Synthesis, Characterization, and Antimicrobial Activity of Magnesium-Doped Hydroxyapatite Suspensions


 ,
,  ,
, 
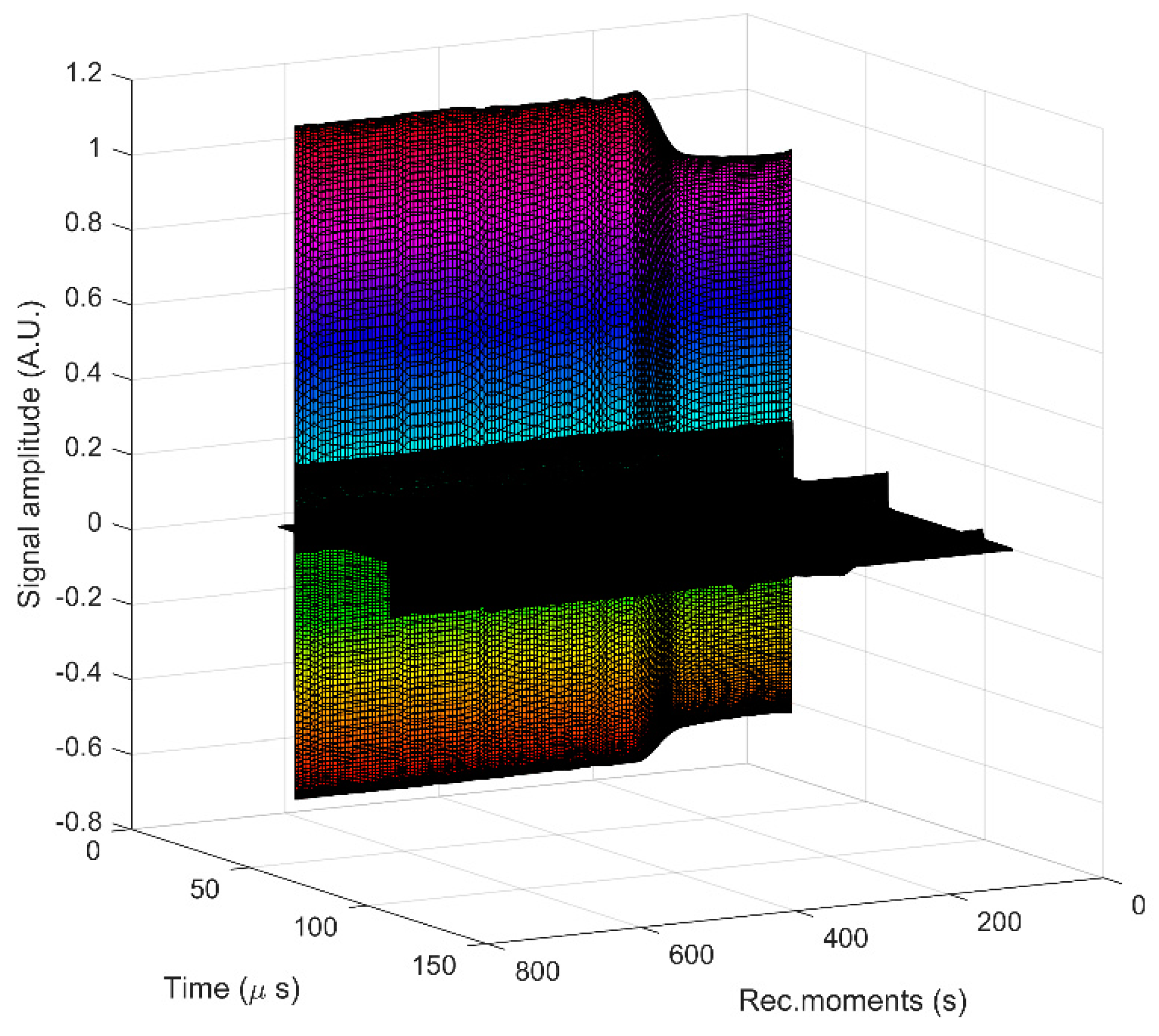{kind=link}
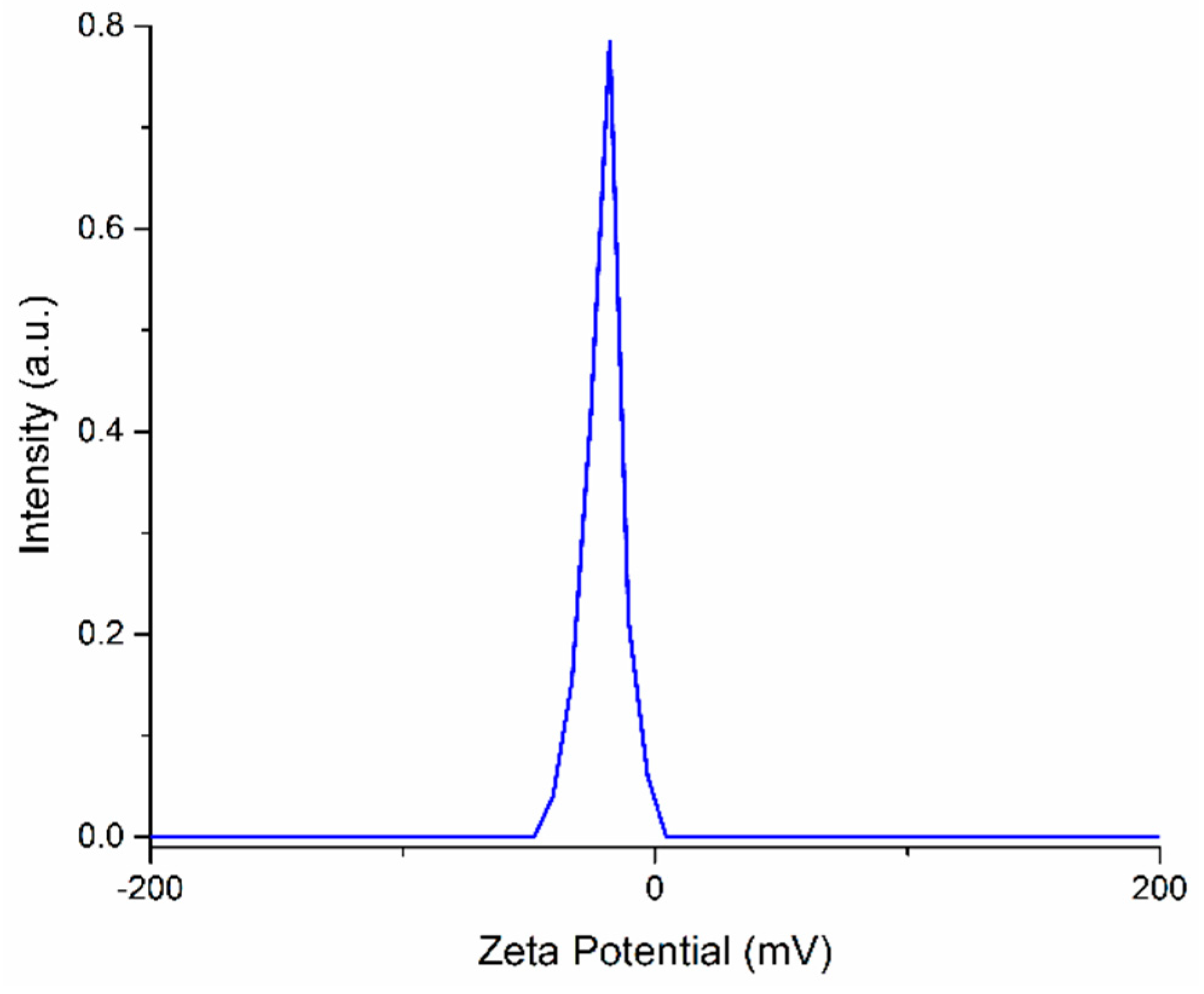{kind=link}
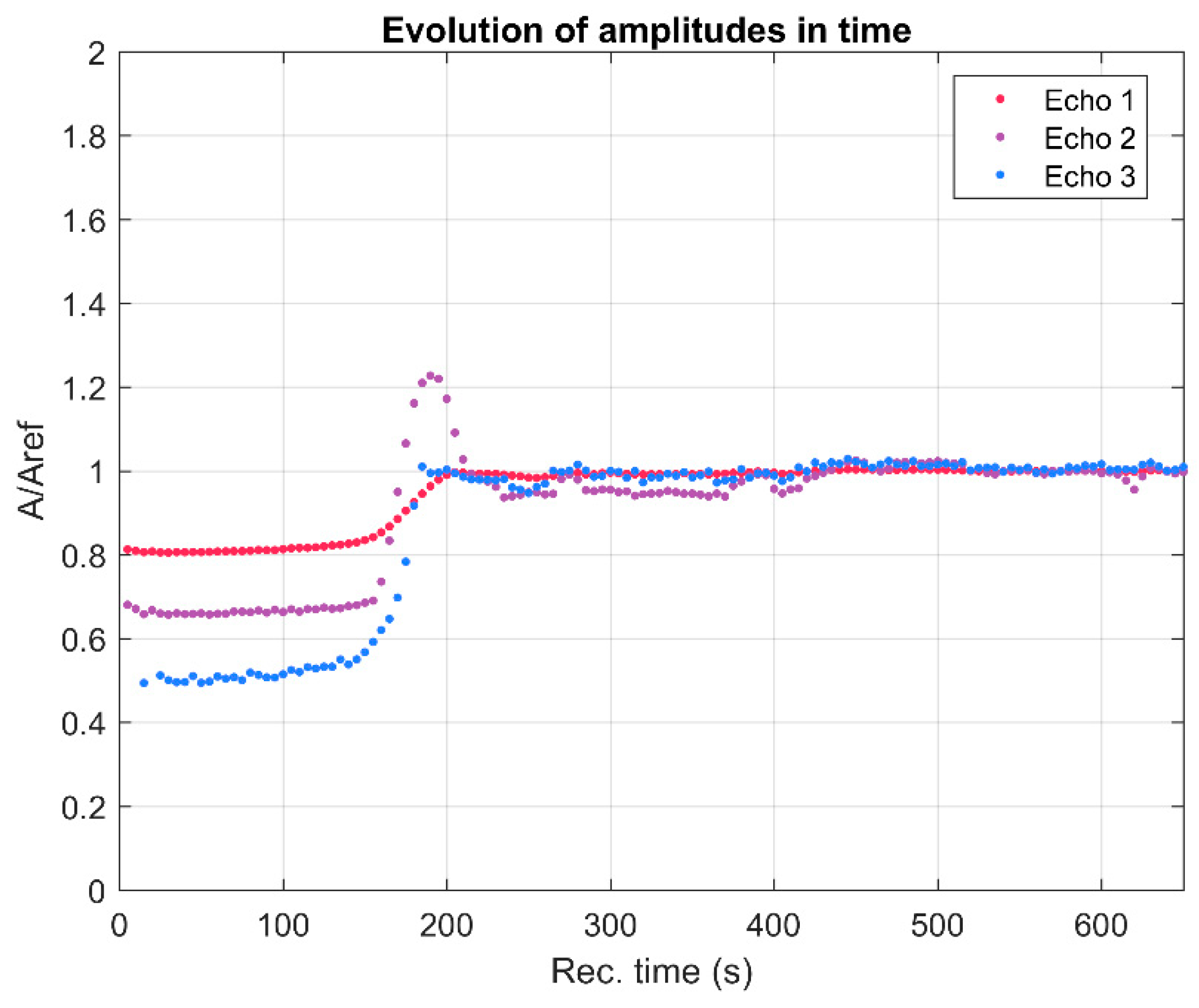{kind=link}
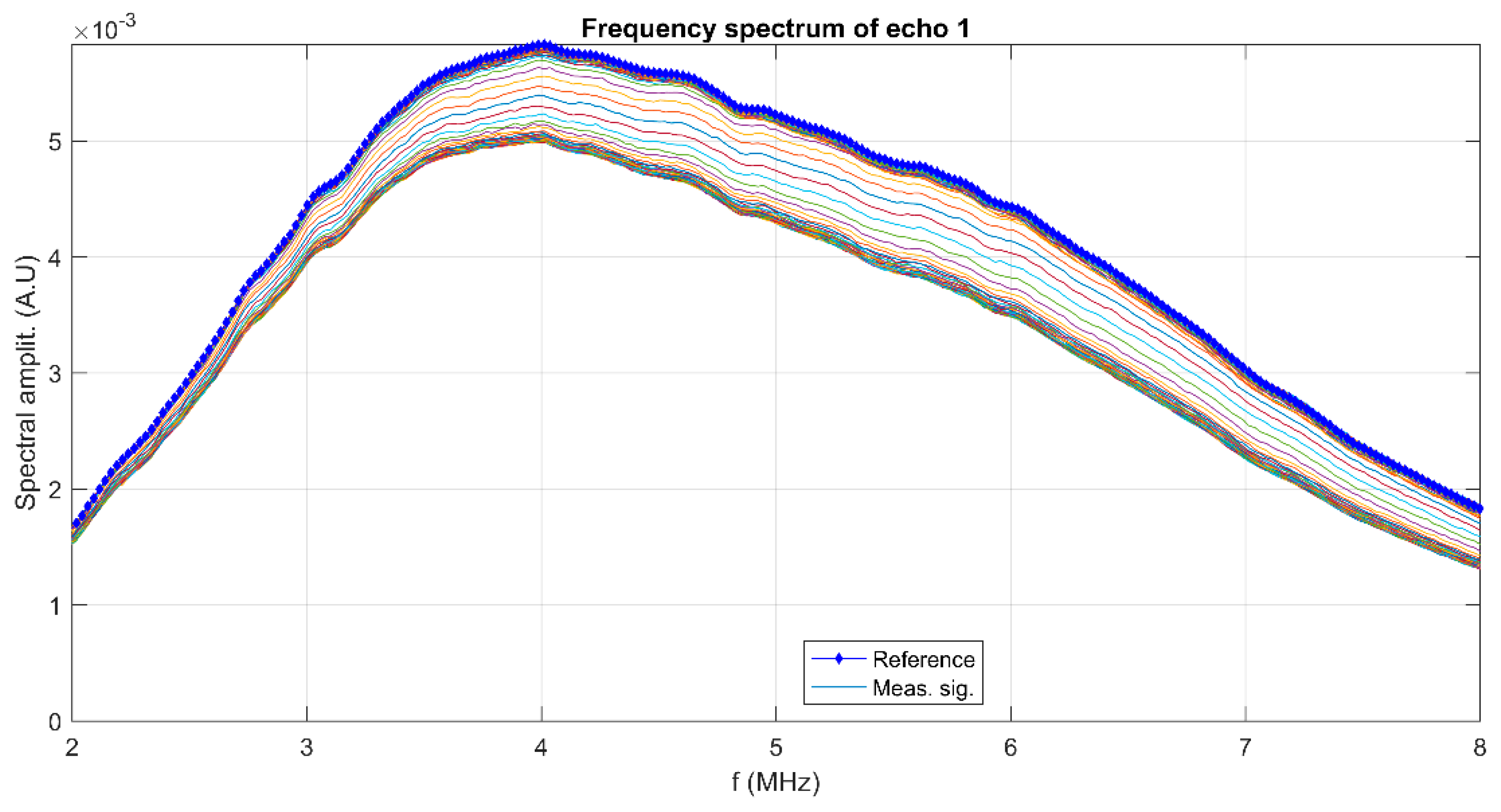{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.1.1. Materials
2.1.2. Magnesium-Doped Hydroxyapatite (MgHAp) Solution
2.2. Characterization Methods
2.3. Antimicrobial Assays
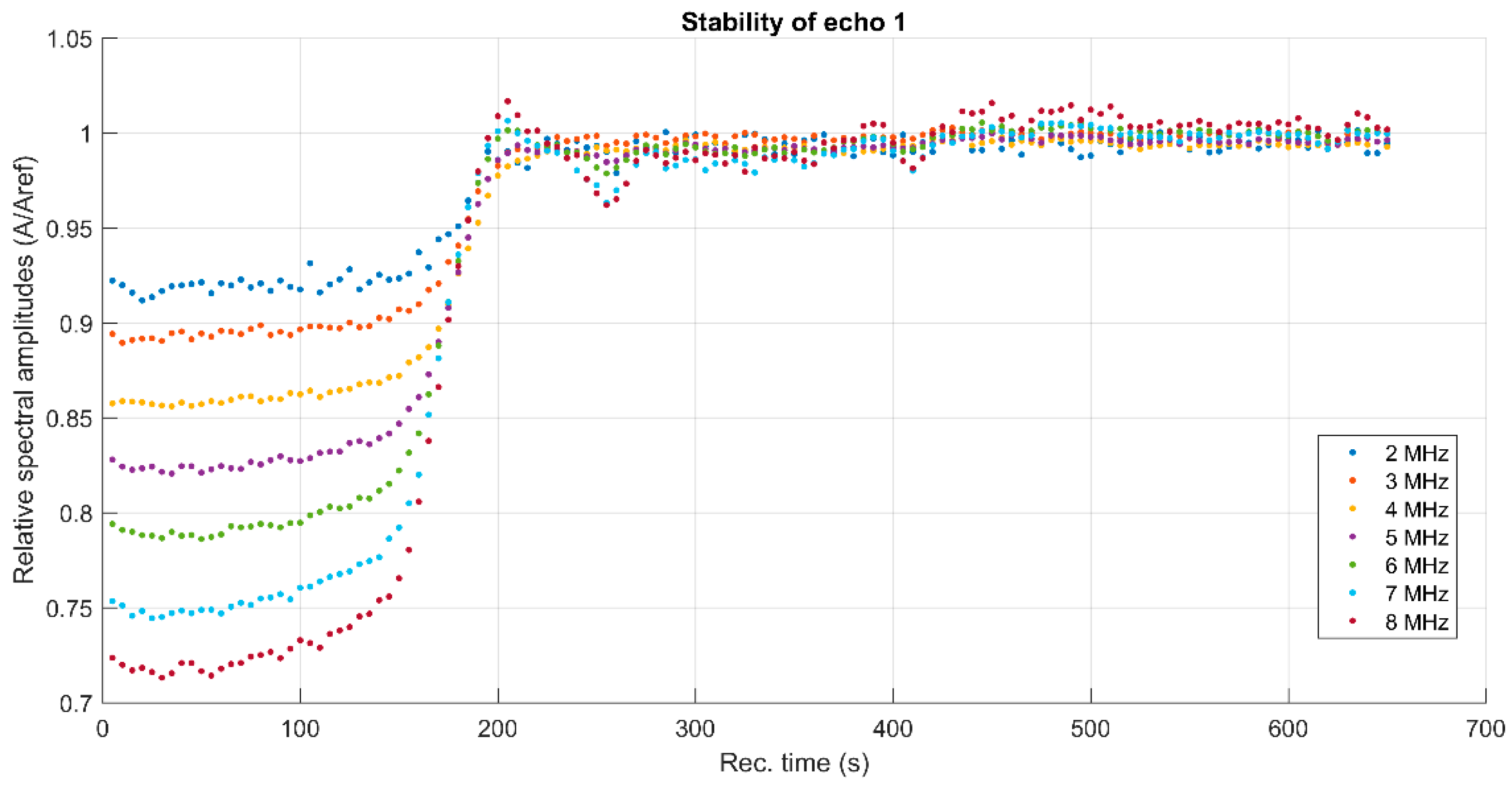
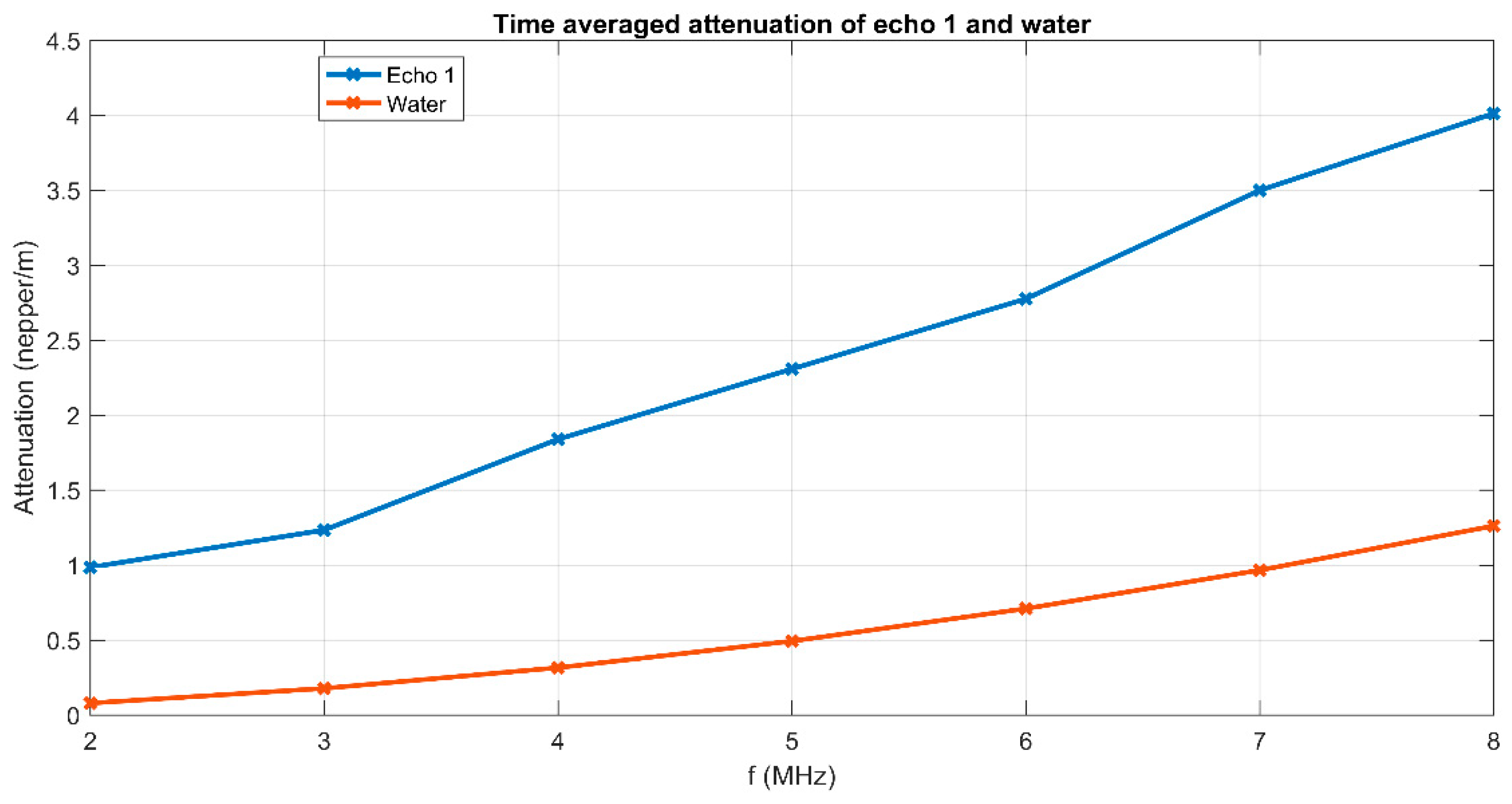
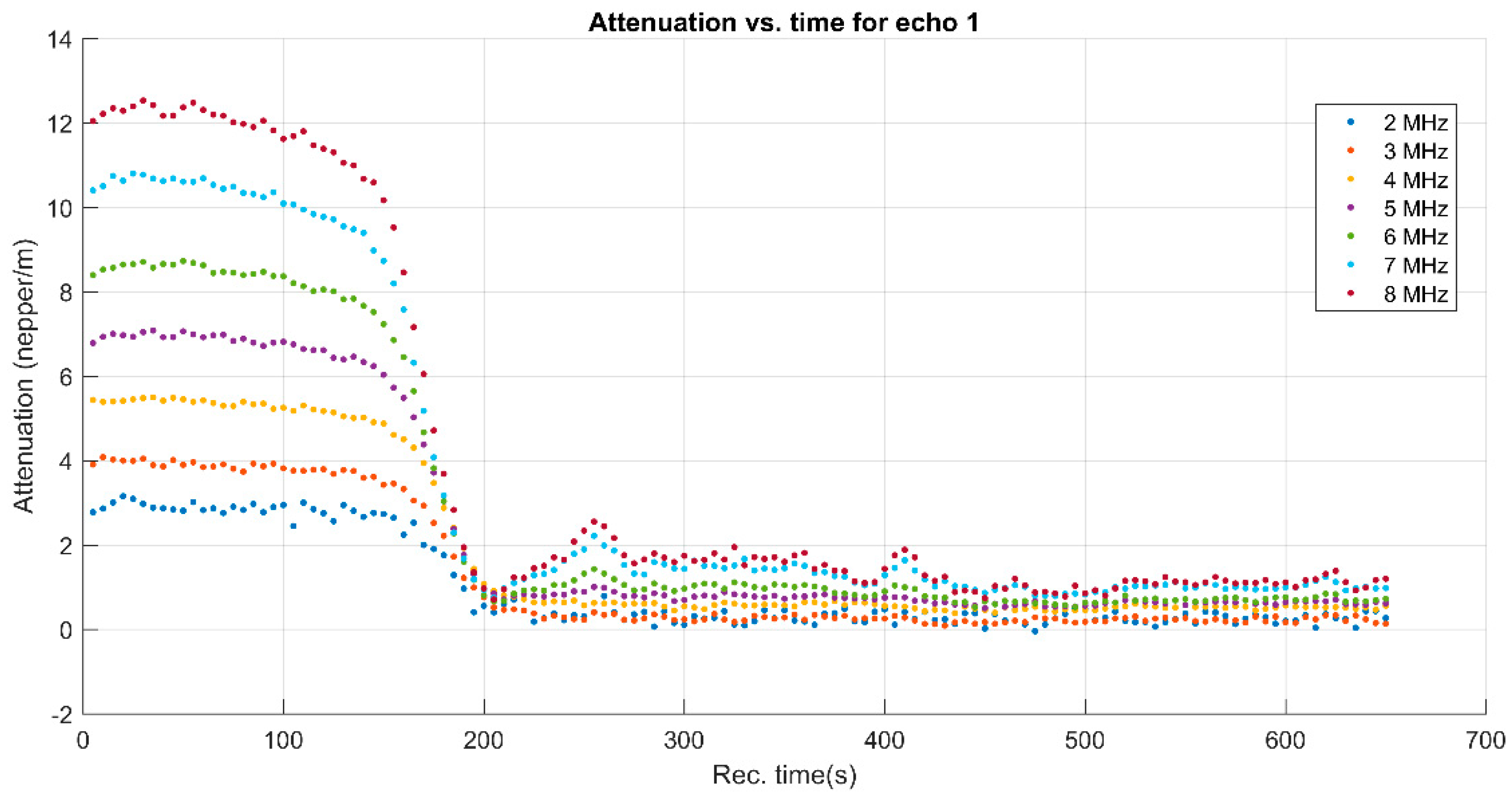
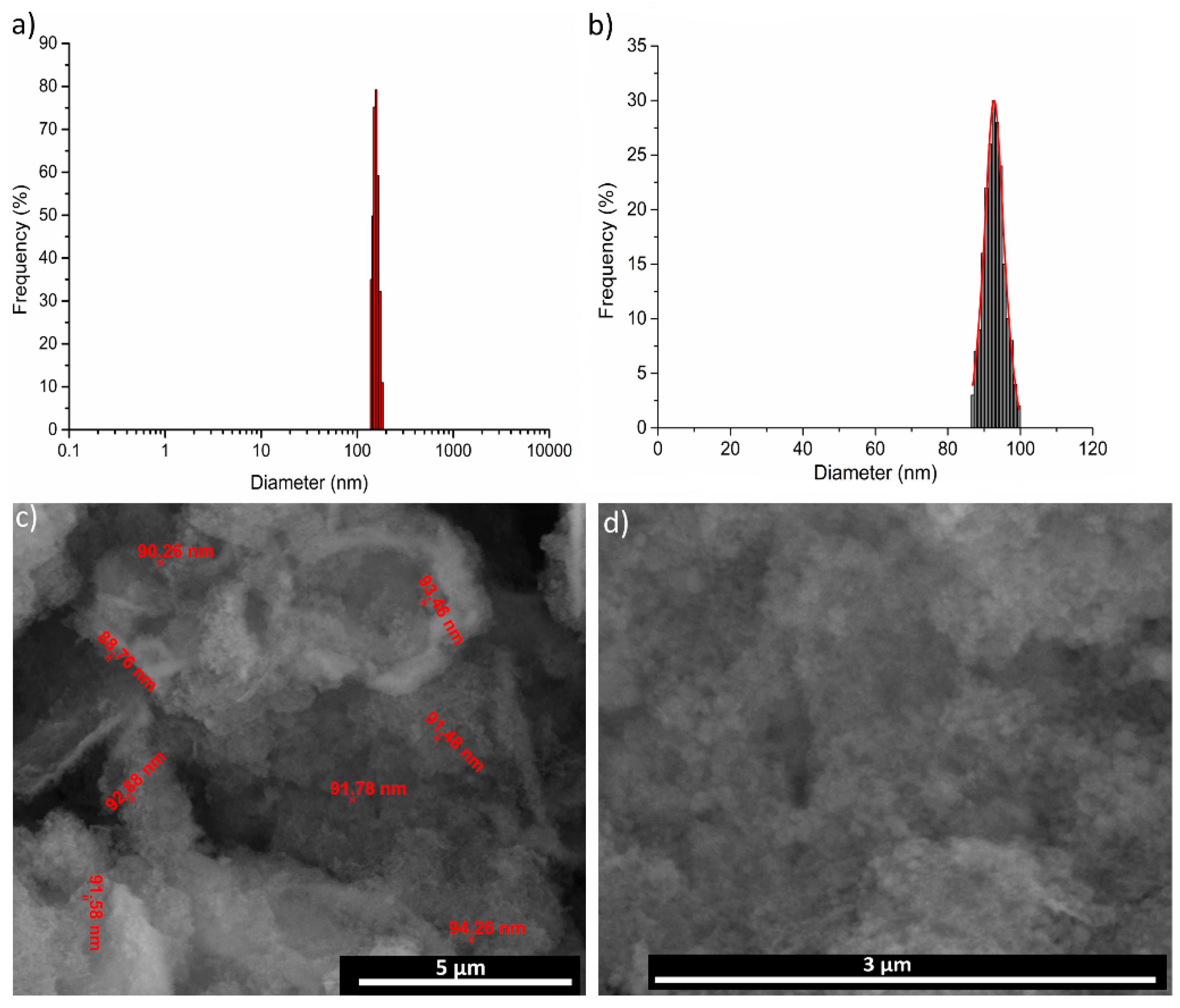
3. Results and Discussions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campana, V.; Milano, G.; Pagano, E.; Barba, M.; Cicione, C.; Salonna, G.; Lattanzi, W.; Logroscino, G. Bone substitutes in orthopaedic surgery: From basic science to clinical practice. J. Mater. Sci. Mater. Med. 2014, 25, 2445–2461. [Google Scholar] [CrossRef] [PubMed]
- Suciu, O.; Bereteu, L.; Drăgănescu, G. Determination of mechanical properties of hydroxyapatite doped with magnesium. AIP Conf. Proc. 2013, 1564, 132. [Google Scholar] [CrossRef]
- Aina, V.; Lusvardi, G.; Annaz, B.; Gibson, I.R.; Imrie, F.E.; Malavasi, G.; Menabue, L.; Cerrato, G.; Martra, G. Magnesium- and strontium-co-substituted hydroxyapatite: The effects of doped-ions on the structure and chemico-physical properties. J. Mater. Sci. Mater. Med. 2012, 23, 2867–2879. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z. Hydroxyapatite and Related Materials; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Qi, G.; Zhang, S.; Khor, K.A.; Lye, S.W.; Zeng, X.; Weng, W.; Liu, C.; Venkatraman, S.S.; Ma, L.L. Osteoblastic cell response on magnesium- incorporated apatite coatings. Appl. Surf. Sci. 2008, 255, 304–307. [Google Scholar] [CrossRef]
- LeGeros, R.Z. Calcium Phosphates in Oral Biology and Medicine; Karger: Basel, Switzerland, 1991. [Google Scholar]
- Zhou, H.; Lee, J. Nanoscale hydroxyapatite particles for bone tissue engineering. Acta Biomater. 2011, 7, 2769–2781. [Google Scholar] [CrossRef] [PubMed]
- Carrodeguas, R.G.; De Aza, S. α-Tricalcium phosphate: Synthesis, properties and biomedical applications. Acta Biomater. 2011, 7, 3536–3546. [Google Scholar] [CrossRef] [PubMed]
