Simultaneous Determination of Streptomycin and Oxytetracycline Using a Oracet-Blue/Silver-Nanoparticle/Graphene-Oxide/Modified Screen-Printed Electrode



Abstract

1. Introduction
2. Material and Methods
2.1. Reagents and Apparatus
2.2. Preparation of Samples
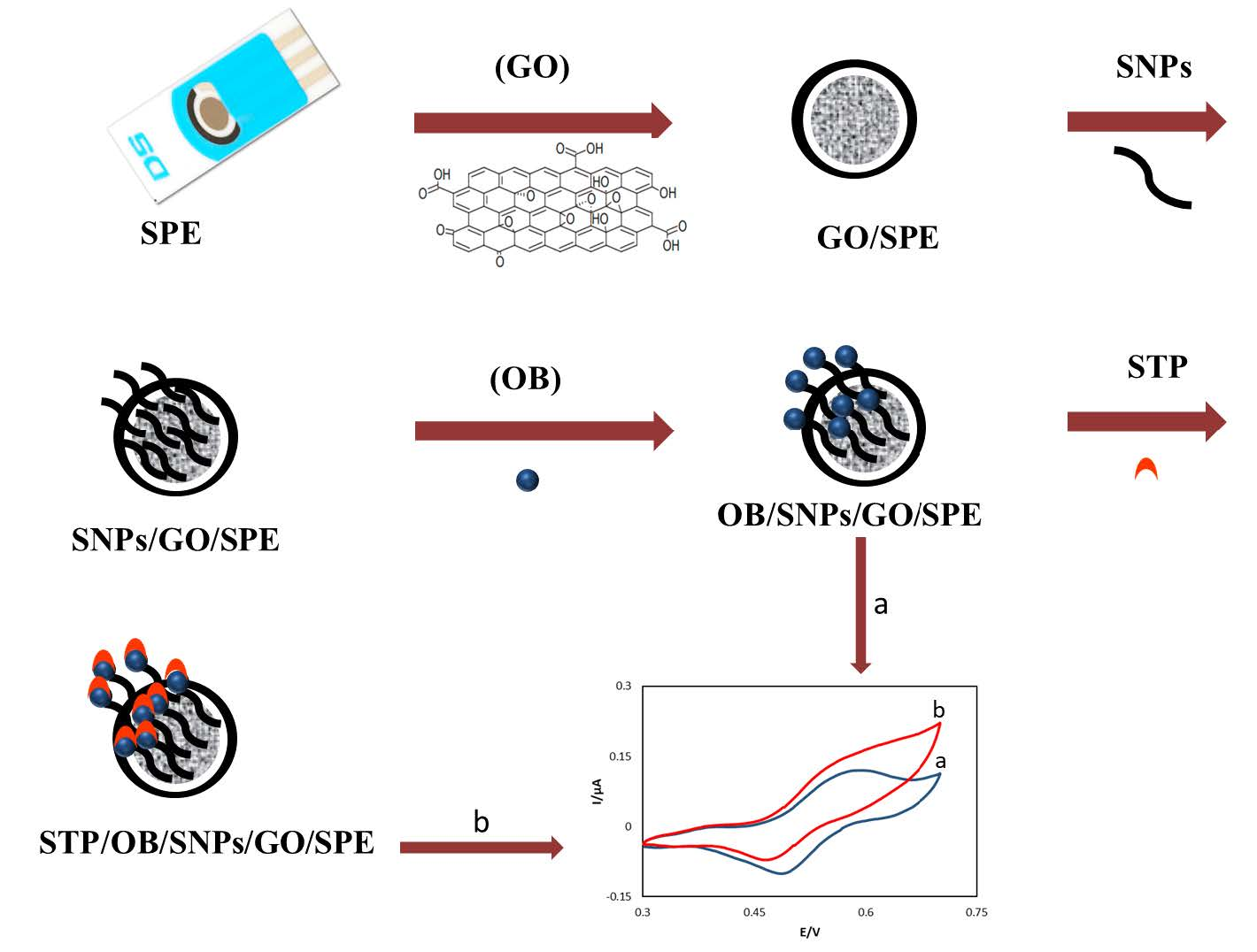
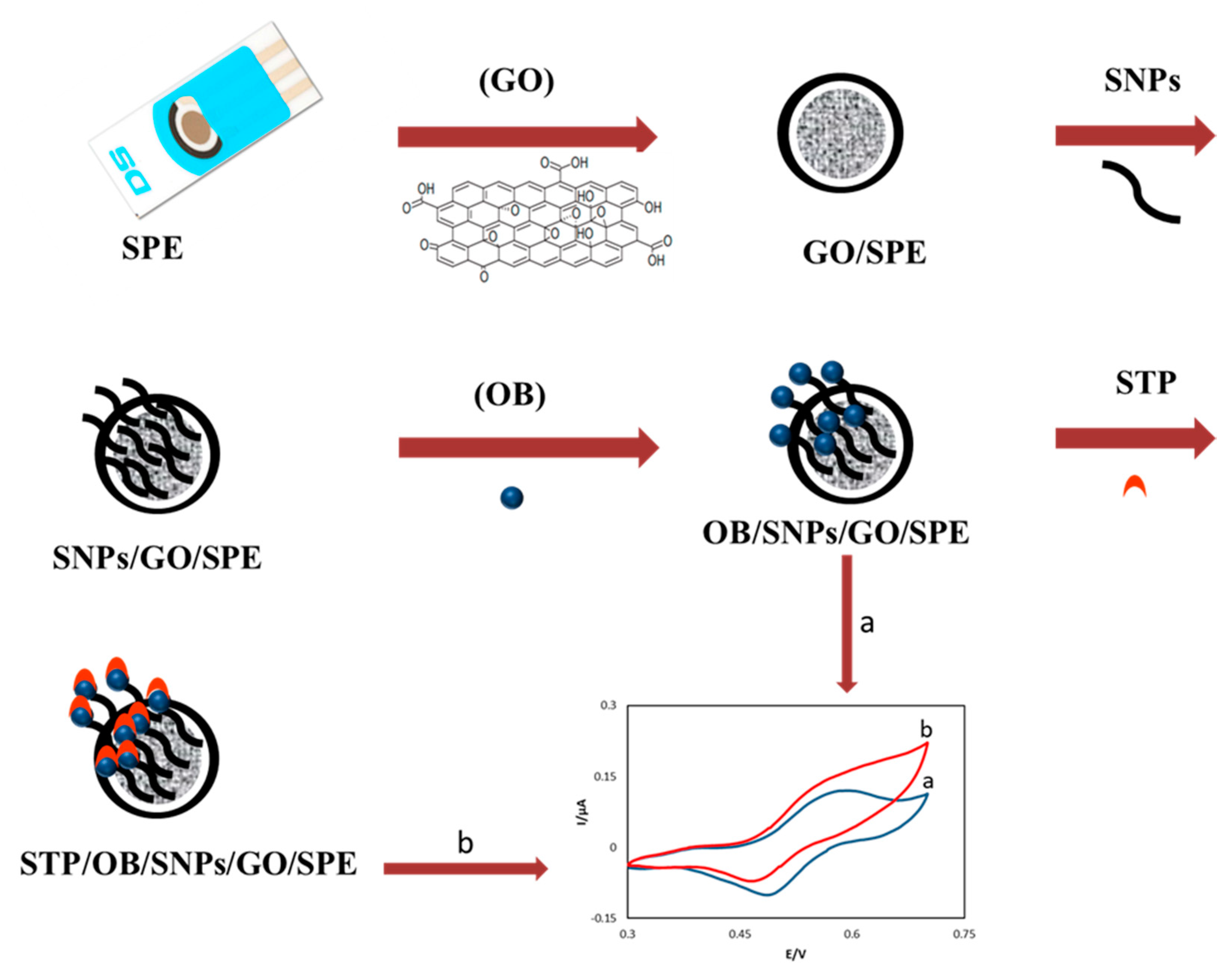
2.3. Electrode Preparation
3. Results
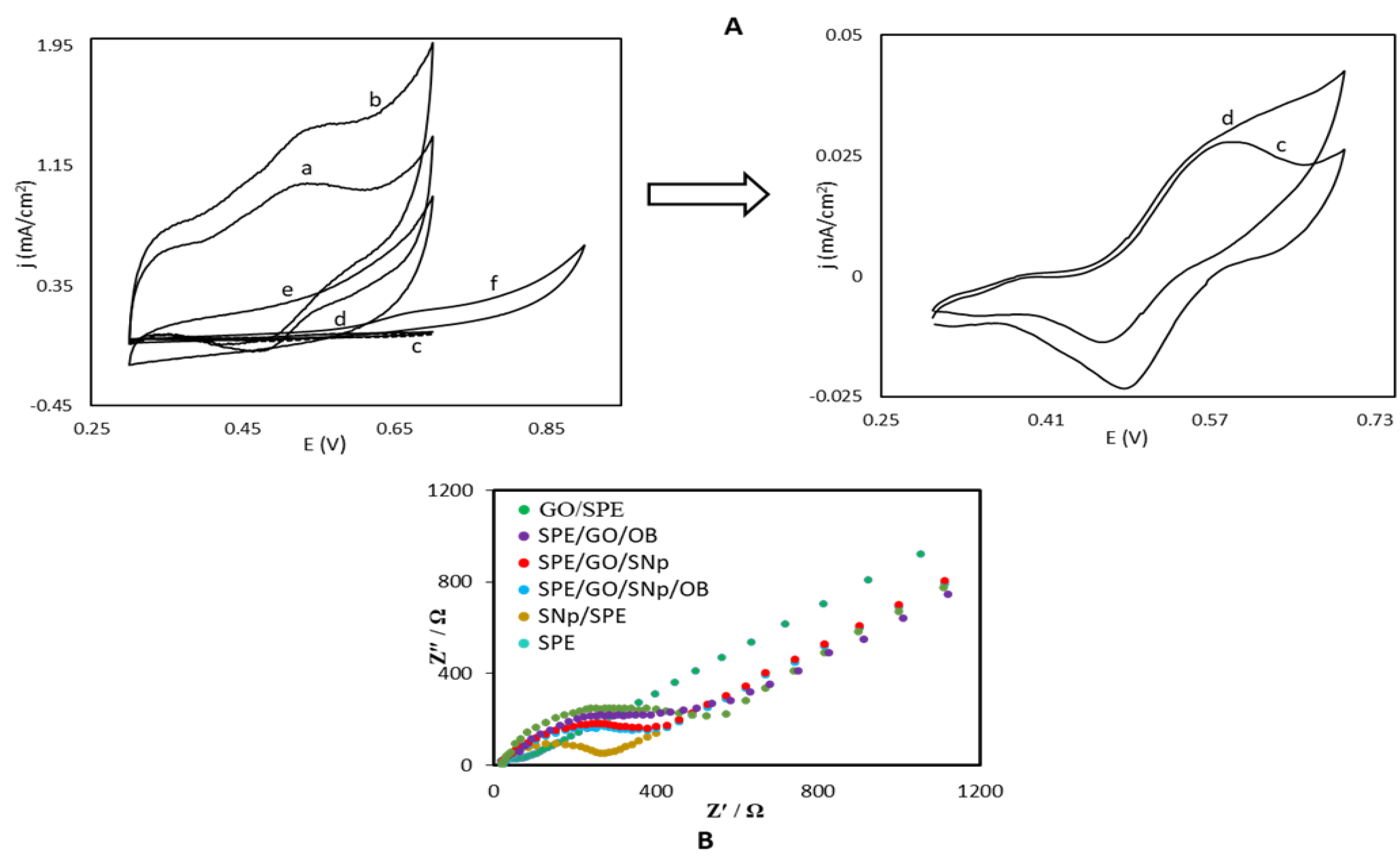
3.1. Electrochemical Charaterization of the OB/SNPs/GO/SPE Electrode
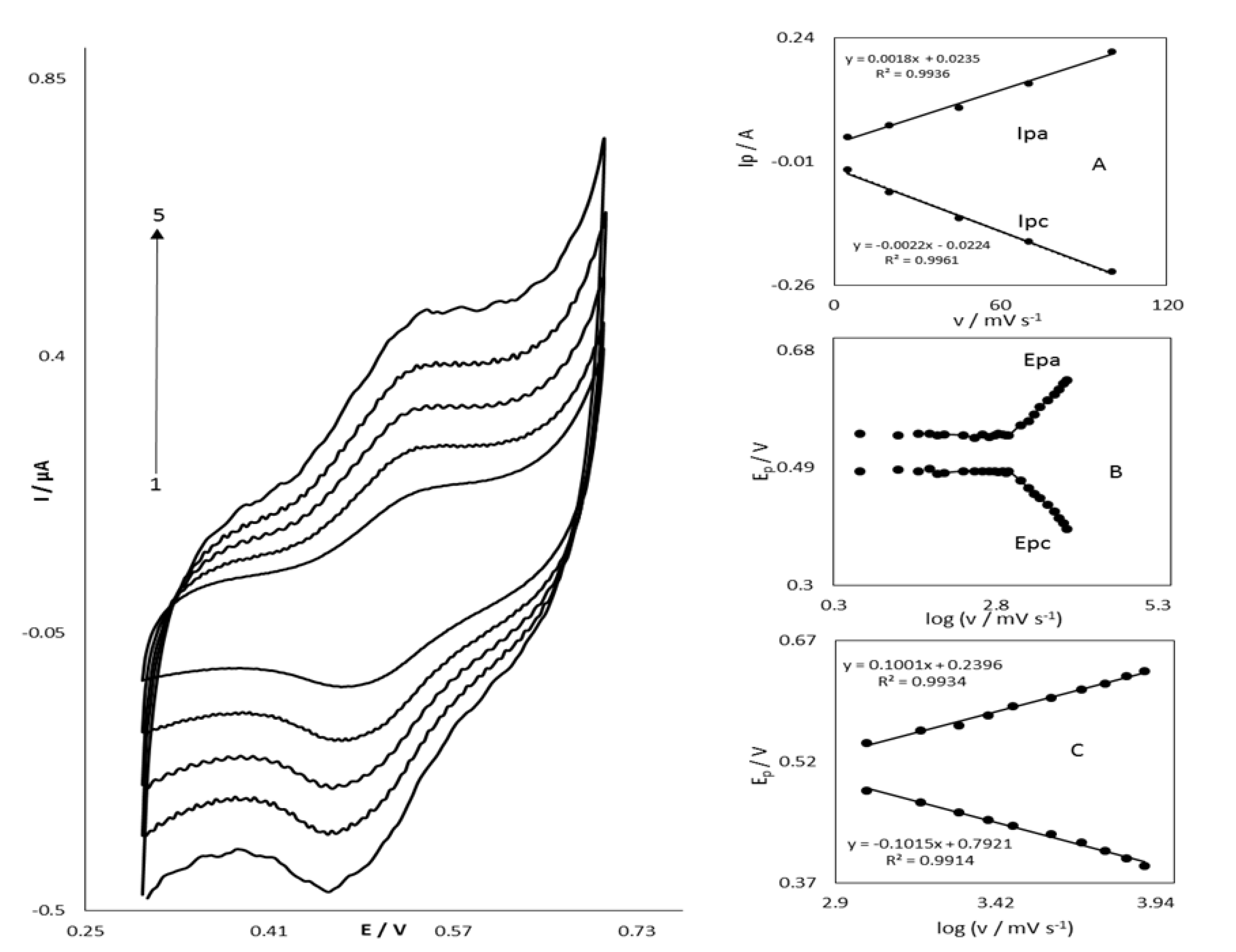
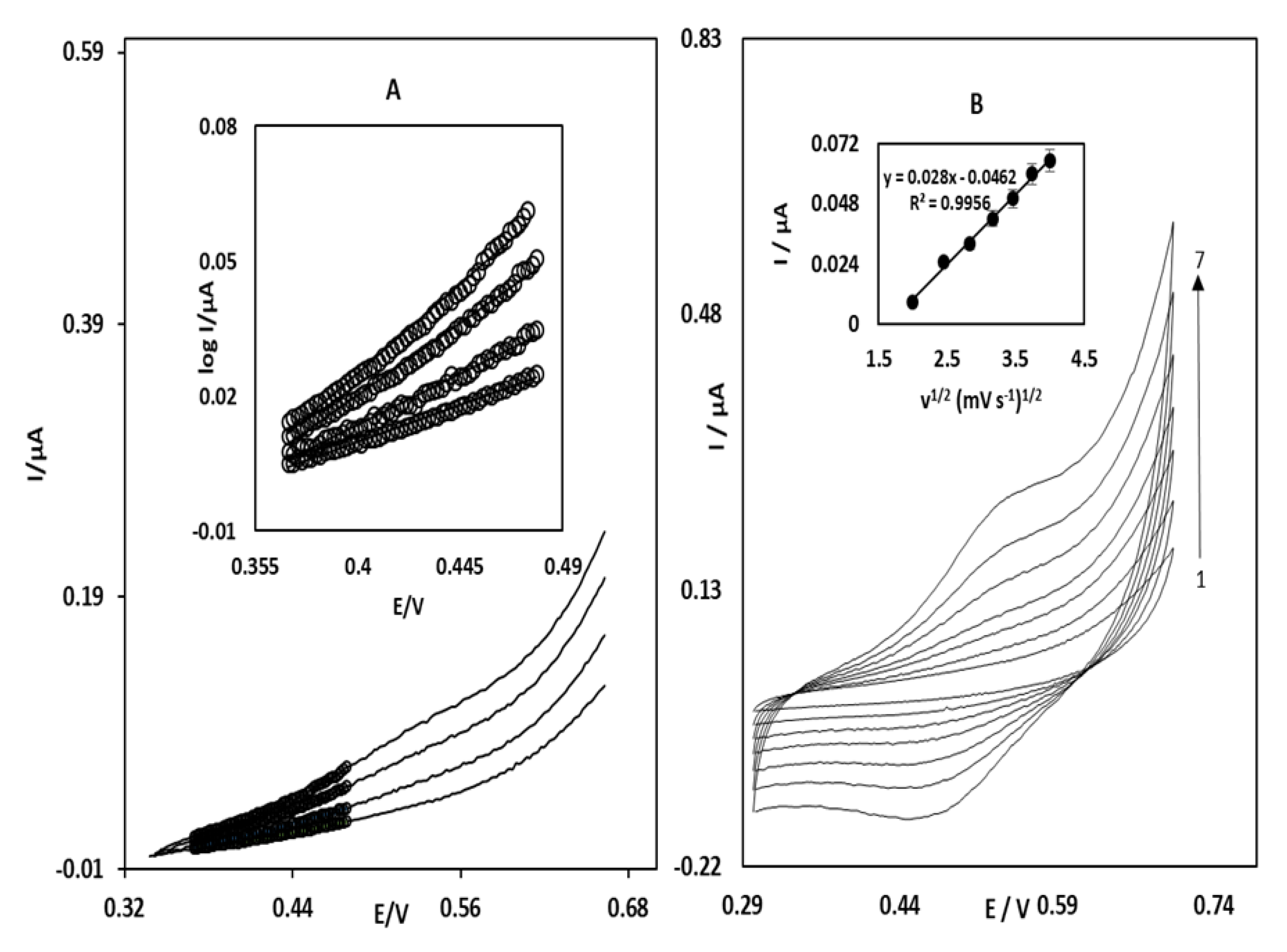
3.2. Electrocatalytic Oxidation of STR at the OB/SNPs/GO/SPE
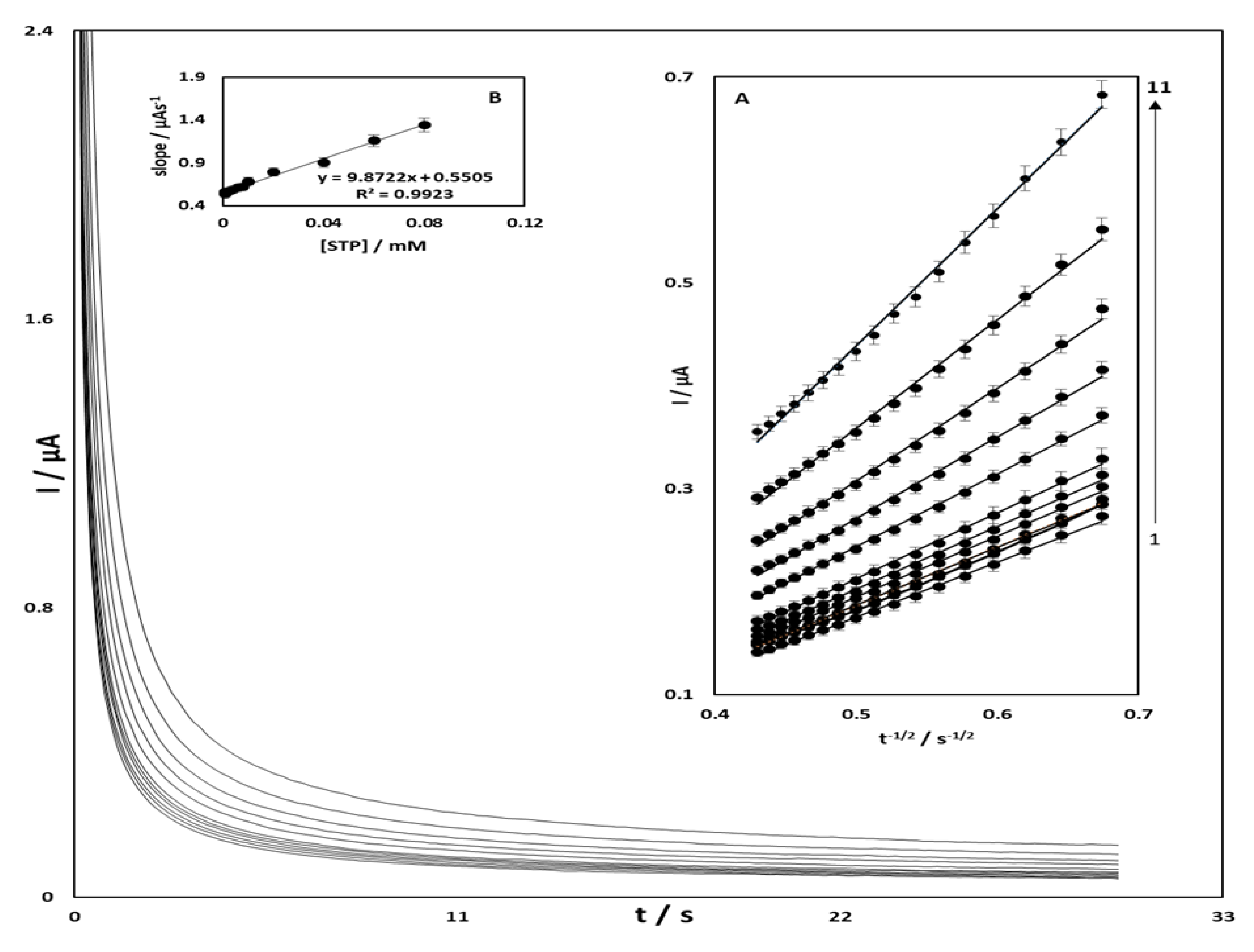
3.3. Chronoamperometric Studies of STR Oxidation at OB/SNPs/GO/SPE
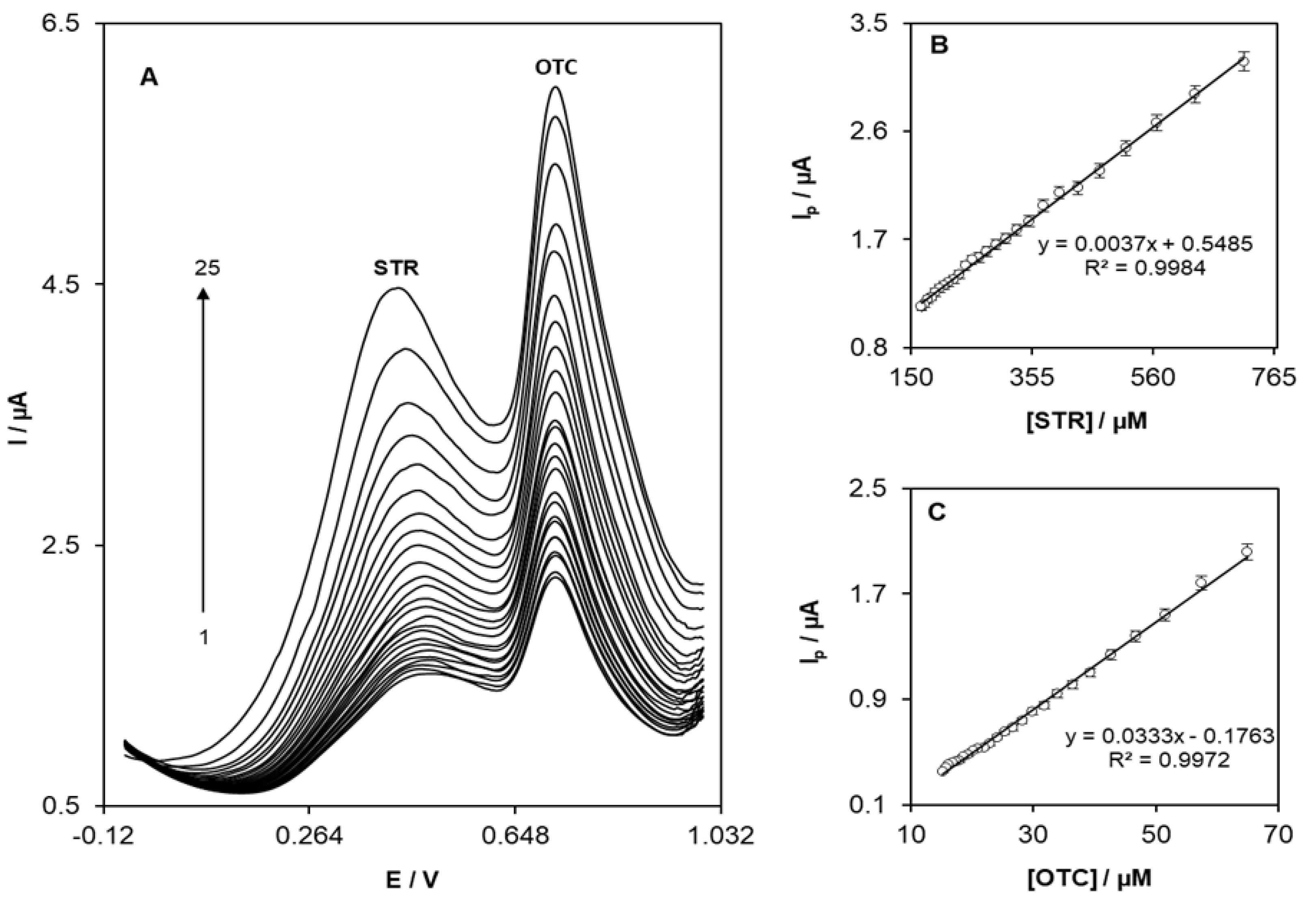
3.4. Simultaneous Determination of STR and OTC
4. Discussions
4.1. Analysis of Impedance Results
4.2. Evaluation of Sensitivity and Stability of the Prepared Electrode
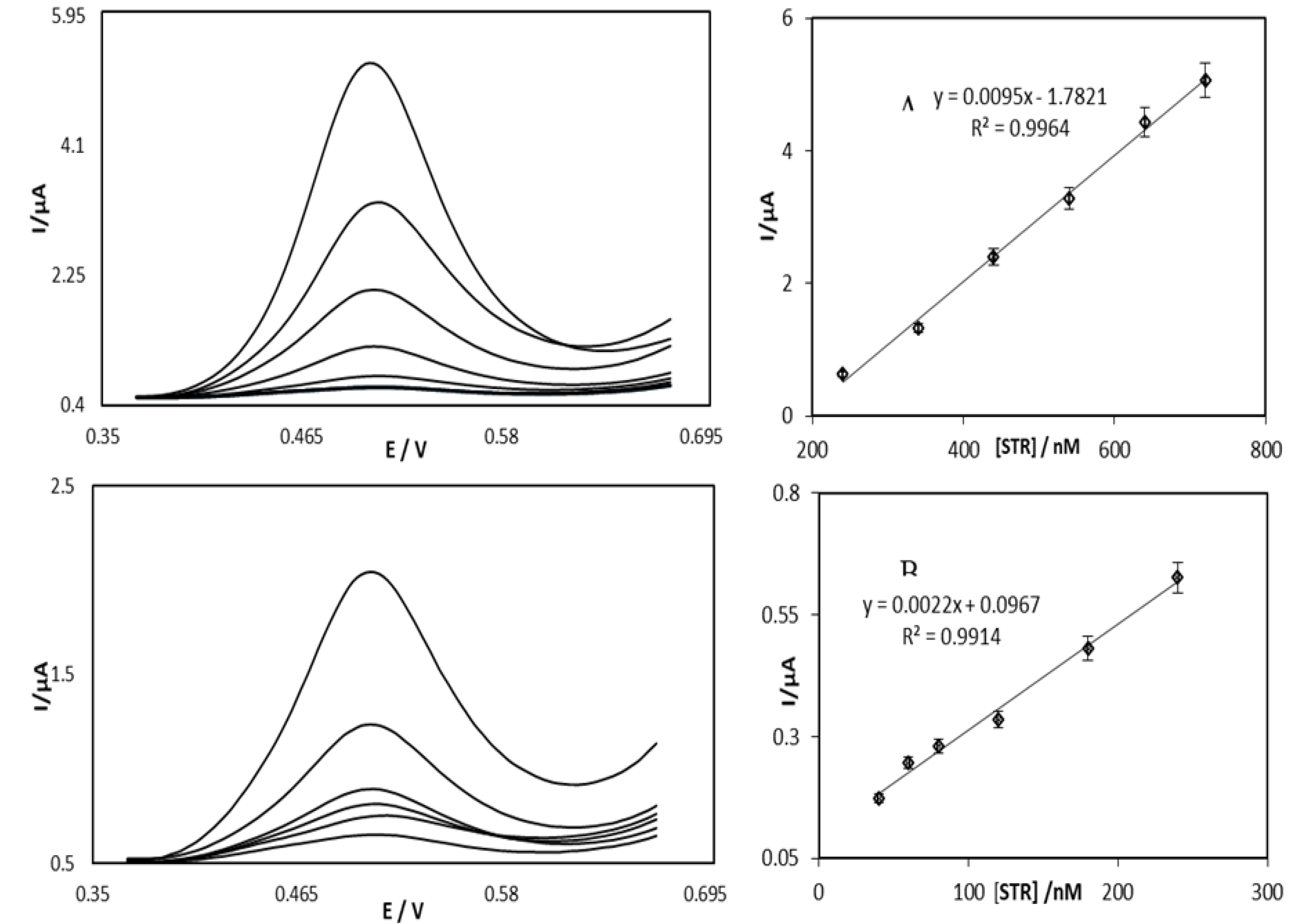
4.3. Application
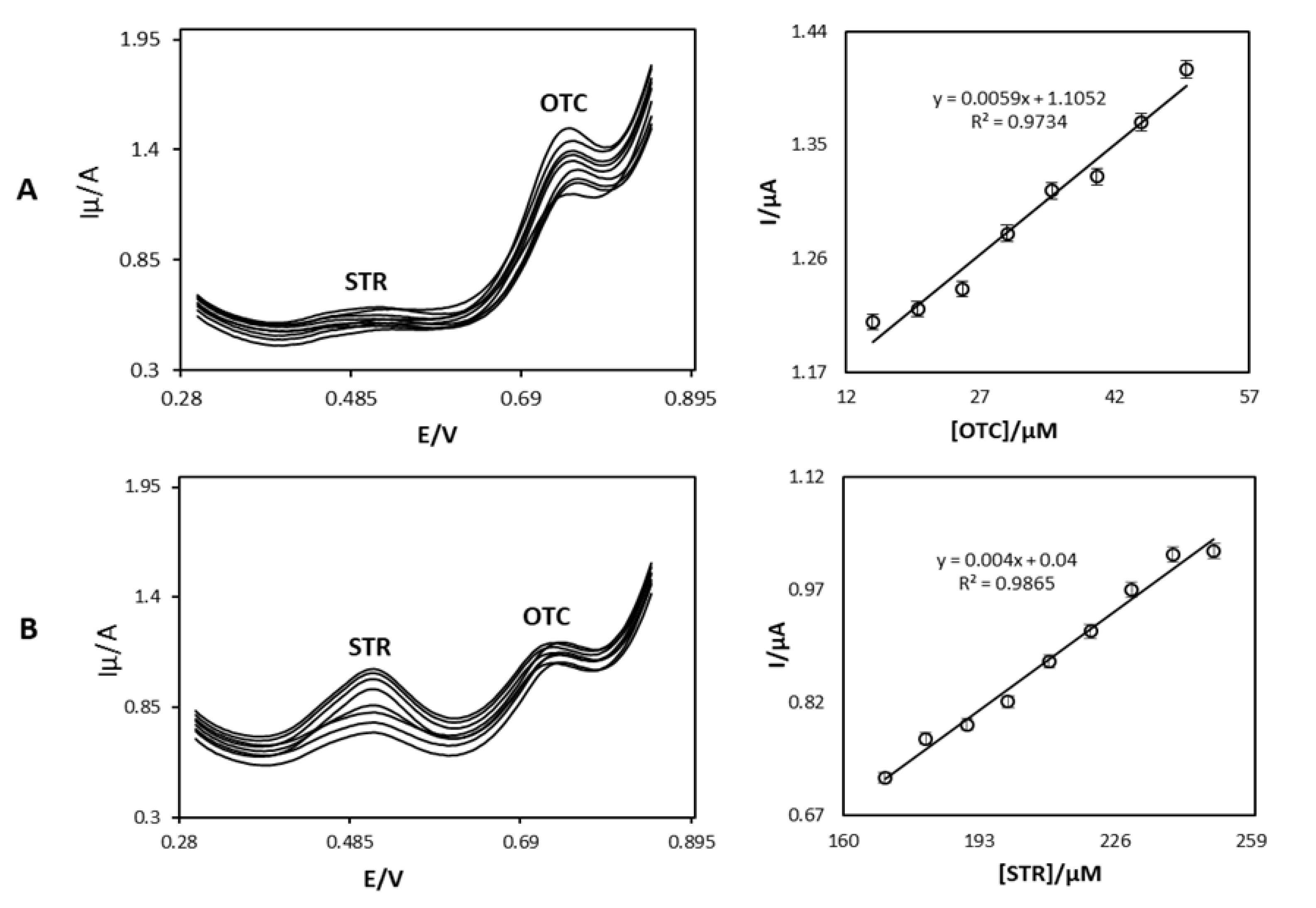
4.4. Reliability and Interference study of Proposed Nanosensor
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Maia, P.P.; Amaya-Farfán, J.; Rath, S.; Reyes, F.G. Simultaneous Determination of Streptomycin and Oxytetracycline in Agricultural Antimicrobials by CZE after an Experimental Design. Pharm. Biomed. Anal. 2007, 43, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Summary of evaluations performed by the Joint FAO/WHO Expert Committee on Food Additives. Available online: http://www.inchem.org/documents/jecfa/jeceval/jec_2031.htm (accessed on 6 March 2020).
