Actinide and Lanthanide Adsorption onto Hierarchically Porous Carbons Beads: A High Surface Affinity for Pu

 ,
,  ,
,
Abstract
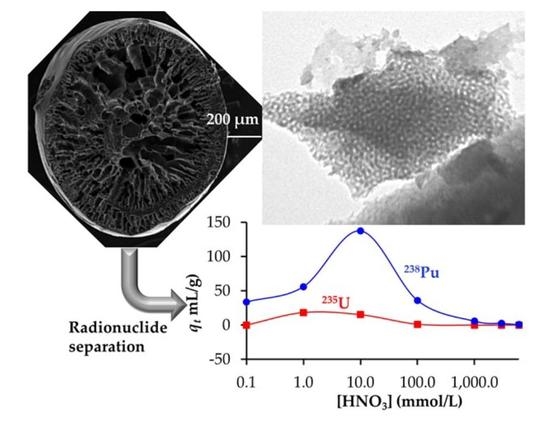
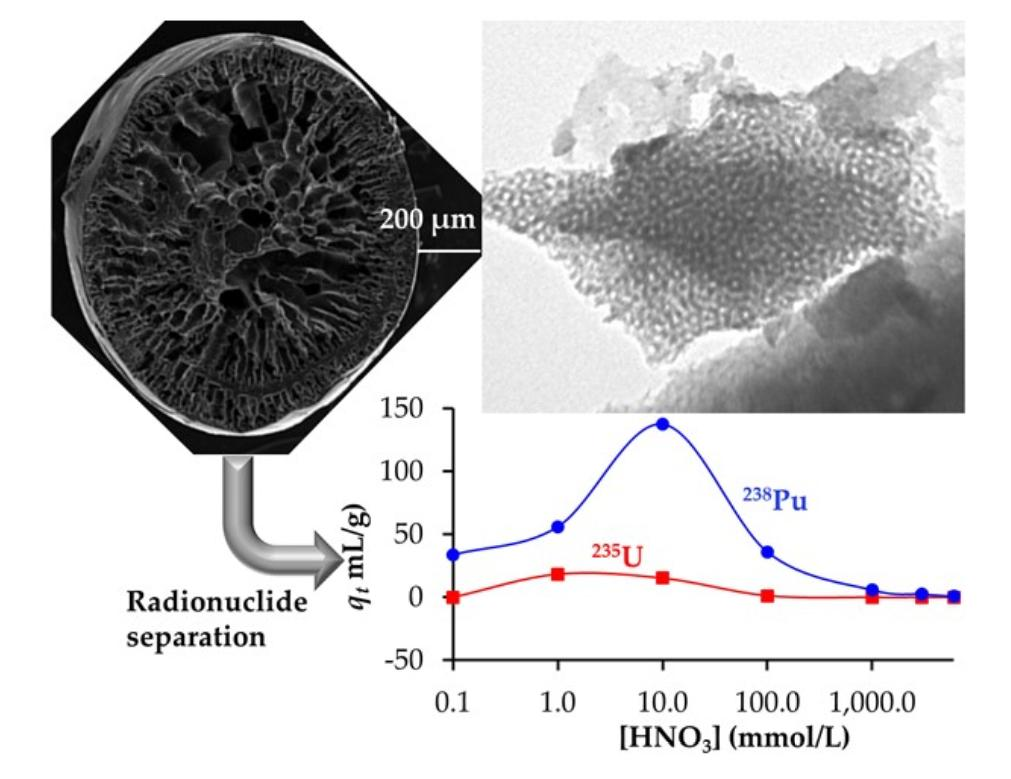
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Bead Synthesis
2.3. Characterization
3. Results
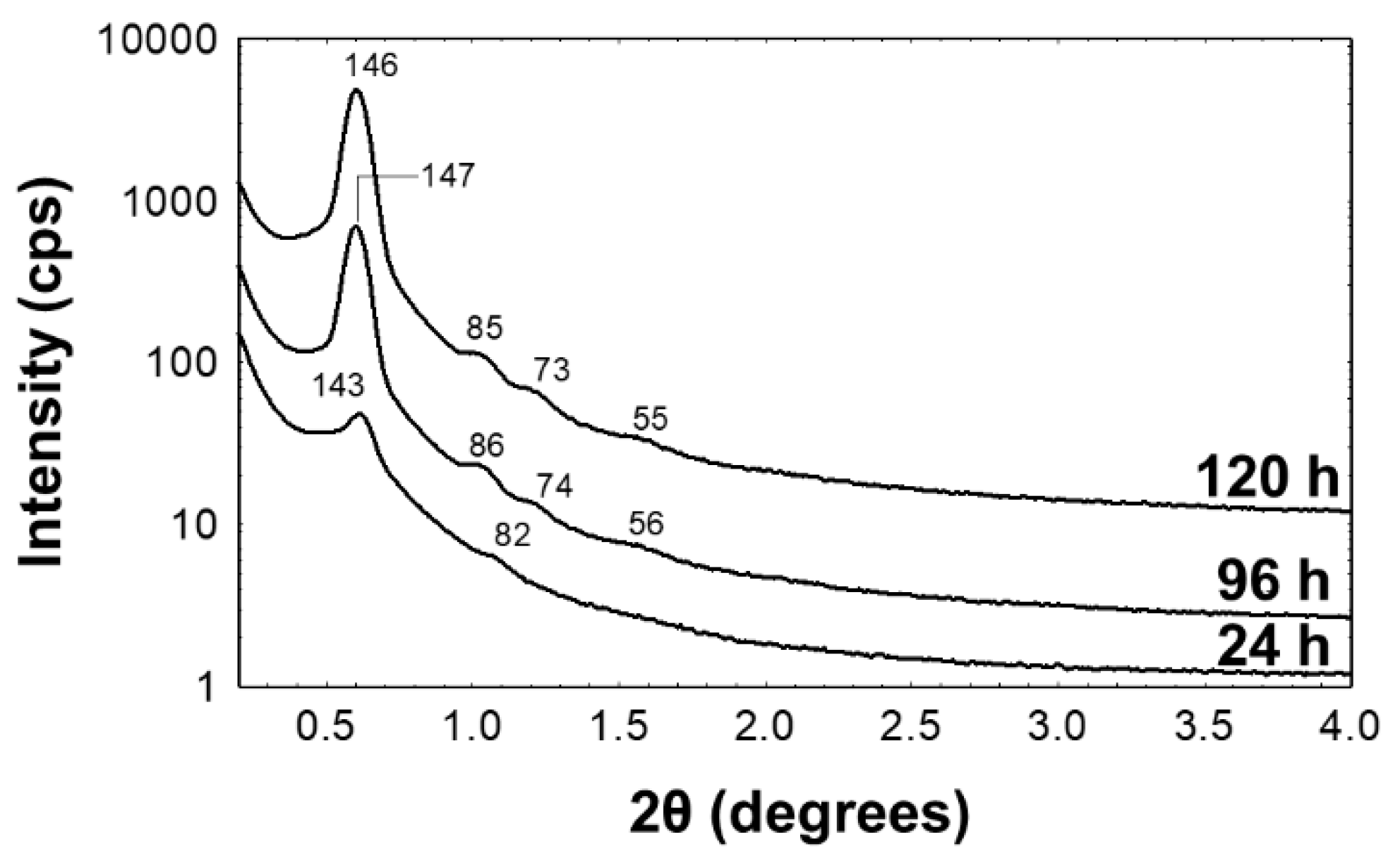
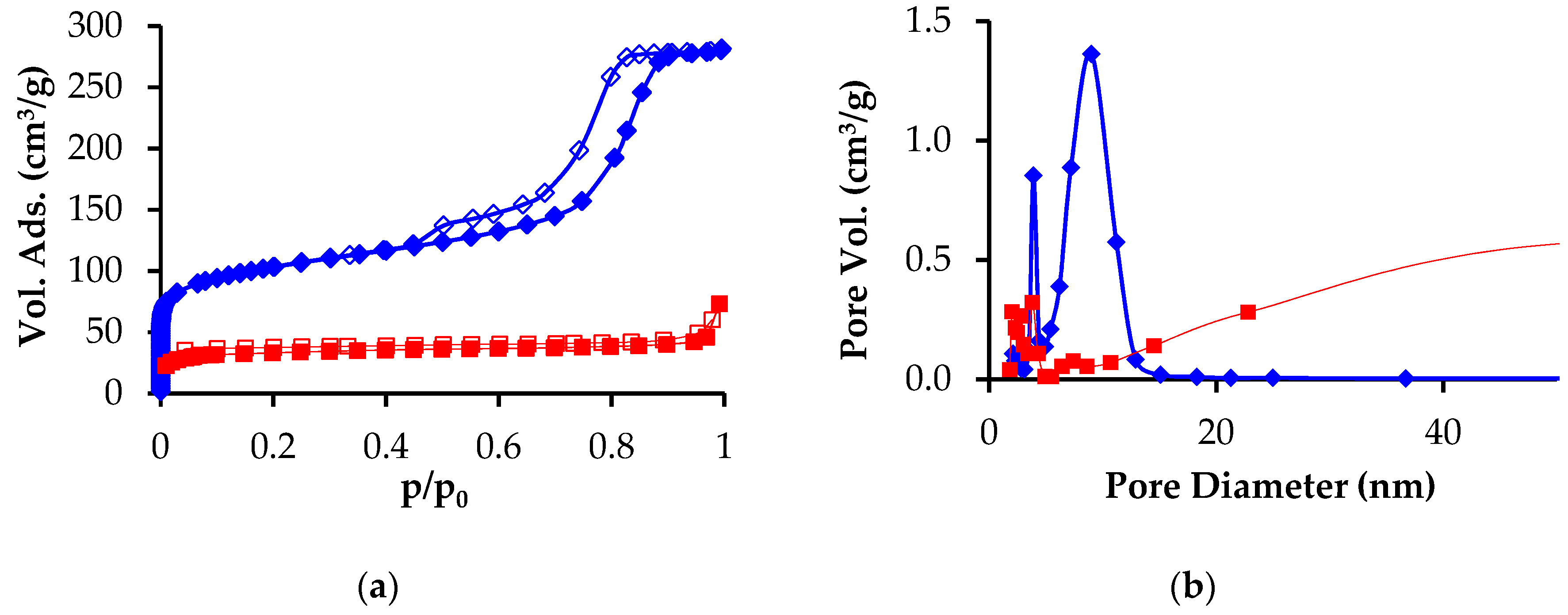
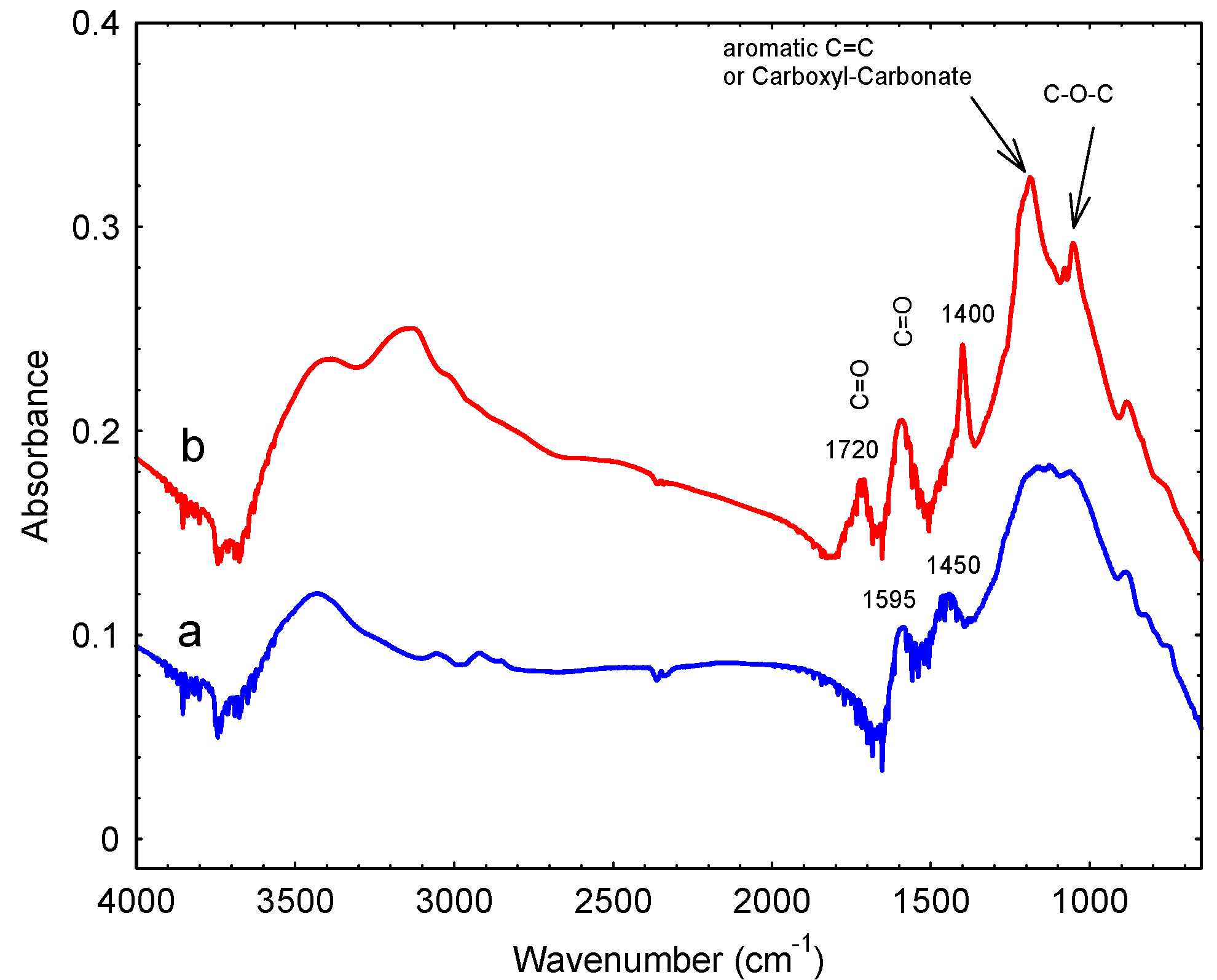
3.1. Preparation and Characterization
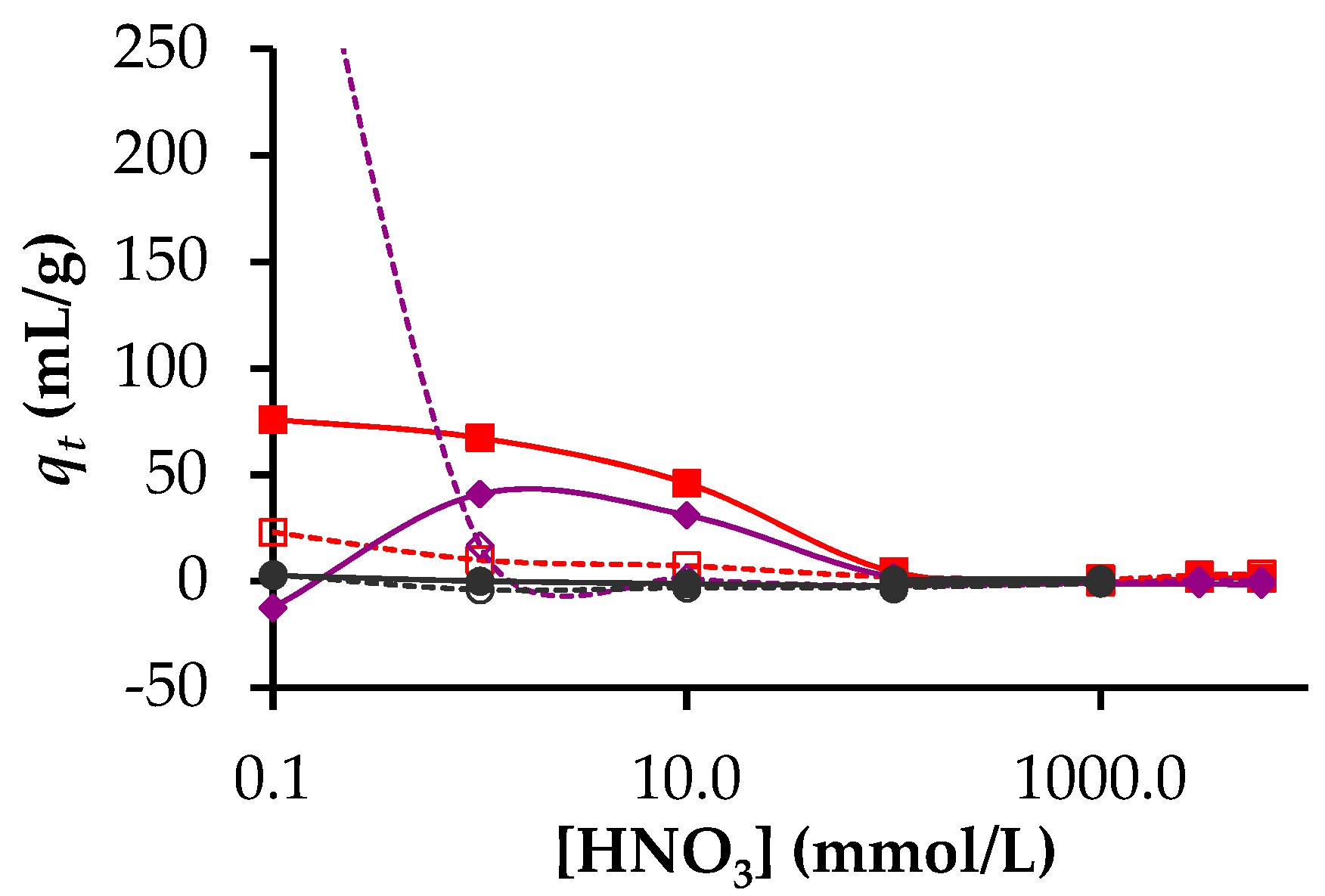
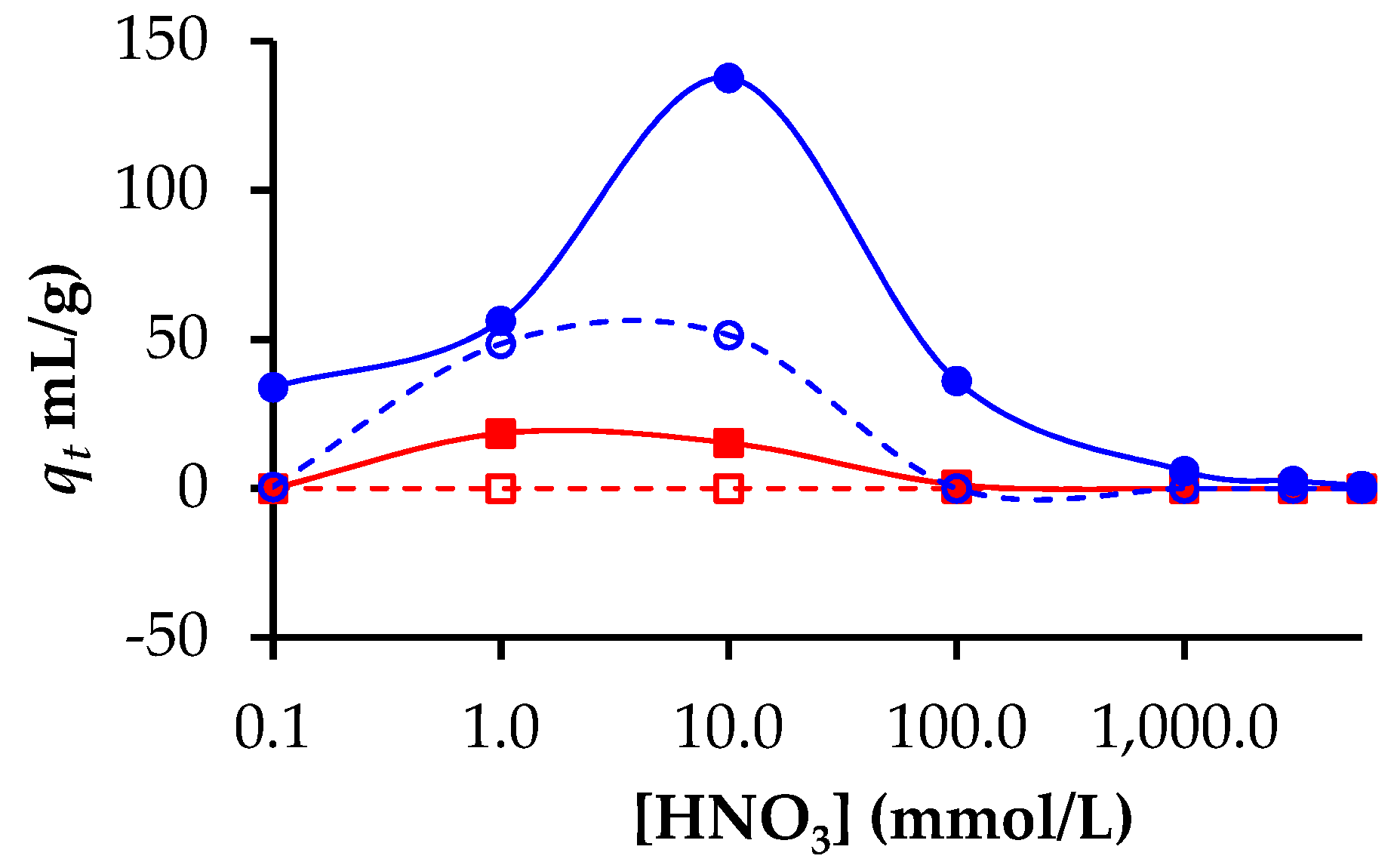
3.2. Adsorption of Lanthanides and Actinides
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mohan, D.; Pittman, C.U., Jr. Arsenic removal from water/wastewater using adsorbents—A critical review. J. Hazard. Mater. 2007, 142, 1–53. [Google Scholar] [CrossRef]
- Cheremisinoff, P.N.; Angelo, C.M. Carbon Adsorption Applications; Ann Arbor Science Publishers, Inc.: Ann Arbor, MI, USA, 1980. [Google Scholar]
- Mauter, M.S.; Elimelech, M. Environmental applications of carbon-based nanomaterials. Environ. Sci. Technol. 2008, 42, 5843–5859. [Google Scholar] [CrossRef]
- Babel, S.; Kurniawan, T.A. Low-cost adsorbents for heavy metals uptake from contaminated water: A review. J. Hazard. Mater. 2003, 97, 219–243. [Google Scholar] [CrossRef]
- Lu, S.; Xu, J.; Zhang, C.; Niu, Z. Adsorption and desorption of radionuclide europium (III) on multiwalled carbon nanotubes studied by batch techniques. J. Radioanal. Nucl. Chem. 2011, 287, 893–898. [Google Scholar] [CrossRef]