- Adzila, S.; Ramesh, S.; Sopyan, I. Properties of magnesium doped nanocrystalline hydroxyapatite synthesize by mechanochemical method. ARPN J. Eng. Appl. Sci. 2016, 11, 14097–14100. [Google Scholar]
- Fu, Y.F.; Chen, D.M. Influence of Sr2+ on strontium substituted hydroxyapatite’s (Sr-doped HA) cytotoxicity. J. Oral Tissue Eng. 2005, 2, 76–78. [Google Scholar]
- Simon, V.; Muresan, D.; Popa, C.; Simon, S. Microscopic analyses of sintered titanium-hydroxyapatite implant materials. J. Optoelectron. Adv. Mater. 2005, 7, 2823–2826. [Google Scholar]
- Xiu, Z.; Lü, M.; Liu, S.; Zhou, G.; Su, B.; Zhang, H. Barium hydroxyapatite nanoparticles synthesized by citric acid sol-gel combustion method. Mater. Res. Bull. 2005, 40, 1617–1622. [Google Scholar] [CrossRef]
- de Lima, I.R.; Alves, G.G.; Soriano, C.A.; Campaneli, A.P.; Gasparoto, T.H.; Ramos, E.S., Jr.; de Sena, L.A.; Rossi, A.M.; Granjeiro, J.M. Understanding the impact of divalent cation substitution on hydroxyapatite: An in vitro multiparametric study on biocompatibility. J. Biomed. Mater. Res. A 2011, 98, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Ergun, C.; Webster, T.J.; Bizios, R.; Doremus, R.H. Hydroxylapatite with substituted magnesium, zinc, cadmium, and yttrium. I. Structure and microstructure. J. Biomed. Mater. Res. 2002, 59, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Predoi, D.; Iconaru, S.L.; Predoi, M.V.; Motelica-Heino, M.; Guegan, R.; Buton, N. Evaluation of antibacterial activity of zinc-doped hydroxyapatite colloids and dispersion stability using ultrasounds. Nanomaterials 2019, 9, 515. [Google Scholar] [CrossRef] [PubMed]
- Predoi, D.; Iconaru, S.L.; Predoi, M.V. Bioceramic layers with antifungal properties. Coatings 2018, 8, 276. [Google Scholar] [CrossRef]
- Nagyné-Kovács, T.; Studnicka, L.; Kincses, A.; Spengler, G.; Molnár, M.; Tolner, M.; Lukács, I.E.; Szilágyi, I.M.; Pokol, G. Synthesis and characterization of Sr and Mg-doped hydroxyapatite by a simple precipitation method. Ceram. Int. 2018, 44, 22976–22982. [Google Scholar] [CrossRef]
- Ciobanu, C.S.; Iconaru, S.L.; Popa, C.L.; Motelica-Heino, M.; Predoi, D. Evaluation of samarium doped hydroxyapatite, ceramics for medical application: Antimicrobial activity. J. Nanomater. 2015, 2015, 849216. [Google Scholar] [CrossRef]
- Ciobanu, C.S.; Iconaru, S.L.; Massuyeau, F.; Constantin, L.V.; Costescu, A.; Predoi, D. Synthesis, structure, and luminescent properties of europium-doped hydroxyapatite nanocrystalline powders. J. Nanomater. 2012, 2012, 942801. [Google Scholar] [CrossRef]
