Detwinning Mechanism for Nanotwinned Cubic Boron Nitride with Unprecedented Strength: A First-Principles Study


Abstract

1. Introduction
2. Methods
3. Results and Discussions
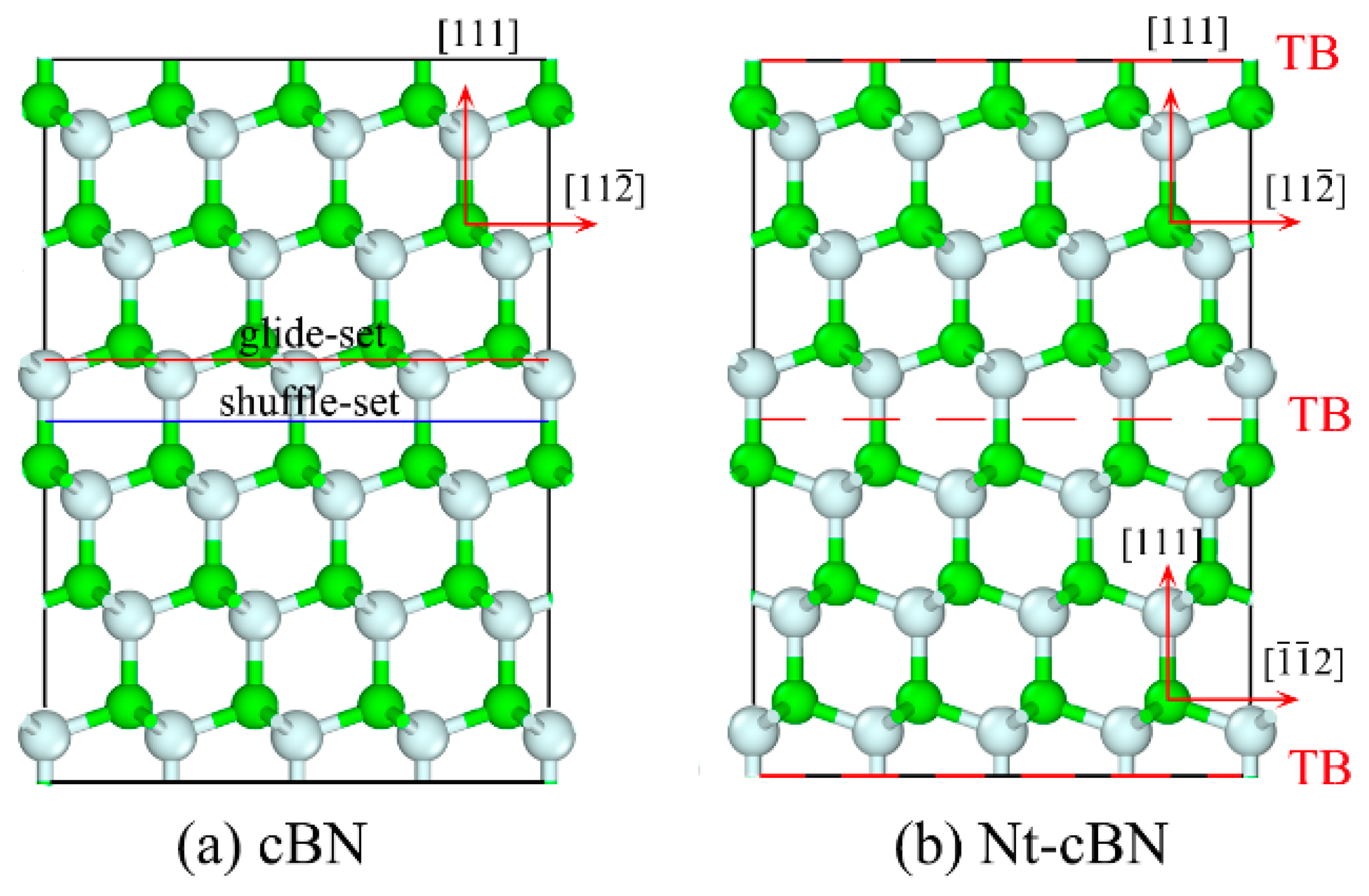
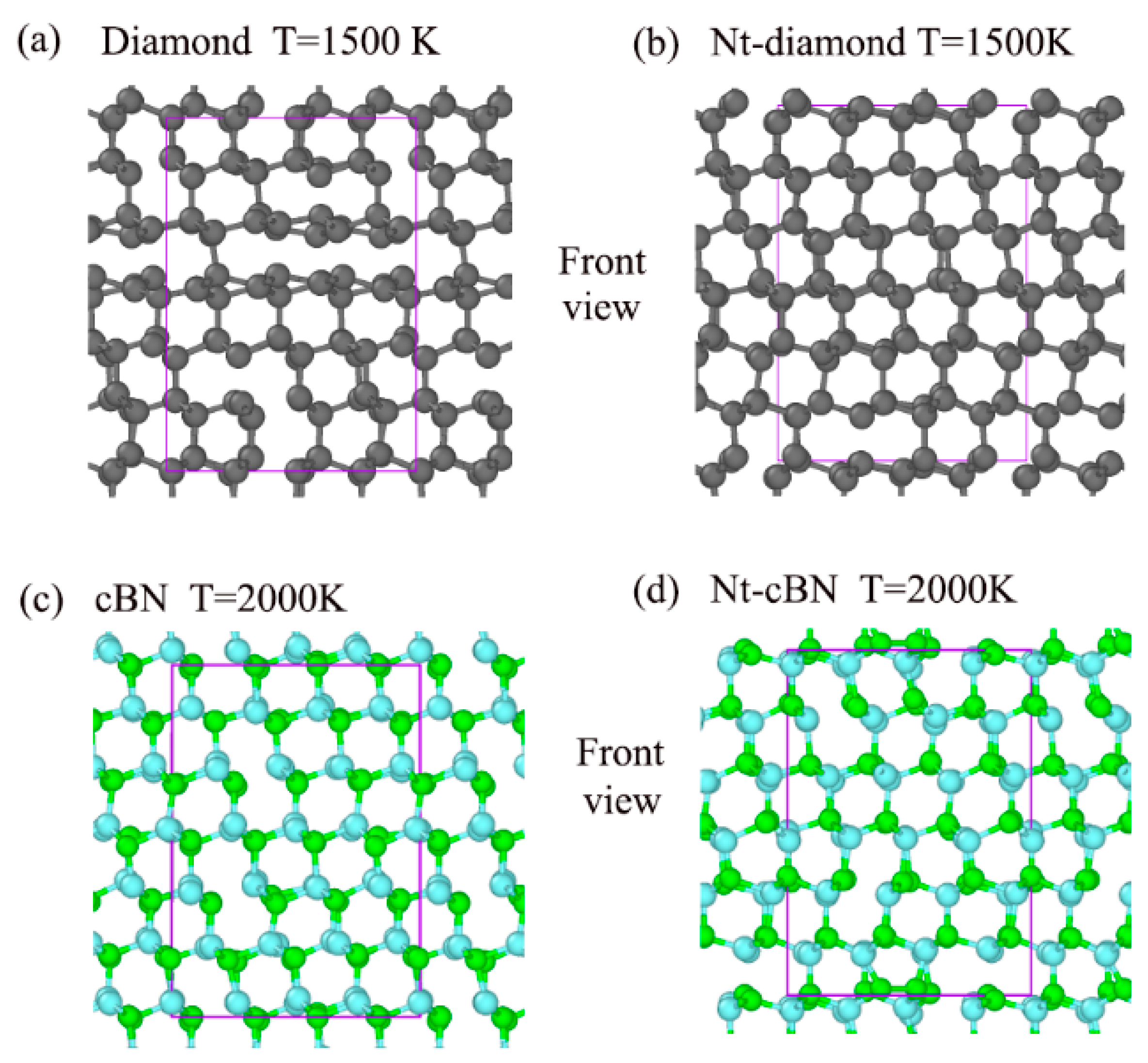
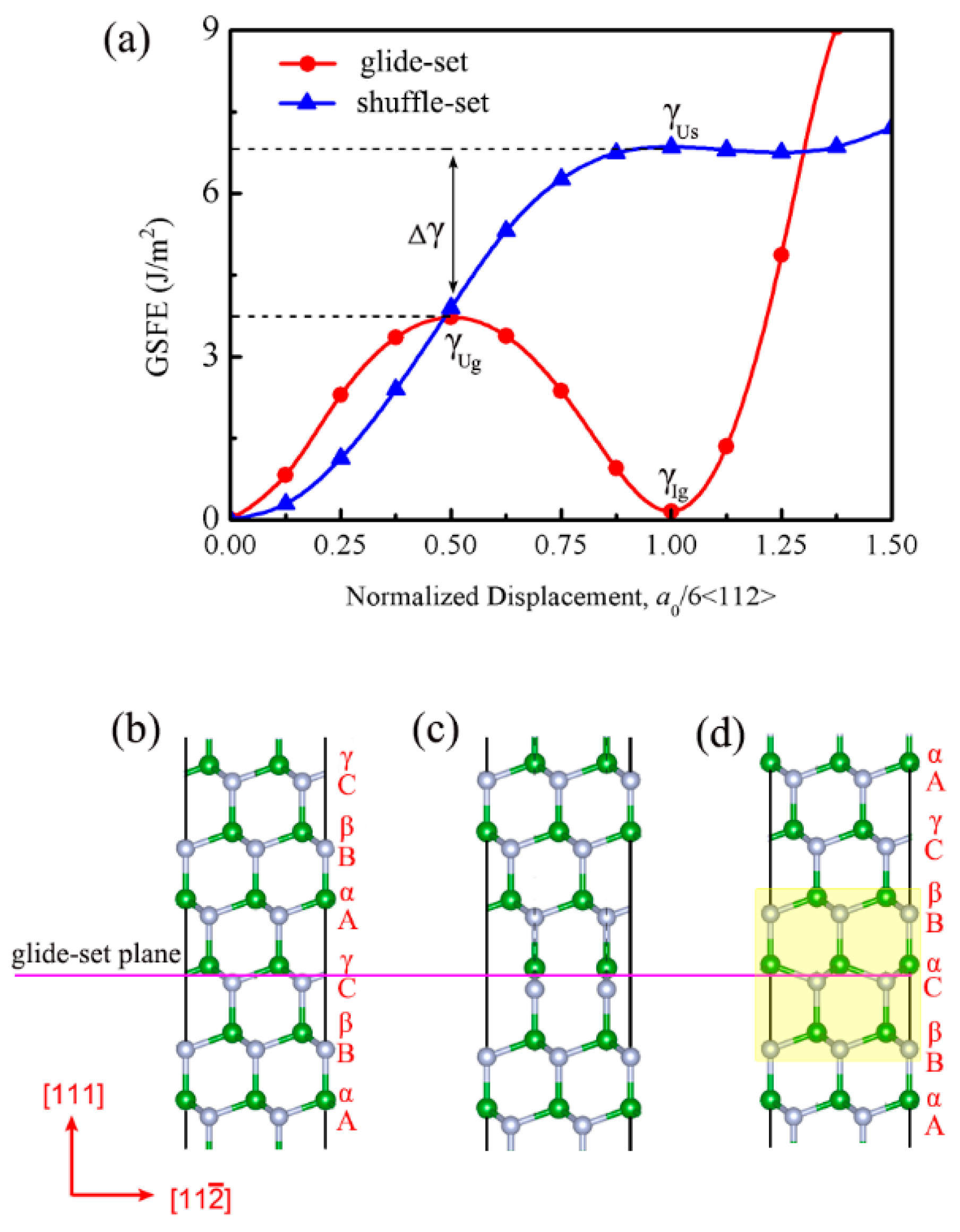
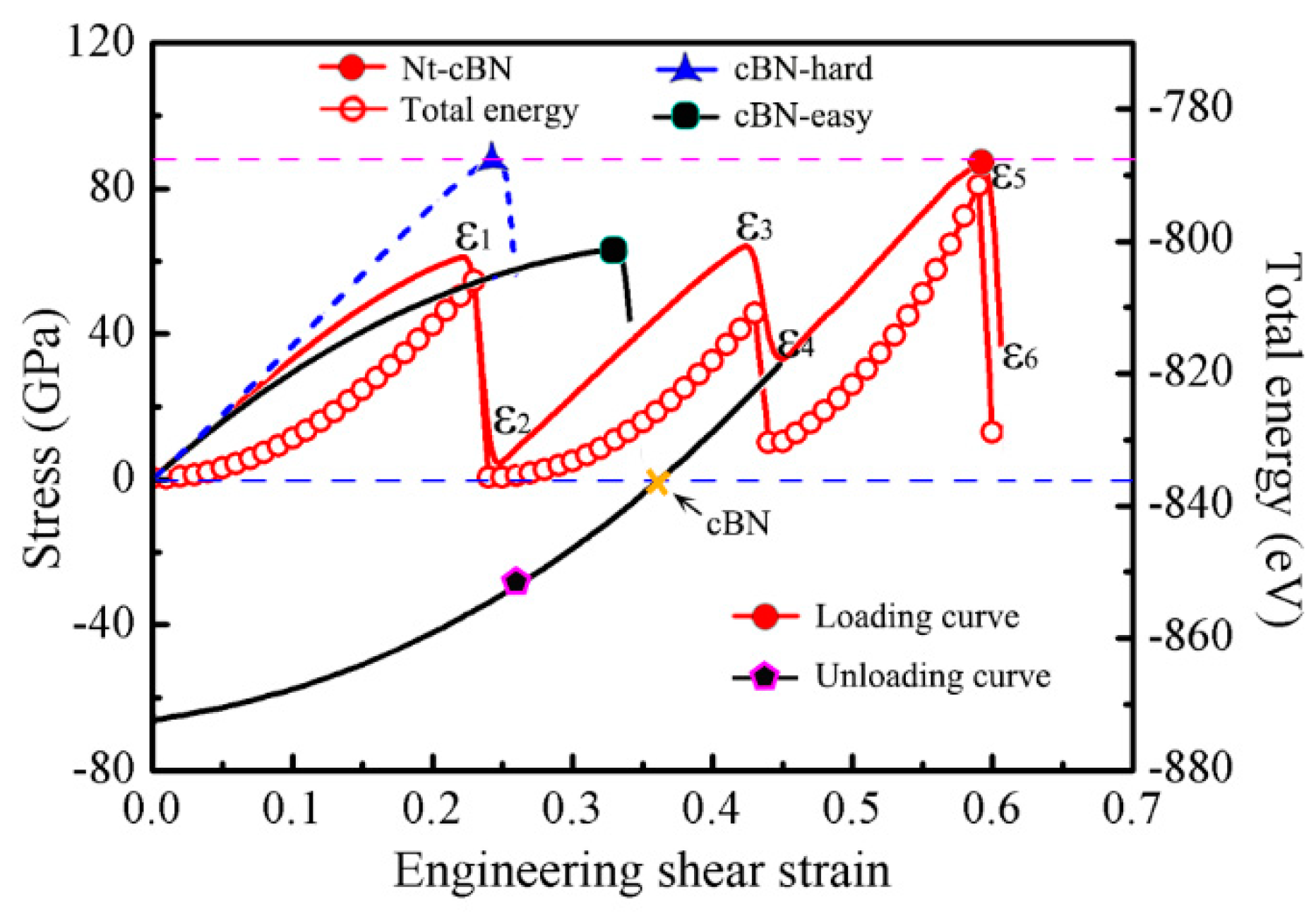
3.1. Excellent Stability of Nanotwinned Structure
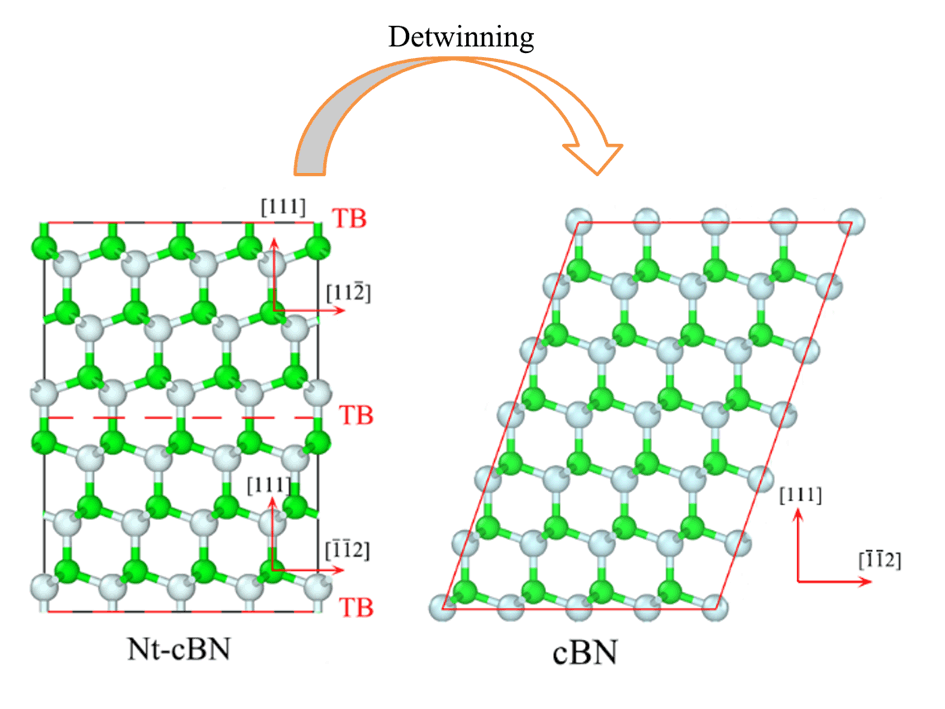
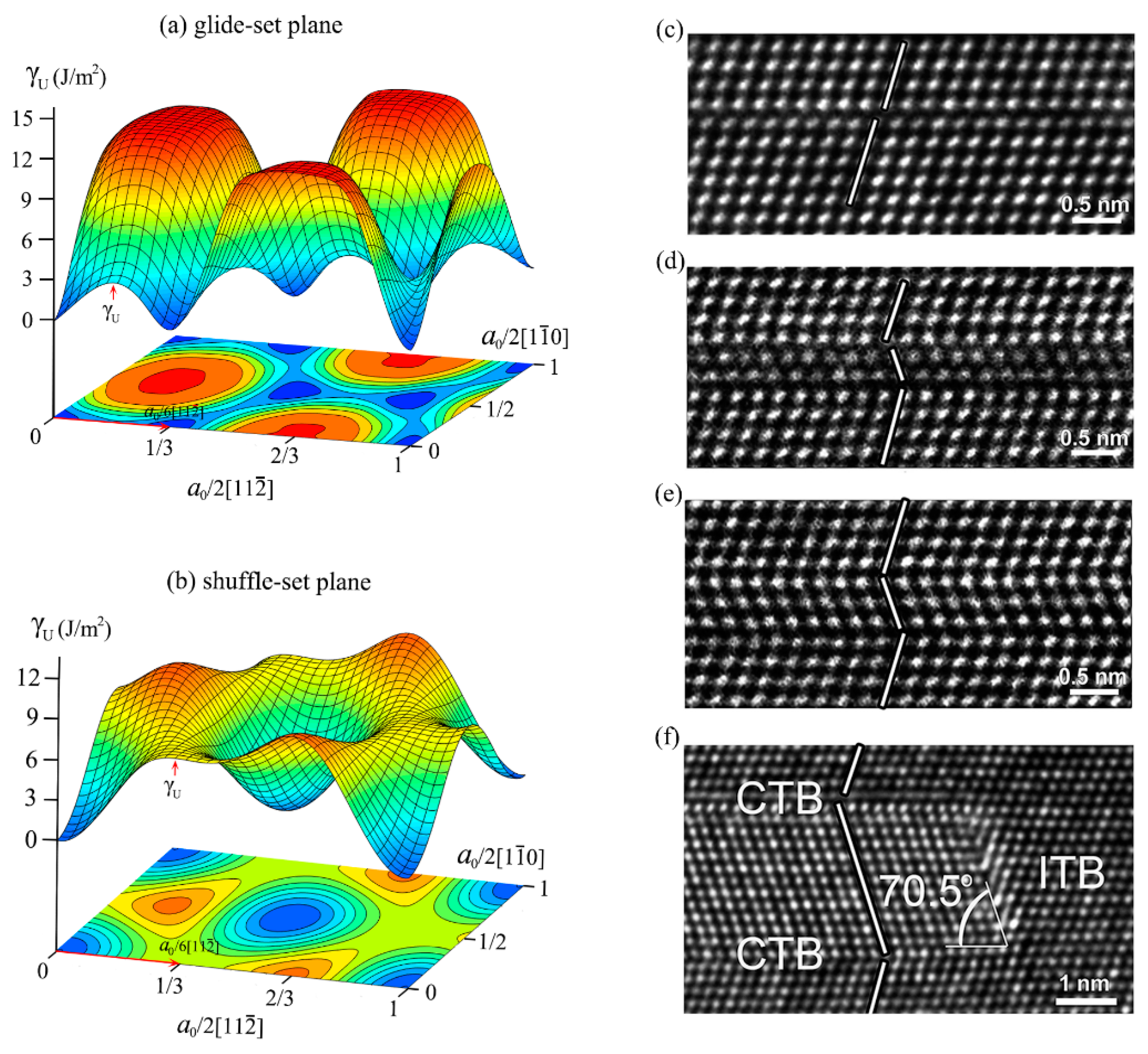
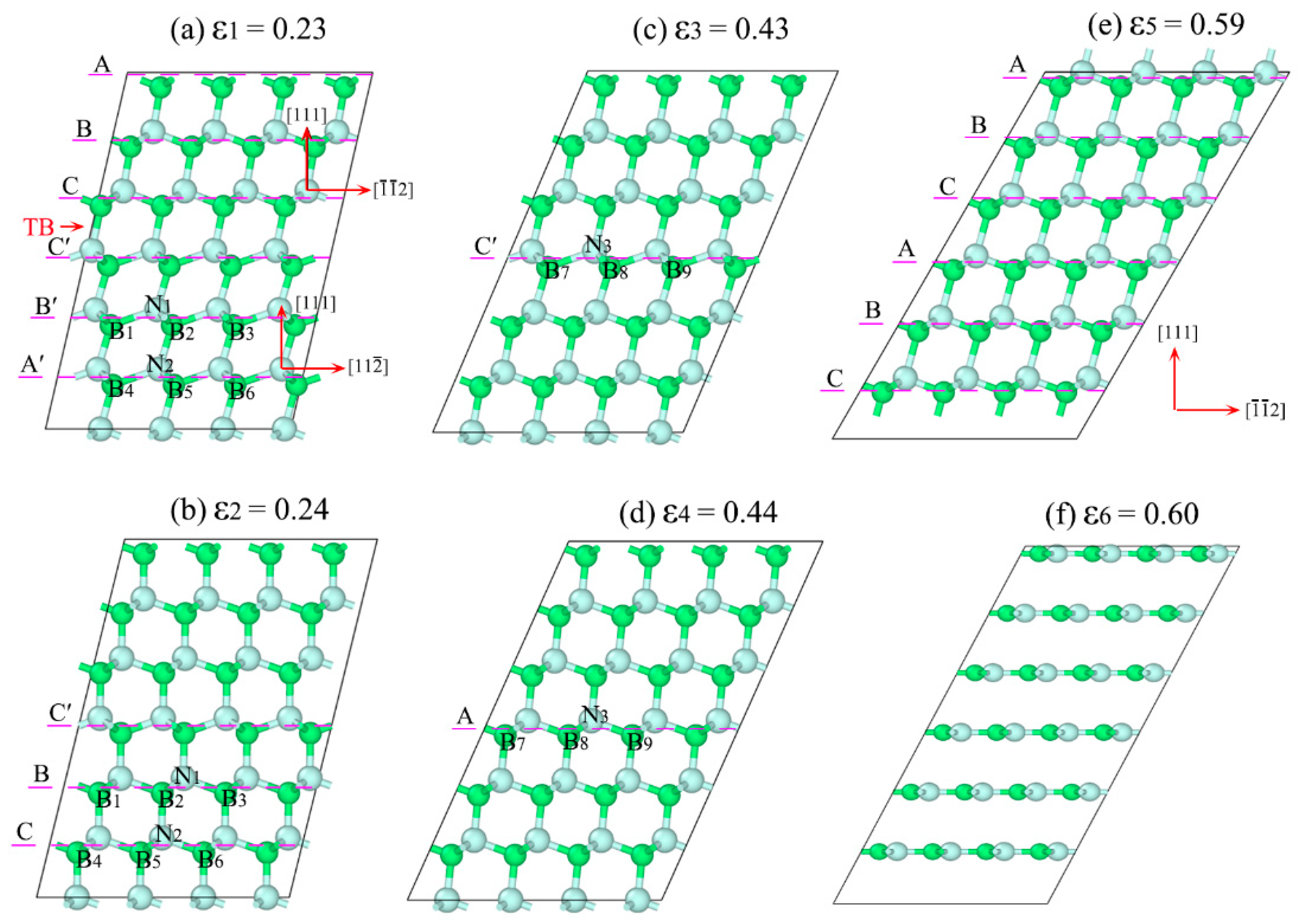
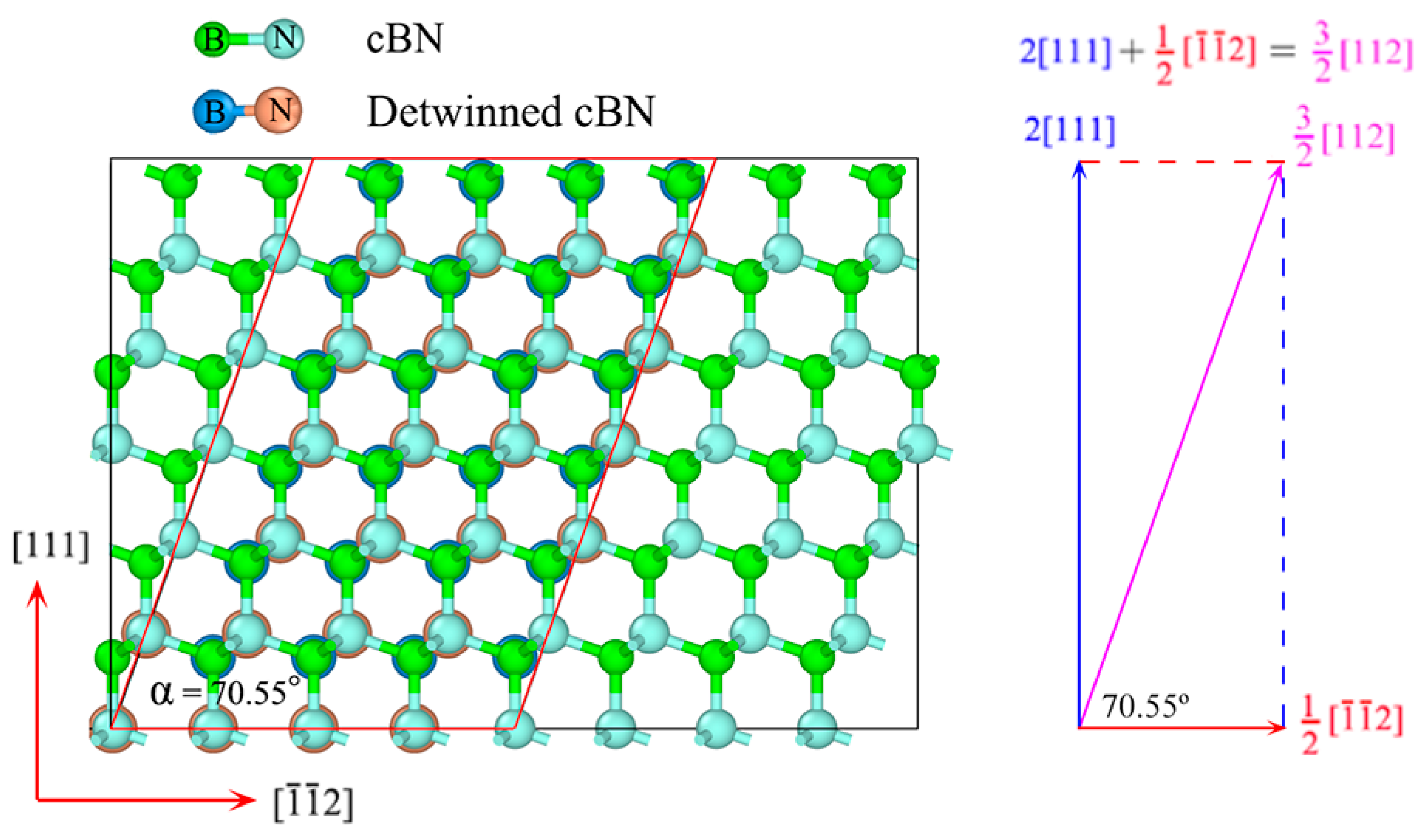
3.2. Detwinning Mechanism of nt-cBN
4. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, C.; Li, Q.; Ma, Y.; Chen, C. Extraordinary indentation strain stiffening produces superhard tungsten nitrides. Phys. Rev. Lett. 2017, 119, 115503. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Peng, X.; Fu, T.; Zhao, Y.; Feng, C.; Lin, Z.; Li, Q. Nanoindentation of ultra-hard cbn films: A molecular dynamics study. Appl. Surf. Sci. 2017, 392, 215–224. [Google Scholar] [CrossRef]
- An, Q.; Goddard, W.A., 3rd. Atomistic origin of brittle failure of boron carbide from large-scale reactive dynamics simulations: Suggestions toward improved ductility. Phys. Rev. Lett. 2015, 115, 105501. [Google Scholar] [CrossRef] [PubMed]
