2. Materials and Methods
Nanoparticle synthesis: SiO
2 nanoparticles (average diameter: 45 nm) were synthesized according to the procedure reported by Stöber et al. with slight modifications [
13]. In brief, 35 mL of ethanol (99.8%, Carl Roth, Karlsruhe, Germany), 5 mL of double-distilled water and 1 mL of ammonium hydroxide (25% aqueous solution, Sigma-Aldrich, St. Louis, MO, USA) were mixed and heated to 70 °C. Subsequently, tetraethyl orthosilicate (>99%, Sigma-Aldrich, St. Louis, MO, USA) was added at once, and the reaction mixture was stirred for one hour while being kept at 70 °C. The reaction was stopped by centrifugation at 9000 rpm for 10 min, and the SiO
2-NPs were washed repeatedly with ethanol and finally, were stored in ethanol at 4 °C.
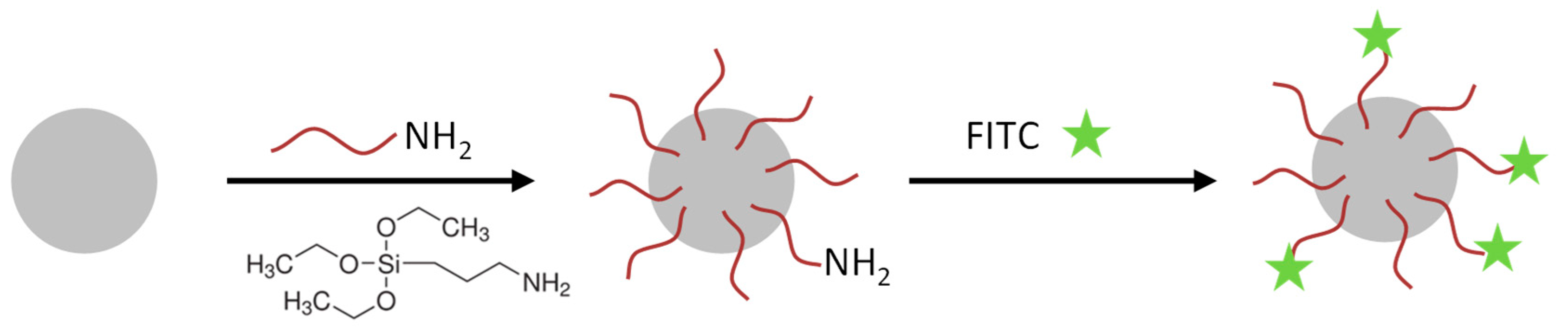
Nanoparticle functionalization and EGFR-antibody coupling: The SiO
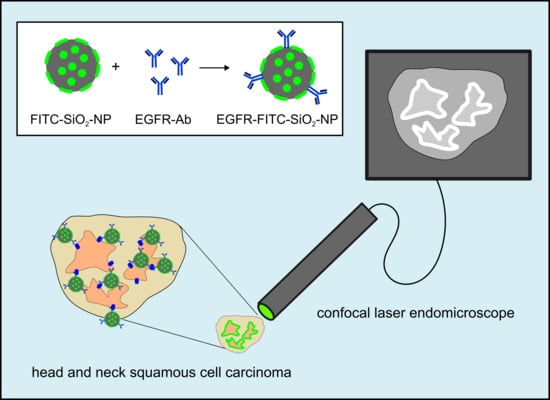
2-NPs were functionalized with amino groups by the treatment with (3-Aminopropyl)triethoxysilan (APTES, Sigma-Aldrich, St. Louis, MO, USA) (see
Figure 1). These amino groups were used for conjugation of the fluorophore 5(6)-carboxyfluorescein N-hydroxysuccinimide ester (FITC, Sigma-Aldrich, St. Louis, MO, USA) and in a second reaction the EGFR-antibody (
Figure 2).