- Tite, T.; Popa, A.-C.; Balescu, L.M.; Bogdan, I.M.; Pasuk, I.; Ferreira, J.M.F.; Stan, G.E. Cationic substitutions in hydroxyapatite: Current status of the derived biofunctional effects and their in vitro interrogation methods. Materials 2018, 11, 2081. [Google Scholar] [CrossRef]
- Li, M.; Xiao, X.; Liu, R.; Chen, C.; Huang, L. Structural characterization of zinc-substituted hydroxyapatite prepared by hydrothermal. J. Mater. Sci. Mater. Med. 2008, 19, 797–803. [Google Scholar] [CrossRef]
- Fadeev, I.V.; Shvorneva, L.I.; Barinov, S.M.; Orlovskii, V.P. Synthesis and structure of magnesium-substituted hydroxyapatite. Inorg. Mater. 2003, 39, 947–950. [Google Scholar] [CrossRef]
- Landi, E.; Tampieri, A.; Mattioli-Belmonte, M.; Celotti, G. Biomimetic Mg-and Mg, CO3-substituted hydroxyapatites: Synthesis characterization and in vitro behavior. J. Eur. Ceram. Soc. 2006, 26, 2593–2601. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2008; pp. 305–306. ISBN 978-0-13-175553-6. [Google Scholar]
- Drouet, C.; Carayon, M.-T.; Combes, C.; Rey, C. Surface enrichment of biomimetic apatites with biologically-active ions Mg2+ and Sr2+ a preamble to the activation of bone repair materials. Mater. Sci. Eng. C 2008, 28, 1544–1550. [Google Scholar] [CrossRef]
- Kannan, S.; Goetz-Neunhoeffer, F.; Neubauer, J.; Pina, S.; Torres, P.M.C.; Ferreira, J.M.F. Synthesis and structural characterization of strontium- and magnesium-co-substituted beta-tricalcium phosphate. Acta Biomater. 2010, 6, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Laurencin, D.; Almora-Barrios, N.; de Leeuw, N.H.; Gervais, C.; Bonhomme, C.; Mauri, F.; Chrzanowski, W.; Knowles, J.C.; Newport, R.J.; Wong, A.; et al. Magnesium incorporation into hydroxyapatite. Biomaterials 2011, 32, 1826–1837. [Google Scholar] [CrossRef] [PubMed]
- Percival, M. Bone health & osteoporosis. Appl. Nutr. Sci. Rep. 1999, 5, 1–6. [Google Scholar]
- Singh, J.; Singh, H.; Batra, U. Magnesium doped hydroxyapatite: Synthesis, characterization and bioactivity evaluation. In Biomaterials Science: Processing, Properties, and Applications V: Ceramic Transactions; Narayan, R., Bose, S., Bandyopadhyay, A., Eds.; Wiley: Hoboken, NJ, USA, 2015; Chapter 15; Volume 254, pp. 161–174. [Google Scholar] [CrossRef]