- Brazhkin, V.V.; Lyapin, A.G.; Hemley, R.J. Harder than diamond: Dreams and reality. Philos. Mag. A 2002, 82, 231–253. [Google Scholar] [CrossRef]
- Huang, C.; Peng, X.; Yang, B.; Xiang, H.; Sun, S.; Chen, X.; Li, Q.; Yin, D.; Fu, T. Anisotropy effects in diamond under nanoindentation. Carbon 2018, 132, 606–615. [Google Scholar] [CrossRef]
- Huang, C.; Peng, X.; Yang, B.; Chen, X.; Li, Q.; Yin, D.; Fu, T. Effects of strain rate and annealing temperature on tensile properties of nanocrystalline diamond. Carbon 2018, 136, 320–328. [Google Scholar] [CrossRef]
- Yang, B.; Peng, X.; Huang, C.; Yin, D.; Xiang, H.; Fu, T. Higher strength and ductility than diamond: Nanotwinned diamond/cubic boron nitride multilayer. ACS Appl. Mater. Int. 2018, 10, 42804–42811. [Google Scholar] [CrossRef]
- Yang, B.; Peng, X.; Huang, C.; Wang, Z.; Yin, D.; Fu, T. Strengthening and toughening by partial slip in nanotwinned diamond. Carbon 2019, 150, 1–7. [Google Scholar] [CrossRef]
- Tanigaki, K.; Ogi, H.; Sumiya, H.; Kusakabe, K.; Nakamura, N.; Hirao, M.; Ledbetter, H. Observation of higher stiffness in nanopolycrystal diamond than monocrystal diamond. Nat. Commun. 2013, 4, 2343. [Google Scholar] [CrossRef]