- Knecht, B.G.; Strasser, A.; Dietrich, R.; Märtlbauer, E.; Niessner, R.; Weller, M.G. Automated Microarray System for the Simultaneous Detection of Antibiotics in Milk. Anal. Chem. 2004, 76, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tang, D.; Zhang, B.; Que, X.; Yang, H.; Chen, G. Au (III)-promoted Magnetic Molecularly Imprinted Polymer Nanospheres for Electrochemical Determination of Streptomycin Residues in Food. Biosens. Bioelectron. 2013, 41, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Oguri, S.; Miki, Y. Determination of Amikacin in Human Plasma by High-Performance Capillary Electrophoresis with Fluorescence Detection. Chromatogr. B Biomed. Sci. Appl. 1996, 686, 205–210. [Google Scholar] [CrossRef]
- Kowalski, P.; Oledzka, I.; Okoniewski, P.; Switala, M.; Lamparczyk, H. Determination of Streptomycin in Eggs yolk by Capillary Electrophoresis. Chromatographia 1999, 50, 101–104. [Google Scholar] [CrossRef]
- Caturla, M.C.; Cusido, E.; Westerlund, D. High-performance Liquid Chromatography Method for the Determination of Aminoglycosides based on Automated pre-column Derivatization with o-Phthalaldehyde. Chromatogr. A 1992, 593, 69–72. [Google Scholar] [CrossRef]
- McLaughlin, L.G.; Henion, J.D. Determination of Aminoglycoside Antibiotics by Reversed-phase Ion-pair High-performance Liquid Chromatography Coupled with Pulsed Amperometry and Ion spray Mass Spectrometry. Chromatogr. A 1992, 591, 195–206. [Google Scholar] [CrossRef]