- Su, Y.; Li, D.; Su, Y.; Lu, C.; Niu, L.; Lian, J.; Li, G. Improvement of the biodegradation property and biomineralization ability of magnesium–hydroxyapatite composites with dicalcium phosphate dihydrate and hydroxyapatite coatings. ACS Biomater. Sci. Eng. 2016, 2, 818–828. [Google Scholar] [CrossRef]
- Adzila, S.; Murad, M.C.; Sopyan, I. Doping metal into calcium phosphate phase for better performance of bone implant materials. Recent Pat. Mater. Sci. 2012, 5, 18–47. [Google Scholar] [CrossRef]
- Tampieri, A.; Celotti, G.; Landi, E.; Sandri, M. Magnesium doped hydroxyapatite: Synthesis and characterization. Key Eng. Mater. 2004, 264, 2051–2054. [Google Scholar] [CrossRef]
- Brooks, E.K.; Ehrensberger, M.T. Bio-Corrosion of Magnesium Alloys for Orthopaedic Applications. J. Funct. Biomater. 2017, 8, 38. [Google Scholar] [CrossRef]
- Driver, M. Coatings for Biomedical Applications; Woodhead Publishing Limited: Cambridge, UK, 2012. [Google Scholar]
- TenHuisen, K.S.; Brown, P.W. Effects of magnesium on the formation of calcium-deficient hydroxyapatite from CaHPO4 · 2H2O and Ca4(PO4)2O. J. Biomed. Mater. Res. 1997, 36, 306–314. [Google Scholar] [CrossRef]
- Bigi, A.; Falini, G.; Foresti, E.; Gazzano, M.; Ripamonti, A.; Roveri, N. Magnesium influence on hydroxyapatite crystallization. J. Inorg. Biochem. 1993, 49, 69–78. [Google Scholar] [CrossRef]
- Kumta, P.N.; Sfeir, C.; Lee, D.-H.; Olton, D.; Choi, D. Nanostructured calcium phosphates for biomedical applications: Novel synthesis and characterization. Acta Biomater. 2005, 1, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, M. Crystallographic behaviour of fluoridated hydroxyapatites containing magnesium and carbonate ions. Biomaterials 1991, 12, 831–835. [Google Scholar] [CrossRef]
- Bigi, A.; Falini, G.; Gazzano, M. Rietveld structure refinements of calcium hydroxylapatite containing magnesium. Acta Crystallogr. Sect. B Struct. Sci. 1996, 52, 87–92. [Google Scholar] [CrossRef]
- Gibson, I.R.; Bonfield, W. Preparation and characterization of magnesium/carbonate co-substituted hydroxyapatites. J. Mater. Sci. Mater. Med. 2002, 13, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, C.; Wen, J.; Li, X.; Fu, L. Synthesis and structural characteristics of magnesium and zinc doped hydroxyapatite whiskers. Ceram. Silik. 2017, 61, 244–249. [Google Scholar] [CrossRef][Green Version]
- Predoi, D.; Iconaru, S.L.; Predoi, M.V. Dextran-coated zinc-doped hydroxyapatite for biomedical applications. Polymers 2019, 11, 886. [Google Scholar] [CrossRef]