- Schierz, A.; Zanker, H. Aqueous suspensions of carbon nanotubes: Surface oxidation, colloidal stability and uranium sorption. Environ. Pollut. 2009, 157, 1088–1094. [Google Scholar] [CrossRef]
- Sundararajan, M.; Ghosh, S.K. Designing novel materials through functionalization of carbon nanotubes for application in nuclear waste management: Speciation of uranyl. J. Phys. Chem. A 2011, 115, 6732–6737. [Google Scholar] [CrossRef]
- Belloni, F.; Kuetahyali, C.; Rondinella, V.V.; Carbol, P.; Wiss, T.; Mangione, A. Can carbon nanotubes play a role in the field of nuclear waste management? Environ. Sci. Technol. 2009, 43, 1250–1255. [Google Scholar] [CrossRef]
- Perevalov, S.A.; Molochnikova, N.P. Sorption of Pu in various oxidation states onto multiwalled carbon nanotubes. J. Radioanal. Nucl. Chem. 2009, 281, 603–608. [Google Scholar] [CrossRef]
- Myasoedova, G.V.; Molochnikova, N.P.; Tkachev, A.G.; Tugolukov, E.N.; Mishchenko, S.V.; Myasoedov, B.F. Sorption preconcentration of radionuclides on Taunit carbon nanostructural material. Radiochemistry 2009, 51, 156–158. [Google Scholar] [CrossRef]
- Accorsi, G.; Armaroli, N.; Parisini, A.; Meneghetti, M.; Marega, R.; Prato, M.; Bonifazi, D. Wet adsorption of a luminescent EuIII complex on carbon nanotubes sidewalls. Adv. Funct. Mater. 2007, 17, 2975–2982. [Google Scholar] [CrossRef]