- Kaale, E.; Leonard, S.; Van Schepdael, A.; Roets, E.; Hoogmartens, J. Capillary Electrophoresis Analysis of Gentamicin sulphate with UV Detection after Pre-capillary Derivatization with 1, 2-phthalic dicarboxaldehyde and Mercaptoacetic acid. Chromatogr. A 2000, 895, 67–79. [Google Scholar] [CrossRef]
- Flurer, C.L. The Analysis of Aminoglycoside Antibiotics by Capillary Electrophoresis. Pharm. Biomed. Anal. 1995, 13, 809–816. [Google Scholar] [CrossRef]
- Edder, P.; Cominoli, A.; Corvi, C. Determination of Streptomycin Residues in Food by Solid-phase Extraction and Liquid Chromatography with post-column Derivatization and Fluorometric Detection. Chromatogr. A 1999, 830, 345–351. [Google Scholar] [CrossRef]
- Salvatore, M.J.; Feygin, I.; Katz, S.E. Agarose gel Electrophoresis System for the Separation of Antibiotics used in Animal Agriculture. Analyst 1993, 118, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, K.; Camarada, M.B.; Rajesh, R.; Venkatesan, R.; Ju, H.; Dharuman, V.; Wen, Y. Layer-by-layer Assembled Gold nanoparticles/lower-generation (Gn≤ 3) Polyamidoamine Dendrimers-grafted Reduced Graphene oxide nanohybrids with 3D Fractal Architecture for Fast, Ultra-trace, and Label-free Electrochemical Gene nanobiosensors. Biosens. Bioelectron. 2018, 120, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhang, L.; Duan, X.; Xu, J.; Zhou, W.; Wen, Y. Differential pulse Striping Voltammetric Determination of Molluscicide Niclosamide using Three Different Carbon Nanomaterials Modified Electrodes. Electrochim. Acta 2014, 127, 86–94. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, Y.; Xu, J.; Wen, Y.; Zhang, J.; Ding, W. Nanohybrid Sensor based on Carboxyl Functionalized Graphene Dispersed Palygorskite for Voltammetric Determination of Niclosamide. Appl. Clay Sci. 2017, 143, 57–66. [Google Scholar] [CrossRef]