- Mochalin, V.N.; Shenderova, O.; Ho, D.; Gogotsi, Y. The properties and applications of nanodiamonds. Nat. Nanotechnol. 2011, 7, 11–23. [Google Scholar] [CrossRef]
- Lu, K.; Lu, L.; Suresh, S. Strengthening materials by engineering coherent internal boundaries at the nanoscale. Science 2009, 324, 349–352. [Google Scholar] [CrossRef]
- Wang, Z.; Saito, M.; McKenna, K.P.; Gu, L.; Tsukimoto, S.; Shluger, A.L.; Ikuhara, Y. Atom-resolved imaging of ordered defect superstructures at individual grain boundaries. Nature 2011, 479, 380–383. [Google Scholar] [CrossRef]
- Wang, Z.; Saito, M.; McKenna, K.P.; Ikuhara, Y. Polymorphism of dislocation core structures at the atomic scale. Nat. Commun. 2014, 5, 3239. [Google Scholar] [CrossRef]
- Hall, E.O. The deformation and aging of mild steel. Proc. Phys. Soc. Lond. Ser. B 1951, 64, 495–502. [Google Scholar]
- Petch, N.J. The cleavage strength of polycrystals. J. Iron Steel Inst. 1953, 174, 25–28. [Google Scholar]
- Fu, T.; Peng, X.; Huang, C.; Weng, S.; Zhao, Y.; Wang, Z.; Hu, N. Strain rate dependence of tension and compression behavior in nano-polycrystalline vanadium nitride. Ceram. Int. 2017, 43, 11635–11641. [Google Scholar] [CrossRef]
- Yip, S. The strongest size. Nature 1998, 86, 713–720. [Google Scholar] [CrossRef]