- Sang, L.-C.; Vinu, A.; Coppens, M.-O. Ordered mesoporous carbon with tunable, unusually large pore size and well-controlled particle morphology. J. Mater. Chem. 2011, 21, 7410–7417. [Google Scholar] [CrossRef]
- Knox, J.H.; Kaur, B.; Millward, G.R. Structure and performance of porous graphitic carbon in liquid chromatography. J. Chromatogr. 1986, 352, 3–25. [Google Scholar] [CrossRef]
- Enzel, P.; Bein, T. Poly (acrylonitrile) chains in zeolite channels: Polymerization and pyrolysis. Chem. Mater. 1992, 4, 819–824. [Google Scholar] [CrossRef][Green Version]
- Wu, C.G.; Bein, T. Conducting carbon wires in ordered, nanometer-sized channels. Science 1994, 266, 1013–1015. [Google Scholar] [CrossRef]
- Ryoo, R.; Joo, S.H.; Jun, S. Synthesis of highly ordered carbon molecular sieves via template-mediated structural transformation. J. Phys. Chem. B 1999, 103, 7743–7746. [Google Scholar] [CrossRef]
- Lu, A.-H.; Schueth, F. Nanocasting: A versatile strategy for creating nanostructured porous materials. Adv. Mater. 2006, 18, 1793–1805. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.; Hyeon, T. Recent progress in the synthesis of porous carbon materials. Adv. Mater. 2006, 18, 2073–2094. [Google Scholar] [CrossRef]
- Tao, Y.; Endo, M.; Inagaki, M.; Kaneko, K. Recent progress in the synthesis and applications of nanoporous carbon films. J. Mater. Chem. 2011, 21, 313–323. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, Z.; Mokaya, R. Templated nanoscale porous carbons. Nanoscale 2010, 2, 639–659. [Google Scholar] [CrossRef]