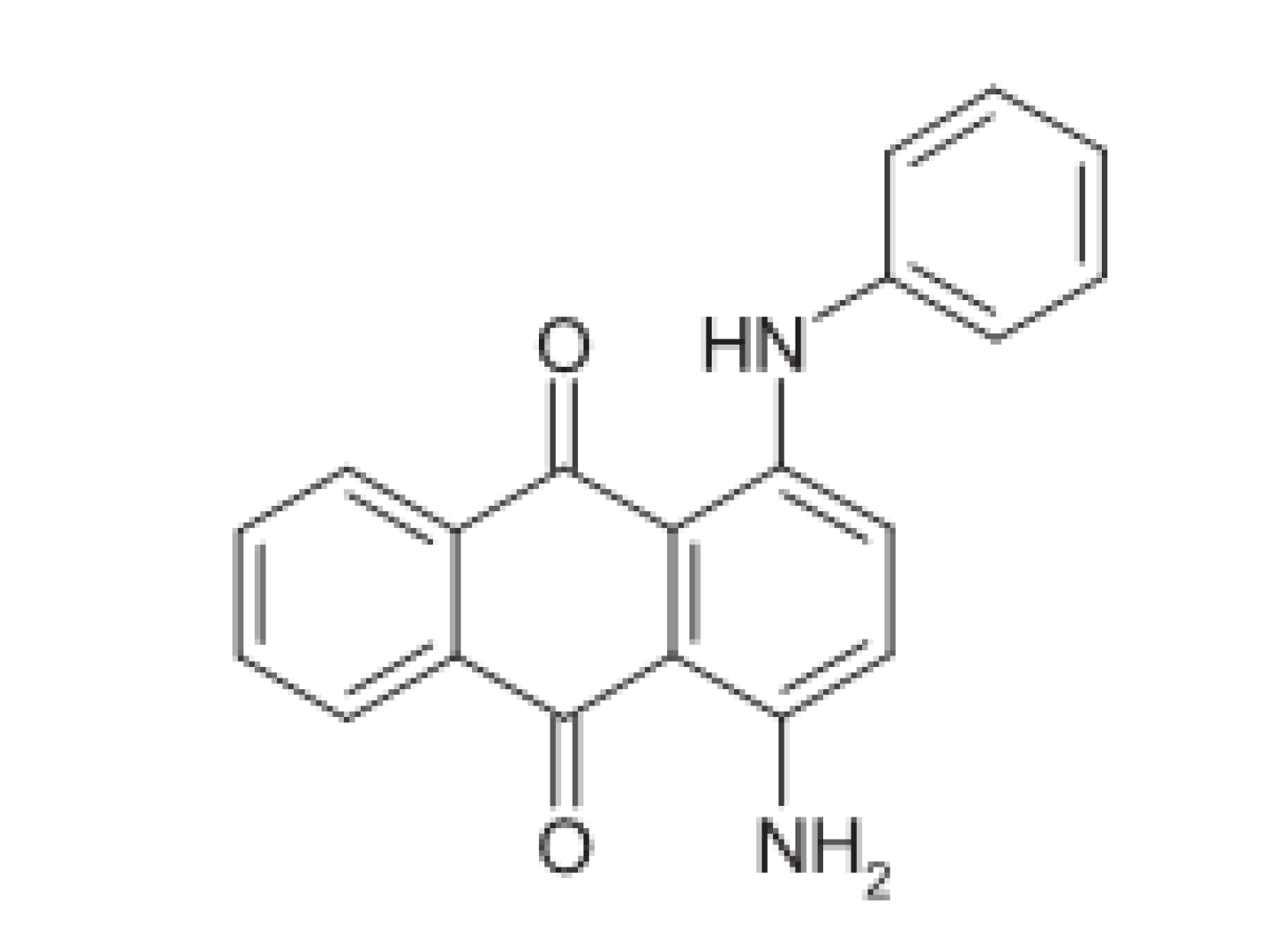
- Zare, H.R.; Nasirizadeh, N. Fabrication, Characterization and Analytical Performance of the Hydroxylamine Sensor based on an Oracet blue Multi-walled Carbon Nanotubes Film Deposited on an Electrode Surface. Braz. Chem. Soc. 2012, 23, 1070–1077. [Google Scholar] [CrossRef][Green Version]
- Zare, H.R.; Rajabzadeh, N.; Nasirizadeh, N.; Ardakani, M.M. Voltammetric Studies of an Oracet blue Modified Glassy Carbon Electrode and its Application for the Simultaneous Determination of Dopamine, Ascorbic acid and Uric acid. Electroanal. Chem. 2006, 589, 60–69. [Google Scholar] [CrossRef]
- Hajihosseini, S.; Nasirizadeh, N.; Hejazi, M.S.; Yaghmaei, P. An Electrochemical DNA Biosensor based on Oracet Blue as a Label for Detection of Helicobacter pylori. Biol. Macromol. 2016, 91, 911–917. [Google Scholar] [CrossRef]
- Moghadam, M.R.; Akbarzadeh, S.; Nasirizadeh, N. Electrochemical Sensor for the Determination of Thiourea using a Glassy Carbon Electrode Modified with a Self-Assembled Monolayer of an Oxadiazole Derivative and with Silver Nanoparticles. Microchim. Acta 2016, 183, 1069–1077. [Google Scholar] [CrossRef]
- Ju, H.; Shen, C. Electrocatalytic Reduction and Determination of Dissolved Oxygen at a Poly (nile blue) modified Electrode. Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal. 2001, 13, 789–793. [Google Scholar] [CrossRef]