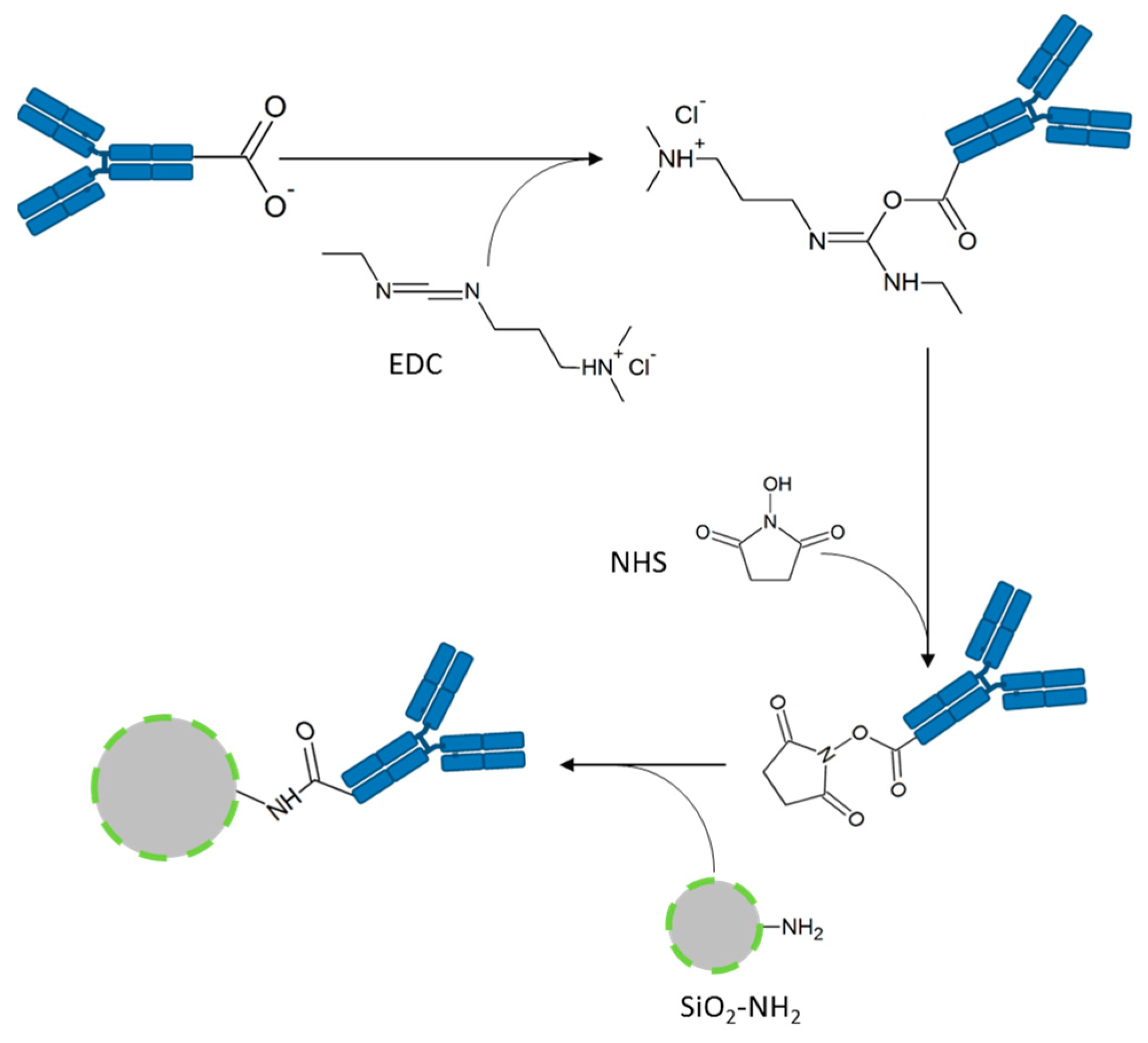
The attachment of the EGFR antibody [EGF receptor (D38B1) XP
® rabbit mAb, #4267, (EGFR-Ab) or EGF receptor (D38B1) XP
® rabbit mAb (Alexa Fluor
® 555 conjugate), #5108, (AF555-EGFR-Ab), both Cell Signaling Technology, Danvers, MA, USA] to the SiO
2 particles is performed by an 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, Thermo Fisher Scientific, Rockford, IL, USA)-N-hydroxysuccinimide (NHS, Thermo Fisher Scientific, Rockford, IL, USA)-coupling using the terminal carboxy group of the antibodies and the primary amino groups located on the particles’ surface (
Figure 2). The resulting amide bond links the antibody covalently to the particle. The EGFR-antibody is aimed at binding the EGF receptor overexpressed on the tumor cells to mark these cells with the FITC-conjugated SiO
2-NPs.