- Ozaki, J.; Endo, N.; Ohizumi, W.; Igarashi, K.; Nakahara, M.; Oya, A.; Yoshida, S.; Iizuka, T. Novel preparation method for the production of mesoporous carbon fiber from a polymer blend. Carbon 1997, 35, 1031–1033. [Google Scholar] [CrossRef]
- Liu, C.Y.; Li, L.X.; Song, H.H.; Chen, X.H. Facile synthesis of ordered mesoporous carbons from F108/resorcinol–formaldehyde composites obtained in basic media. Chem. Commun. 2007, 757–759. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Gu, D.; Zhang, F.Q.; Shi, Y.F.; Cheng, L.; Feng, D.; Wu, Z.X.; Chen, Z.X.; Wan, Y.; Stein, A.; et al. A family of highly ordered mesoporous polymer resin and carbon structures from organic—Organic self-assembly. Chem. Mater. 2006, 18, 4447–4464. [Google Scholar] [CrossRef]
- Liang, C.; Li, Z.; Dai, S. Mesoporous carbon materials: Synthesis and modification. Angew. Chem. Int. Ed. 2008, 47, 3696–3717. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.A.; Mhlanga, S.D.; Coville, N.J. Carbon spheres. Mater. Sci. Eng. R 2010, 70, 1–28. [Google Scholar] [CrossRef]
- Tranter, T.J.; Mann, N.R.; Todd, T.A.; Sebesta, F. Evaluation of a novel solid phase extraction composite for the removal of actinides from acidic nuclear waste solutions. Czech. J. Phys. 2003, 53, A589–A594. [Google Scholar] [CrossRef]