- Laviron, E. General Expression of the Linear Potential Sweep Voltammogram in the case of Diffusionless Electrochemical Systems. Electroanal. Chem. Interfacial Electrochem. 1979, 101, 19–28. [Google Scholar] [CrossRef]
- Zare, H.R.; Shekari, Z.; Nasirizadeh, N.; Jafari, A.A. Fabrication, electrochemical Characteristics and Electrocatalytic Activity of 4-((2-hydroxyphenylimino) methyl) benzene-1, 2-diol Electrodeposited on a Carbon Nanotube Modified Glassy Carbon Electrode as a Hydrazine Sensor. Catal. Sci. Technol. 2012, 2, 492–501. [Google Scholar] [CrossRef]
- Nasirizadeh, N.; Shekari, Z.; Zare, H.R.; Shishehbore, M.R.; Fakhari, A.R.; Ahmar, H. Electrosynthesis of an Imidazole Derivative and it’s Application as a Bifunctional Electrocatalyst for Simultaneous Determination of Ascorbic acid, Adrenaline, Acetaminophen, and Tryptophan at a Multi-wall Carbon Nanotubes Modified Electrode Surface. Biosens. Bioelectron. 2013, 41, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Nasirizadeh, N.; Zare, H.R.; Fakhari, A.R.; Ahmar, H.; Ahmadzadeh, M.R.; Naeimi, A. A Study of the Electrochemical Behavior of an Oxadiazole Derivative Electrodeposited on Multi-wall Carbon Nanotube-Modified Electrode and its Application as a Hydrazine sensor. Solid State Electrochem. 2011, 15, 2683–2693. [Google Scholar] [CrossRef]
- Zare, H.R.; Nasirizadeh, N.; Ardakani, M.M. Electrochemical Properties of a Tetrabromo-p-benzoquinone Modified Carbon Paste Electrode Application to the Simultaneous Determination of Ascorbic acid, Dopamine and Uric acid. Electroanal. Chem. 2005, 577, 25–33. [Google Scholar]