- Sumiya, H.; Irifune, T. Hardness and deformation microstructures of nano-polycrystalline diamonds synthesized from various carbons under high pressure and high temperature. J. Mater. Res. 2011, 22, 2345–2351. [Google Scholar] [CrossRef]
- Irifune, T.; Kurio, A.; Sakamoto, S.; Inoue, T.; Sumiya, H. Materials: Ultrahard polycrystalline diamond from graphite. Nature 2003, 421, 599–600. [Google Scholar] [CrossRef]
- Solozhenko, V.L.; Kurakevych, O.O.; Le Godec, Y. Creation of nanostuctures by extreme conditions: High-pressure synthesis of ultrahard nanocrystalline cubic boron nitride. Adv. Mater. 2012, 24, 1540–1544. [Google Scholar] [CrossRef]
- Dubrovinskaia, N.; Solozhenko, V.L.; Miyajima, N.; Dmitriev, V.; Kurakevych, O.O.; Dubrovinsky, L. Superhard nanocomposite of dense polymorphs of boron nitride: Noncarbon material has reached diamond hardness. Appl. Phys. Lett. 2007, 90, 101912. [Google Scholar] [CrossRef]
- Hall, E.O. The Deformation and Ageing of Mild Steel. Discussion of Results. Proc. Phys. Soc. Lond. Sect. B 1951, 64, 747. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Yu, D.; Ma, Y.; Wang, Y.; Jiang, Y.; Hu, W.; Tang, C.; Gao, Y.; Luo, K.; et al. Ultrahard nanotwinned cubic boron nitride. Nature 2013, 493, 385–388. [Google Scholar] [CrossRef]