- Rietveld, H. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Popa, N.C. The (hkl) dependence of diffraction-line broadening caused by strain and size for all Laue groups in Rietveld refinement. J. Appl. Crystallogr. 1998, 31, 176–180. [Google Scholar] [CrossRef]
- Greenwood, R.; Kendall, K. Selection of suitable dispersants for aqueous suspensions of zirconia and titania powders using acoustophoresis. J. Eur. Ceram. Soc. 1999, 19, 479–488. [Google Scholar] [CrossRef]
- O’Brien, R.W.; Midmore, B.R.; Lamb, A.; Hunter, R.J. Electroacoustic studies of moderately concentrated colloidal suspensions. Faraday Discuss. Chem. Soc. 1990, 90, 301–312. [Google Scholar] [CrossRef]
- Hanaor, D.A.H.; Michelazzi, M.; Leonelli, C.; Sorrell, C.C. The effects of carboxylic acids on the aqueous dispersion and electrophoretic deposition of ZrO2. J. Eur. Ceram. Soc. 2012, 32, 235–244. [Google Scholar] [CrossRef]
- Fairhurst, D. An Overview of the Zeta Potential Part 3: Uses and Applications, American Pharmaceutical Review. 2013. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/139288-An-Overview-of-the-Zeta-Potential-Part-3-Uses-and-Applications/ (accessed on 2 August 2019).
- Ciobanu, C.S.; Iconaru, S.L.; Pasuk, I.; Vasile, B.S.; Lupu, A.R.; Hermenean, A.; Dinischiotu, A.; Predoi, D. Structural properties of silver doped hydroxyapatite and their biocompatibility. Mater. Sci. Eng. C 2013, 33, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Popa, C.L.; Deniaud, A.; Michaud-Soret, I.; Guégan, R.; Motelica-Heino, M.; Predoi, P. Structural and biological assessment of zinc doped hydroxyapatite nanoparticles. J. Nanomater. 2016, 2016, 1062878. [Google Scholar] [CrossRef]
- Markovic, M.; Fowler, B.O.; Tung, M.S. Preparation and comprehensive characterization of a calcium hydroxyapatite reference material. J. Res. Natl. Inst. Stand. Technol. 2004, 109, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, A.C.; Miculescu, M.; Machedon-Pisu, T.; Maidaniuc, A.; Ciocoiu, R.C.; Ionita, M.; Pasuk, I.; Stan, G.E.; Miculescu, F. Internal and external surface features of newly developed porous ceramics with random interconnected 3D channels by a fibrous sacrificial porogen method. Appl. Surf. Sci. 2019, 489, 226–238. [Google Scholar] [CrossRef]