- Andrieux, C.P.; Saveant, J.M. Heterogeneous (chemically modified electrodes, polymer electrodes) vs. Homogeneous Catalysis of Electrochemical Reactions. Electroanal. Chem. Interfacial Electrochem. 1978, 93, 163–168. [Google Scholar] [CrossRef]
- Antoniadou, S.; Jannakoudakis, A.D.; Theodoridou, E. Electrocatalytic Reactions on Carbon Fibre Electrodes Modified by Hemine II. Electro-Oxid. Hydrazine. Synth. Met. 1989, 30, 295–304. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Fundamentals and Applications. Electrochem. Methods 2001, 2, 580–632. [Google Scholar]
- Denuault, G.; Mirkin, M.V.; Bard, A.J. Direct Determination of Diffusion Coefficients by Chronoamperometry at Microdisk Electrodes. Electroanal. Chem. Interfacial Electrochem. 1991, 308, 27–38. [Google Scholar] [CrossRef]
- Skoog, D.A.; Holler, F.J.; Nieman, T.A. Principles of Instrumental Analysis, 5th ed.; Harcourt Brace: Philadelphia, PA, USA, 1998; p. 571. [Google Scholar]
- Van Bruijnsvoort, M.; Ottink, S.J.; Jonker, K.M.; de Boer, E. Determination of Streptomycin and Dihydrostreptomycin in Milk and Honey by Liquid Chromatography with Tandem Mass Spectrometry. Chromatogr. A 2004, 1058, 137–142. [Google Scholar] [CrossRef]
- Granja, R.H.; Niño, A.M.; Zucchetti, R.A.; Niño, R.E.; Patel, R.; Salerno, A.G. Determination of Streptomycin Residues in Honey by Liquid Chromatography–Tandem Mass Spectrometry. Anal. Chim. Acta 2009, 637, 64–67. [Google Scholar] [CrossRef]
- Taghdisi, S.M.; Danesh, N.M.; Ramezani, M.; Abnous, K. Electrochemical Aptamer based assay for the Neonicotinoid Insecticide Acetamiprid based on the use of an Unmodified Gold Electrode. Microchim. Acta 2017, 184, 499–505. [Google Scholar] [CrossRef]