- Lu, K. Stabilizing nanostructures in metals using grain and twin boundary architectures. Nat. Rev. Mater. 2016, 1, 16019. [Google Scholar] [CrossRef]
- Huang, Q.; Yu, D.L.; Xu, B.; Hu, W.T.; Ma, Y.M.; Wang, Y.B.; Zhao, Z.S.; Wen, B.; He, J.L.; Liu, Z.Y.; et al. Nanotwinned diamond with unprecedented hardness and stability. Nature 2014, 510, 250. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhou, H.; Lu, Q.; Gao, H.; Lu, L. Extra strengthening and work hardening in gradient nanotwinned metals. Science 2018, 362, 559. [Google Scholar] [CrossRef]
- Zhou, X.; Li, X.Y.; Lu, K. Enhanced thermal stability of nanograined metals below a critical grain size. Science 2018, 360, 526–530. [Google Scholar] [CrossRef]
- Pan, Q.; Zhou, H.; Lu, Q.; Gao, H.; Lu, L. History-independent cyclic response of nanotwinned metals. Nature 2017, 551, 214–217. [Google Scholar] [CrossRef]
- You, Z.; Lu, L. Deformation and fracture mechanisms of nanotwinned metals. Natl. Sci. Rev. 2017, 4, 519–521. [Google Scholar] [CrossRef]
- Li, X.; Yin, S.; Oh, S.H.; Gao, H. Hardening and toughening mechanisms in nanotwinned ceramics. Scr. Mater. 2017, 133, 105–112. [Google Scholar] [CrossRef]
- Taheri Mousavi, S.M.; Zou, G.; Zhou, H.; Gao, H. Anisotropy governs strain stiffening in nanotwinned-materials. Nat. Commun. 2018, 9, 1586. [Google Scholar] [CrossRef]
