann, M.; Zaragoza, G.; Vizcarra, A.; Kesternich, V.; Hernández-Saravia, L.P. A Novel Platform of Using Copper (II) Complex with Triazole-Carboxilated Modified as Bidentated Ligand SPCE for the Detection of Hydrogen Peroxide in Milk. J. Electroanal. Chem. 2020, 879, 114763. [Google Scholar] [CrossRef]
- Atacan, K.; Özacar, M. Construction of a Non-Enzymatic Electrochemical Sensor Based on CuO/g-C3N4 Composite for Selective Detection of Hydrogen Peroxide. Mater. Chem. Phys. 2021, 266, 124527. [Google Scholar] [CrossRef]
- Apetrei, I.M.; Apetrei, C. Application of Voltammetric E-Tongue for the Detection of Ammonia and Putrescine in Beef Products. Sens. Actuators B Chem. 2016, 234, 371–379. [Google Scholar] [CrossRef]
- Kaçar, C.; Erden, P.E.; Dalkiran, B.; İnal, E.K.; Kiliç, E. Amperometric Biogenic Amine Biosensors Based on Prussian Blue, Indium Tin Oxide Nanoparticles and Diamine Oxidase– or Monoamine Oxidase–Modified Electrodes. Anal. Bioanal. Chem. 2020, 412, 1933–1946. [Google Scholar] [CrossRef]
- Liang, C.H.; Ho, K.J.; Huang, L.Y.; Tsai, C.H.; Lin, S.Y.; Mau, J.L. Antioxidant Properties of Fruiting Bodies, Mycelia, and Fermented Products of the Culinary-Medicinal King Oyster Mushroom, Pleurotus Eryngii (Higher Basidiomycetes), with High Ergothioneine Content. Int. J. Med. Mushrooms 2013, 15, 267–275. [Google Scholar] [CrossRef]
- Halliwell, B.; Cheah, I.K.; Tang, R.M.Y. Ergothioneine — A Diet-Derived Antioxidant with Therapeutic Potential. FEBS Lett. 2018, 592, 3357–3366. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Khan, A.A.; Muhammad, M.J.; Muhammad, I.; Jan, I.; Samin, G.; Zahid, A.; Fozia; Muhammad, I.; Wang, P.; Lu, L.; et al. Modulation of Agronomic and Nutritional Response of Pleurotus Eryngii Strains by Utilizing Glycine Betaine Enriched Cotton Waste. J. Sci. Food Agric. 2019, 99, 6911–6921. [Google Scholar] [CrossRef] [PubMed]
- Król, K.; Gantner, M.; Tatarak, A.; Hallmann, E. The Content of Polyphenols in Coffee Beans as Roasting, Origin and Storage Effect. Eur. Food Res. Technol. 2020, 246, 33–39. [Google Scholar] [CrossRef]
- Halász, A.; Baráth, Á.; Simon-Sarkadi, L.; Holzapfel, W. Biogenic Amines and Their Production by Microorganisms in Food. Trends Food Sci. Technol. 1994, 5, 42–49. [Google Scholar] [CrossRef]
- Treviño, E.; Beil, D.; Steinhart, H. Formation of Biogenic Amines during the Maturity Process of Raw Meat Products, for Example of Cervelat Sausage. Food Chem. 1997, 60, 521–526. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mugo, S.M.; Dhanjai; Lu, W.; Robertson, S. A Multipurpose and Multilayered Microneedle Sensor for Redox Potential Monitoring in Diverse Food Analysis. Biosensors 2022, 12, 1001. https://doi.org/10.3390/bios12111001
Mugo SM, Dhanjai, Lu W, Robertson S. A Multipurpose and Multilayered Microneedle Sensor for Redox Potential Monitoring in Diverse Food Analysis. Biosensors. 2022; 12(11):1001. https://doi.org/10.3390/bios12111001
Chicago/Turabian StyleMugo, Samuel M., Dhanjai, Weihao Lu, and Scott Robertson. 2022. "A Multipurpose and Multilayered Microneedle Sensor for Redox Potential Monitoring in Diverse Food Analysis" Biosensors 12, no. 11: 1001. https://doi.org/10.3390/bios12111001
APA StyleMugo, S. M., Dhanjai, Lu, W., & Robertson, S. (2022). A Multipurpose and Multilayered Microneedle Sensor for Redox Potential Monitoring in Diverse Food Analysis. Biosensors, 12(11), 1001. https://doi.org/10.3390/bios12111001