- Kim, H.-T.; Lee, C.-H.; Shul, Y.-G.; Moon, J.-K.; Lee, E.-H. Evaluation of PAN–TiO2 composite adsorbent for removal of Pb (II) ion in aqueous solution. Sep. Sci. Technol. 2003, 38, 695–713. [Google Scholar] [CrossRef]
- Griffith, C.S.; Luca, V.; Yee, P.; Sebesta, F. Separation of cesium and strontium from acidic radioactive waste simulants using a microporous tungstate/polyacrylonitrile (PAN) composite adsorbent. Sep. Sci. Technol. 2005, 40, 1781–1796. [Google Scholar] [CrossRef]
- Moon, J.-K.; Kim, K.-W.; Jung, C.-H.; Shul, Y.-G.; Lee, E.-H. Preparation of organic-inorganic composite adsorbent beads for removal of radionuclides and heavy metal ions. J. Radioanal. Nucl. Chem. 2000, 246, 299–307. [Google Scholar] [CrossRef]
- Griffith, C.S.; Sebesta, F.; Hanna, J.V.; Yee, P.; Drabarek, E.; Smith, M.E.; Luca, V. Tungsten bronze-based nuclear waste form ceramics. Part 2: Conversion of granular microporous tungstate–polyacrylonitrile (PAN) composite adsorbents to leach resistant ceramics. J. Nucl. Mater. 2006, 358, 151–163. [Google Scholar] [CrossRef]
- Geller, B.E. Status and prospects for development of polyacrylonitrile fibre production. A review. Fibre Chem. 2002, 34, 151–161. [Google Scholar] [CrossRef]
- Sizgek, G.D.; Griffith, C.S.; Sizgek, E.; Luca, V. Mesoporous zirconium titanium oxides. Part 3. Synthesis and adsorption properties of unfunctionalized and phosphonate-functionalized hierarchical polyacrylonitrile-F-127-templated beads. Langmuir 2009, 25, 11874–11882. [Google Scholar] [CrossRef] [PubMed]