- Li, B.; Sun, H.; Chen, C. Reply to ‘anisotropy governs strain stiffening in nanotwinned-materials’. Nat. Commun. 2018, 9, 1585. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Guan, J.; Zhu, Z.; Tomanek, D. Phase coexistence and metal-insulator transition in few-layer phosphorene: A computational study. Phys. Rev. Lett. 2014, 113, 046804. [Google Scholar] [CrossRef]
- Yang, B.; Peng, X.; Xiang, H.; Yin, D.; Huang, C.; Sun, S.; Fu, T. Generalized stacking fault energies and ideal strengths of MC systems (M = Ti, Zr, Hf) doped with si/al using first principles calculations. J. Alloys Compd. 2018, 739, 431–438. [Google Scholar] [CrossRef]
- Roundy, D.; Krenn, C.R.; Cohen, M.L.; Morris, J.W., Jr. Ideal shear strengths of fcc aluminum and copper. Phys. Rev. Lett. 1999, 82, 2713–2716. [Google Scholar] [CrossRef]
- Roundy, D.; Krenn, C.R.; Cohen, M.L.; Morris, J.W. The ideal strength of tungsten. Philos. Mag. A 2001, 81, 1725–1747. [Google Scholar] [CrossRef]
- Rodney, D.; Ventelon, L.; Clouet, E.; Pizzagalli, L.; Willaime, F. Ab initio modeling of dislocation core properties in metals and semiconductors. Acta Mater. 2017, 124, 633–659. [Google Scholar] [CrossRef]