- Madupalli, H.; Pavan, B.; Tecklenburg, M.M.J. Carbonate substitution in the mineral component of bone: Discriminating the structural changes, simultaneously imposed by carbonate in A and B sites of apatite. J. Solid State Chem. 2017, 255, 27–35. [Google Scholar] [CrossRef]
- Spence, G.; Phillips, S.; Campion, C.; Brooks, R.; Rushton, N. Bone formation in a carbonate-substituted hydroxyapatite implant is inhibited by zoledronate: The importance of bioresorption to osteoconduction. J. Bone Jt. Surg. Br. 2008, 90, 1635–1640. [Google Scholar] [CrossRef]
- Sima, L.E.; Stan, G.E.; Morosanu, C.O.; Melinescu, A.; Ianculescu, A.; Melinte, R.; Neamtu, J.; Petrescu, S.M. Differentiation of mesenchymal stem cells onto highly adherent radio frequency-sputtered carbonated hydroxylapatite thin films. J. Biomed. Mater. Res. A 2010, 95, 1203–1214. [Google Scholar] [CrossRef]
- Germaini, M.M.; Detsch, R.; Grünewald, A.; Magnaudeix, A.; Lalloue, F.; Boccaccini, A.R.; Champion, E. Osteoblast and osteoclast responses to A/B type carbonate-substituted hydroxyapatite ceramics for bone regeneration. Biomed. Mater. Biomed. Mater. 2017, 12, 035008. [Google Scholar] [CrossRef]
- Cumber, S.A.; Lead, J.R. Particle size distributions of silver nanoparticles at environmentally relevant conditions. J. Chromatogr. A 2009, 1216, 9099–9105. [Google Scholar] [CrossRef]
- Huang, J.; Li, Q.; Sun, D.; Lu, Y.; Su, Y.; Yang, X.; Wang, H.; Wang, Y.; Shao, W.; He, N.; et al. Biosynthesis of silver and gold nanoparticles by novel sundried Cinnamomum camphora leaf. Nanotechnology 2007, 18, 105104. [Google Scholar] [CrossRef]
- Moorthi, C.; Kathiresan, K. Application of Plackett Burman factorial design in the development of curcumin loaded Eudragit E 100 nanoparticles. Nano Biomed. Eng. 2013, 5, 28–33. [Google Scholar] [CrossRef]
- Ibrahim, A.I.O.; Moodley, D.S.; Petrik, L.; Patel, N. Use of antibacterial nanoparticles in Endodontics. S. Afr. Dent. J. 2017, 72, 105–112. [Google Scholar]
- Wu, D.; Fan, W.; Kishen, A.; Gutmann, J.L.; Fan, B. Evaluation of the antibacterial efficacy of silver nanoparticles against Enterococcus faecalis biofilm. Int. Endod. J. 2014, 40, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Beyth, N.; Houri-Haddad, Y.; Domb, A.; Khan, W.; Hazan, R. Alternative antimicrobial approach: Nano-antimicrobial materials. Evid. Based Complement. Altern. Med. 2015, 2015, 246012. [Google Scholar] [CrossRef]