- Drisko, G.L.; Chee Kimling, M.; Scales, N.; Ide, A.; Sizgek, E.; Caruso, R.A.; Luca, V. One-pot preparation and uranyl adsorption properties of hierarchically porous zirconium titanium oxide beads using phase separation processes to vary macropore morphology. Langmuir 2010, 26, 17581–17588. [Google Scholar] [CrossRef] [PubMed]
- Ide, A.; Drisko, G.L.; Scales, N.; Luca, V.; Schiesser, C.H.; Caruso, R.A. Monitoring bisphosphonate surface functionalization and acid stability of hierarchically porous titanium zirconium oxides. Langmuir 2011, 27, 12985–12995. [Google Scholar] [CrossRef]
- Griffith, C.S.; Sizgek, G.D.; Sizgek, E.; Scales, N.; Yee, P.J.; Luca, V. Mesoporous zirconium titanium oxides. Part 1: Porosity modulation and adsorption properties of xerogels. Langmuir 2008, 24, 12312–12322. [Google Scholar] [CrossRef]
- Drisko, G.L.; Luca, V.; Sizgek, E.; Scales, N.; Caruso, R.A. Template synthesis and adsorption properties of hierarchically porous zirconium titanium oxides. Langmuir 2009, 25, 5286–5293. [Google Scholar] [CrossRef]
- Razavizadeh, B.M.; Mousavi-Khoshdel, M.; Gharibi, H.; Behjatmanesh-Ardakani, R.; Javadian, S.; Sohrabi, B. Thermodynamic studies of mixed ionic/nonionic surfactant systems. J. Colloid Interfaces Sci. 2004, 276, 197–207. [Google Scholar] [CrossRef]