The preparation was conducted as follows: First, two aliquots of 3 mg FITC-SiO2-NPs were washed thrice with 0.9% sodium chloride (NaCl, Fresenius Kabi, Bad Homburg, Germany). The NPs were centrifuged (13,000 rpm, 10 min, room temperature (RT)), the supernatants were removed, and the NPs were dispersed by sonication and vortexing. In the next step, 2 mg/mL FITC-SiO2-NPs were incubated with NHS and EDC according to the manufacturer’s instructions. The EGFR-antibody was added to one aliquot (262.5 ng, EGFR-Ab or AF555-EGFR-Ab) and 15 µL 0.9% NaCl were added to the second aliquot (FITC-SiO2-NPs). Then, the coupling was performed for 2 h at 12 °C in a Thermomixer comfort (Thermo Fisher Scientific, Rockford, IL, USA) at 1400 rpm. The nanoparticles were centrifuged, washed thrice with 0.9% NaCl and dispersed at 6 mg/mL in 0.9% NaCl.
Analytical characterization: The SiO2-NPs were characterized by transmission electron microscopy (TEM) and ζ-potential measurements. TEM images were recorded using a Philips EM420 microscope (Philips, Amsterdam, Netherlands) with an acceleration voltage of 120 kV. Samples for TEM were prepared by dropping a diluted solution of SiO2-NPs in ethanol onto a carbon coated copper grid (Plano, Wetzlar; Germany). ζ-potential measurements were conducted with a Malvern Zetasizer Nano ZS (Malvern Panalytical, Kassel, Germany). The SiO2-NPs were dispersed in 1 mL of ethanol or water, filtered (Millex-GS syringe filter, pore size 0.22 µm, Merck Millipore, Billerica, MA, USA) and transferred to Malvern cuvettes (Disposable capillary cell DTS1061). 15 ζ-potential measurements were carried out twice at 25 °C to determine mean ζ-potential.
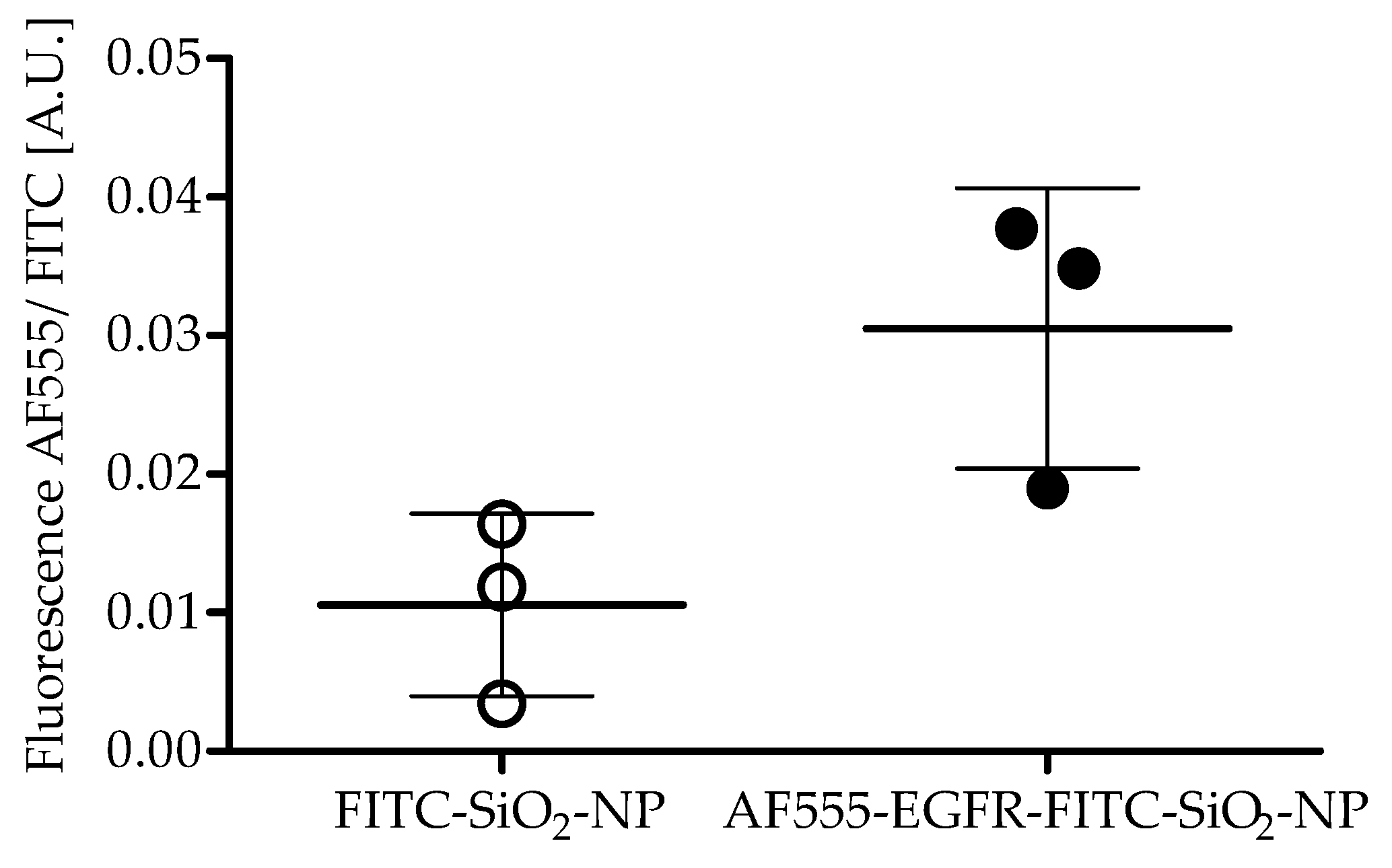
Antibody coupling efficiency: Aliquots of AF555-EGFR-FITC-SiO2-NPs and FITC-SiO2-NPs were measured with a Fluoroskan Ascent Microplate Reader (Thermo Fisher Scientific, Rockford, IL, USA) after AF555-EGFR-Ab conjugation with 485/538 and 538/600 nm filter pairs. The fluorescence of AF555-EGFR-Ab was divided by the FITC fluorescence of the NPs to determine the coupling efficiency. The experiment was repeated three times and a paired t-test was performed.