- Iconaru, S.L.; Prodan, A.M.; Motelica-Heino, M.; Sizaret, S.; Predoi, D. Synthesis and characterization of polysaccharide-maghemite composite nanoparticles and their antibacterial properties. Nanoscale Res. Lett. 2012, 7, 576. [Google Scholar] [CrossRef]
- Vincent, M.; Duval, R.E.; Hartemann, P.; Engels-Deutsch, M. Contact killing and antimicrobial properties of copper. J. Appl. Microbiol. 2018, 124, 1032–1046. [Google Scholar] [CrossRef]
- Monzavi, A.; Eshraghi, S.; Hashemian, R.; Momen-Heravi, F. In vitro and ex vivo antimicrobial efficacy of nano-MgO in the elimination of endodontic pathogens. Clin. Oral Investig. 2015, 19, 349–356. [Google Scholar] [CrossRef]
- Lellouche, J.; Kahana, E.; Elias, S.; Gedanken, A.; Banin, E. Antibiofilm activity of nanosized magnesium fluoride. Biomaterials 2009, 30, 5969–5978. [Google Scholar] [CrossRef]
- Huang, L.; Li, D.-Q.; Lin, Y.-J.; Wei, M.; Evans, D.G.; Duan, X. Controllable preparation of Nano-MgO and investigation of its bactericidal properties. J. Inorg. Biochem. 2005, 99, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Sabella, S.; Carney, R.P.; Brunetti, V.; Malvindi, A.M.; Al-Juffali, N.; Vecchio, G.; Janes, S.M.; Bakr, O.M.; Cingolani, R.; Stellacci, F.; et al. A general mechanism for in-tracellular toxicity of metal-containing nanoparticles. Nanoscale 2014, 6, 7052–7061. [Google Scholar] [CrossRef] [PubMed]
- Ghobadian, M.; Nabiuni, M.; Parivar, K.; Fathi, M.; Pazooki, J. Toxic effects of magnesium oxide nanoparticles on early developmental and larval stages of zebrafish (Danio rerio). Ecotoxicol. Environ. Saf. 2015, 122, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.H.; Ng, A.M.; Xu, X.; Shen, Z.; Gethings, L.A.; Wong, M.T.; Chan, C.M.; Guo, M.Y.; Ng, Y.H.; Djurišić, A.B.; et al. Mechanisms of antibacterial activity of MgO: Non-ROS mediated toxicity of MgO nanoparticles towards Escherichia coli. Small 2013, 10, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Manke, A.; Wang, L.; Rojanasakul, Y. Mechanisms of nano-particle-induced oxidative stress and toxicity. BioMed Res. Int. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Kalaichelvan, P.T. Ecotoxicity of nanoparticles. ISRN Toxicol. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Vijayakumar, S.; Thanigaivel, S.; Mukherjee, A.; Chandrasekaran, N. Toxicity of magnesium oxide nanoparticles in two fresh water fishes tilapia (Oreochromis mossambicus) and zebrafish (Danio rerio). Int. J. Pharm. Pharm. Sci. 2014, 6, 487–490. [Google Scholar]