- Vinu, A.; Hossian, K.Z.; Srinivasu, P.; Miyahara, M.; Anandan, S.; Gokulakrishnan, N.; Mori, T.; Ariga, K.; Balasubramanian, V.V. Carboxy-mesoporous carbon and its excellent adsorption capability for proteins. J. Mater. Chem. 2007, 17, 1819–1825. [Google Scholar] [CrossRef]
- Fanning, P.E.; Vannice, M.A. A Drifts study of the formation of surface groups on carbon by oxidation. Carbon 1993, 31, 721–730. [Google Scholar] [CrossRef]
- Zawadzki, J. Infrared Spectroscopy in Surface Chemistry of Carbons, in Chemistry and Physics of Carbon; Thrower, P.A., Ed.; Dekker: New York, NY, USA, 1989; p. 147. [Google Scholar]
- Wang, X.; Chen, C.; Hu, W.; Ding, A.; Xu, D.; Zhou, X. Sorption of 243Am (III) to multiwall carbon nanotubes. Environ. Sci. Technol. 2005, 39, 2856–2860. [Google Scholar] [CrossRef]
- Arisaka, M.; Watanabe, M.; Kimura, T. Selective adsorption of trivalent actinides from lanthanides onto activated carbons in acidic aqueous solution. J. Nucl. Mater. 2010, 407, 116–118. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Band Position (cm−1) | Assignment |
|---|---|
| 1053 | Alcohol groups |
| 1186 | Alcohol |
| 1400 | Lactones |
| 1595 | Quinones |
| 1720 | COOH |
| 3147 | COOH |
| Radioisotope | % Removal at 1 mmol HNO3 | % Removal at 10 mmol HNO3 | % Removal at 100 mmol HNO3 | |||
|---|---|---|---|---|---|---|
| Hierarchical beads | PAN beads | Hierarchical beads | PAN beads | Hierarchical beads | PAN beads | |