- Xiang, H.; Li, H.; Fu, T.; Huang, C.; Peng, X. Formation of prismatic loops in aln and gan under nanoindentation. Acta Mater. 2017, 138, 131–139. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, H.; Li, Q.; Gao, B.; Wang, Y.; Li, H.; Chen, C.; Ma, Y. Superhard BC(3) in cubic diamond structure. Phys. Rev. Lett. 2015, 114, 015502. [Google Scholar] [CrossRef]
- Wang, P.; He, D.; Wang, L.; Kou, Z.; Li, Y.; Xiong, L.; Hu, Q.; Xu, C.; Lei, L.; Wang, Q.; et al. Diamond-cbn alloy: A universal cutting material. Appl. Phys. Lett. 2015, 107, 101901. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, H.; Chen, C. Atomistic deformation modes in strong covalent solids. Phys. Rev. Lett. 2005, 94, 145505. [Google Scholar] [CrossRef]
- Telling, R.H.; Pickard, C.J.; Payne, M.C.; Field, J.E. Theoretical strength and cleavage of diamond. Phys. Rev. Lett. 2000, 84, 5160–5163. [Google Scholar] [CrossRef]
- Zheng, S.; Zhang, R.; Huang, R.; Taniguchi, T.; Ma, X.; Ikuhara, Y.; Beyerlein, I.J. Structure and energetics of nanotwins in cubic boron nitrides. Appl. Phys. Lett. 2016, 109, 081901. [Google Scholar] [CrossRef]
- Zhang, R.F.; Veprek, S.; Argon, A.S. Anisotropic ideal strengths and chemical bonding of wurtzite bn in comparison to zincblende bn. Phys. Rev. B 2008, 77, 172103. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Energy (ev) | TBE (mJ/m2) | ||