- Heimann, R.B.; Lehmann, H.D. Recent Research and Patents on Controlling Corrosion of Bioresorbable Mg Alloy Implants: Towards Next Generation Biomaterials. Recent Pat. Mate. Sci. 2017, 10, 2–19. [Google Scholar] [CrossRef]
- Heimann, R.B. Osseoconductive and Corrosion-Inhibiting Plasma-Sprayed Calcium Phosphate Coatings for Metallic Medical Implants. Metals 2017, 7, 468. [Google Scholar] [CrossRef]
- Zhang, E.; Xu, L.; Yu, G.; Pan, F.; Yang, K. In vivo evaluation of biodegradable magnesium alloy bone implants in the first 6 months implantation. J. Biomed. Mater. Res. A 2009, 90, 882–893. [Google Scholar] [CrossRef]
- Cizek, J.; Matejicek, J. Medicine Meets Thermal Spray Technology: A Review of Patents. J. Therm. Spray Technol. 2018, 27, 1251–1279. [Google Scholar] [CrossRef]
- Costescu, A.; Ciobanu, C.S.; Iconaru, S.L.; Ghita, R.V.; Chifiriuc, C.M.; Marutescu, L.G.; Predoi, D. Fabrication, characterization, and antimicrobial activity, evaluation of low silver concentrations in silver-doped hydroxyapatite nanoparticles. J. Nanomater. 2013, 2013, 194854. [Google Scholar] [CrossRef]
- Iconaru, S.L.; Mikael, M.H.; Predoi, D. Study on europium-doped hydroxyapatite nanoparticles by Fourier-transform infrared spectroscopy and their antimicrobial properties. J. Spectrosc. 2013, 2013, 284285. [Google Scholar] [CrossRef]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Predoi, D.; Iconaru, S.L.; Predoi, M.V.; Stan, G.E.; Buton, N. Synthesis, Characterization, and Antimicrobial Activity of Magnesium-Doped Hydroxyapatite Suspensions. Nanomaterials 2019, 9, 1295. https://doi.org/10.3390/nano9091295
Predoi D, Iconaru SL, Predoi MV, Stan GE, Buton N. Synthesis, Characterization, and Antimicrobial Activity of Magnesium-Doped Hydroxyapatite Suspensions. Nanomaterials. 2019; 9(9):1295. https://doi.org/10.3390/nano9091295
Chicago/Turabian StylePredoi, Daniela, Simona Liliana Iconaru, Mihai Valentin Predoi, George E. Stan, and Nicolas Buton. 2019. "Synthesis, Characterization, and Antimicrobial Activity of Magnesium-Doped Hydroxyapatite Suspensions" Nanomaterials 9, no. 9: 1295. https://doi.org/10.3390/nano9091295
APA StylePredoi, D., Iconaru, S. L., Predoi, M. V., Stan, G. E., & Buton, N. (2019). Synthesis, Characterization, and Antimicrobial Activity of Magnesium-Doped Hydroxyapatite Suspensions. Nanomaterials, 9(9), 1295. https://doi.org/10.3390/nano9091295