- Ji, S.; Zhang, F.; Luo, X.; Yang, B.; Jin, G.; Yan, J.; Liang, X. Synthesis of Molecularly Imprinted Polymer Sorbents and Application for the Determination of Aminoglycosides Antibiotics in Honey. Chromatogr. A 2013, 1313, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, B.; Cui, Y.; Chen, H.; Gao, Z.; Tang, D. Multifunctional Gold–silica Nanostructures for Ultrasensitive Electrochemical Immunoassay of Streptomycin Residues. Acs Appl. Mater. Interfaces 2011, 3, 4668–4676. [Google Scholar] [CrossRef] [PubMed]
- Baxter, G.A.; Ferguson, J.P.; O’Conno, M.C.; Elliott, C.T. Detection of Streptomycin Residues in Whole Milk Using an Optical Immunobiosensor. Agricult. Food Chem. 2001, 49, 3204–3207. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Z.; Zhang, H.; Luo, L.; Yao, S. Selective and Sensitive Molecularly Imprinted Sol–Gel Film-based Electrochemical Sensor Combining Mercaptoacetic acid-Modified PbS Nanoparticles with Fe3O4@ Au–multi-walled Carbon Nanotubes–Chitosan. Solid State Electrochem. 2012, 16, 857–867. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Linear Range (μM) | LOD (μM) | Matrices | Ref |
|---|---|---|---|---|
| LC/MS | - | 3, 0.6 | Milk, Honey | [31] |
| LC/MS | - | 4.7 | Honey | [32] |
| Electrochemistry | - | 14.1, 15.3 | Milk, Serum | [33] |
| Electrochemistry | 8.6 × 10−3–3.44 × 10−2 | 1.7 × 10−2 | Food | [4] |
| Electrochemistry | 1.0–33.8 33.8–81.7 | 1.0 | Honey | [34] |
| Electrochemistry | 860.0–8.60.0 × 106 | 5.0 × 109 | Muscle/Kidney/milk/Honey | [35] |
| Electrochemistry | 7.0 × 106 | Milk | [36] | |
| Electrochemistry | 1.0–1.0 × 103 | 1.5 × 10−3 | Injection Sample Solution | [37] |