|---|---|---|---|
| Perfect Crystal | Twin | ||
| cBN | −836.29 | −835.83 | 81.72 |
| Diamond | −872.62 | −872.06 | 101.69 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, B.; Peng, X.; Sun, S.; Huang, C.; Yin, D.; Chen, X.; Fu, T. Detwinning Mechanism for Nanotwinned Cubic Boron Nitride with Unprecedented Strength: A First-Principles Study. Nanomaterials 2019, 9, 1117. https://doi.org/10.3390/nano9081117
Yang B, Peng X, Sun S, Huang C, Yin D, Chen X, Fu T. Detwinning Mechanism for Nanotwinned Cubic Boron Nitride with Unprecedented Strength: A First-Principles Study. Nanomaterials. 2019; 9(8):1117. https://doi.org/10.3390/nano9081117
Chicago/Turabian StyleYang, Bo, Xianghe Peng, Sha Sun, Cheng Huang, Deqiang Yin, Xiang Chen, and Tao Fu. 2019. "Detwinning Mechanism for Nanotwinned Cubic Boron Nitride with Unprecedented Strength: A First-Principles Study" Nanomaterials 9, no. 8: 1117. https://doi.org/10.3390/nano9081117
APA StyleYang, B., Peng, X., Sun, S., Huang, C., Yin, D., Chen, X., & Fu, T. (2019). Detwinning Mechanism for Nanotwinned Cubic Boron Nitride with Unprecedented Strength: A First-Principles Study. Nanomaterials, 9(8), 1117. https://doi.org/10.3390/nano9081117