| 241Am | 40 | 9 | 31 | 7 | 4 | 2 |
| 153Gd | 29 | 14 | 23 | 1 | 2 | 0 |
| 237Np | 0 | 0 | 0 | 0 | 0 | 0 |
| 238U | 13 | 10 | 11 | 9 | 1 | 9 |
| 239Pu | 36 | 50 | 22 | 51 | 27 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luca, V.; Sizgek, D.G.; Sizgek, E.; Arrachart, G.; Rey, C.; Scales, N.; Aly, Z.; Drisko, G.L. Actinide and Lanthanide Adsorption onto Hierarchically Porous Carbons Beads: A High Surface Affinity for Pu. Nanomaterials 2019, 9, 1464. https://doi.org/10.3390/nano9101464
Luca V, Sizgek DG, Sizgek E, Arrachart G, Rey C, Scales N, Aly Z, Drisko GL. Actinide and Lanthanide Adsorption onto Hierarchically Porous Carbons Beads: A High Surface Affinity for Pu. Nanomaterials. 2019; 9(10):1464. https://doi.org/10.3390/nano9101464
Chicago/Turabian StyleLuca, Vittorio, Devlet G. Sizgek, Erden Sizgek, Guilhem Arrachart, Cyrielle Rey, Nicholas Scales, Zaynab Aly, and Glenna L. Drisko. 2019. "Actinide and Lanthanide Adsorption onto Hierarchically Porous Carbons Beads: A High Surface Affinity for Pu" Nanomaterials 9, no. 10: 1464. https://doi.org/10.3390/nano9101464
APA StyleLuca, V., Sizgek, D. G., Sizgek, E., Arrachart, G., Rey, C., Scales, N., Aly, Z., & Drisko, G. L. (2019). Actinide and Lanthanide Adsorption onto Hierarchically Porous Carbons Beads: A High Surface Affinity for Pu. Nanomaterials, 9(10), 1464. https://doi.org/10.3390/nano9101464