Human specimen: The use of human tissue specimens is protected under and is in consent with prior decisions from the ethics committee of the university medical center Mainz. Patients gave informed consent according to national legal guidelines and agreed to scientific use of excess biological material retrieved during treatment.
Cell lines and culture conditions: The human hepatocarcinoma cell line HuH7 [
14] was obtained from RIKEN BioResource Center. A human base of tongue squamous cell carcinoma cell line HNSCCUM-02T was established in our laboratory [
15]. Both cell lines were cultured in Dulbecco’s modified Eagle’s medium: Nutrient mixture F12 (DMEM/F12, Gibco, Thermo Fisher Scientific, Rockford, IL, USA), supplemented with 5% bovine calf serum iron supplemented (FCS, VWR Life Science Seradigm, Radnor, PA, USA), and 2% penicillin-streptomycin (both Sigma-Aldrich, St. Louis, MO, USA) (cell culture medium) at 37 °C in a humidified incubator at 5% (
V/
V) CO
2. Primary human fibroblasts were isolated from nasal concha originating from a conchotomy. The fibroblasts were cultured in DMEM/F12 with 10% FCS and stained with a monoclonal antibody to fibroblast surface protein-ascites (Acris Antibodies Inc., Herford, Germany) for verification. At a confluence of 85% to 90%, the cells were trypsinized with Trypsin-EDTA (T/E, Sigma-Aldrich, St. Louis, MO, USA) and were counted using a hemocytometer (Brand GmbH & Co KG, Wertheim Germany). Depending on the assay performed, different cell counts were seeded and treated.
Nanoparticle preparation: Stocks of EGFR-FITC-SiO2-NPs (1 or 2 mg/mL) were prepared in high-purity H2O (Cayman Chemical Company, Ann Arbor, MI, USA) or 0.9% NaCl. The NP dispersions were sonicated using a Sonorex Super RK 510 H (BANDELIN electronics, Berlin, Germany) for 5 min at 32 W amplitude, prior to each experiment.