| Electrochemistry | 4 × 10−4–2.4 × 10−1 2.4 × 10−1–7.2 × 10−1 | 0.17 × 10−3 | Milk | This Work |
| Sample | Added (µM) | Found (µM) | RSD (%) | Recovery (%) |
|---|---|---|---|---|
| Milk1 | 0 | 19 | 3.42 | - |
| 10 | 29.2 | 3.26 | 100.68 | |
| 20 | 38.7 | 3.08 | 99.23 | |
| 30 | 49.2 | 3.16 | 100.40 | |
| Milk2 | 0 | 62 | 3.49 | - |
| 10 | 72.4 | 4.39 | 100.55 | |
| 20 | 81.9 | 3.03 | 99.87 | |
| 30 | 92.5 | 4.18 | 100.54 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akbarzadeh, S.; Khajesharifi, H.; Thompson, M. Simultaneous Determination of Streptomycin and Oxytetracycline Using a Oracet-Blue/Silver-Nanoparticle/Graphene-Oxide/Modified Screen-Printed Electrode. Biosensors 2020, 10, 23. https://doi.org/10.3390/bios10030023
Akbarzadeh S, Khajesharifi H, Thompson M. Simultaneous Determination of Streptomycin and Oxytetracycline Using a Oracet-Blue/Silver-Nanoparticle/Graphene-Oxide/Modified Screen-Printed Electrode. Biosensors. 2020; 10(3):23. https://doi.org/10.3390/bios10030023
Chicago/Turabian StyleAkbarzadeh, Sanaz, Habibollah Khajesharifi, and Michael Thompson. 2020. "Simultaneous Determination of Streptomycin and Oxytetracycline Using a Oracet-Blue/Silver-Nanoparticle/Graphene-Oxide/Modified Screen-Printed Electrode" Biosensors 10, no. 3: 23. https://doi.org/10.3390/bios10030023
APA StyleAkbarzadeh, S., Khajesharifi, H., & Thompson, M. (2020). Simultaneous Determination of Streptomycin and Oxytetracycline Using a Oracet-Blue/Silver-Nanoparticle/Graphene-Oxide/Modified Screen-Printed Electrode. Biosensors, 10(3), 23. https://doi.org/10.3390/bios10030023