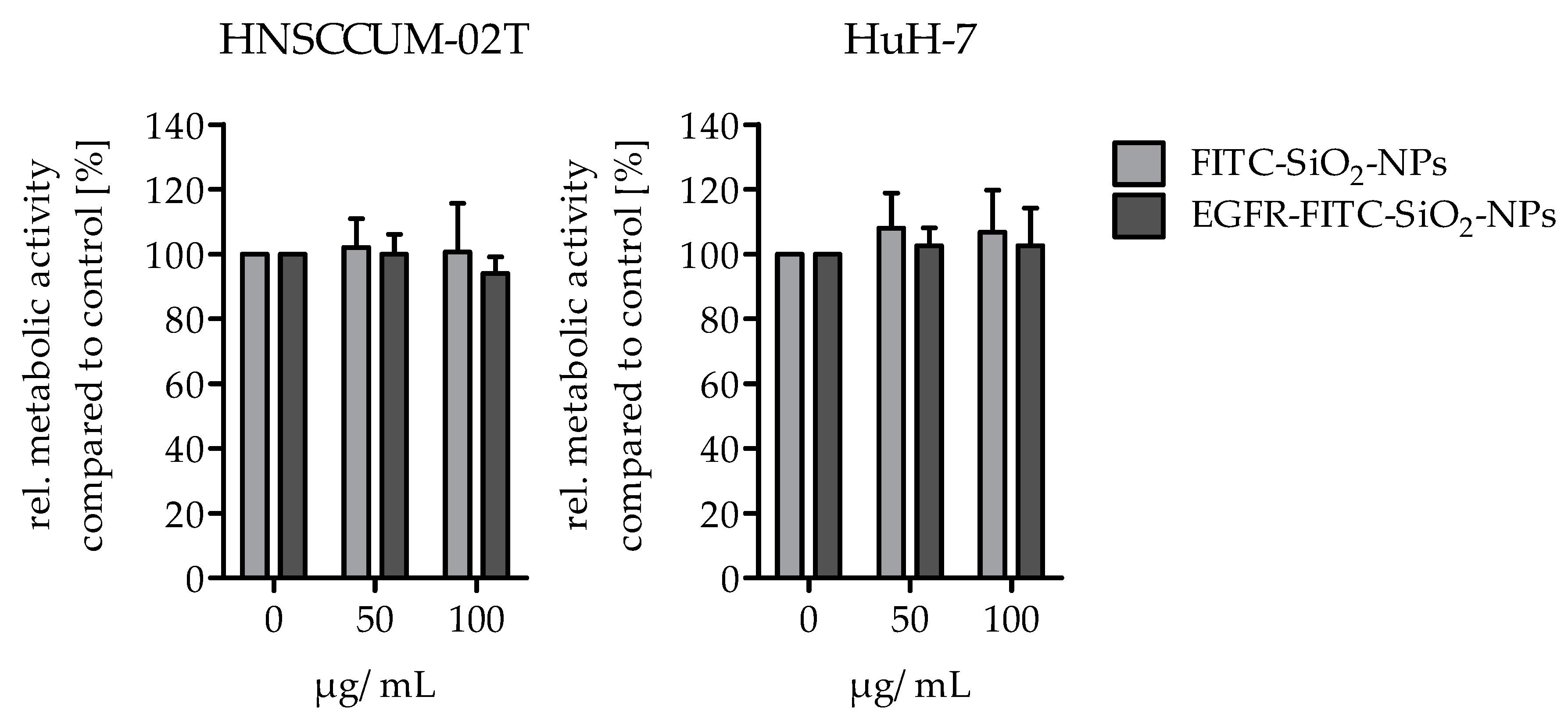
Cellular viability: The alamarBlue® assay was performed to analyze the potential effects of EGFR-FITC-SiO2-NPs on the viability of HuH7 and HNSCCUM-02T cells. Per well, 15,000 cells were seeded in a black 96-well plate with clear bottom (Greiner Bio-One International AG, Kremsmünster, Austria) and cultivated overnight for adherence. The cells were treated with 50 or 100 µg/mL EGFR-FITC-SiO2-NPs and FITC-SiO2-NPs in cell culture medium and controls (0 µg/mL) were treated with 5% 0.9% NaCl in cell culture medium, respectively. For a quantification of the viability, the medium was replaced after 24 h incubation by a medium including alamarBlue® (10% V/V, Thermo Fisher Scientific, Rockford, IL, USA). The cells were incubated for 3 h at 37 °C and then the fluorescence was measured (Fluoroskan Ascent Microplate Reader, Ex: 538 nm, Em: 600 nm, Thermo Fisher Scientific, Rockford, IL, USA). The media were removed, the cells briefly washed with PBS and lysed with lysis buffer [0.07 µM sodium dodecyl sulphate (SDS), 0.05 M tris(hydroxymethyl)aminomethane hydrochloride (Tris), 1.57 M glycerol in water]. After several freeze and thaw cycles the protein content of each sample was determined with DC™ Protein Assay (Bio-Rad, Munich, Germany). Metabolic activity data were normalized to protein content and were expressed as percentage of the respective controls. The experiment was repeated three times.
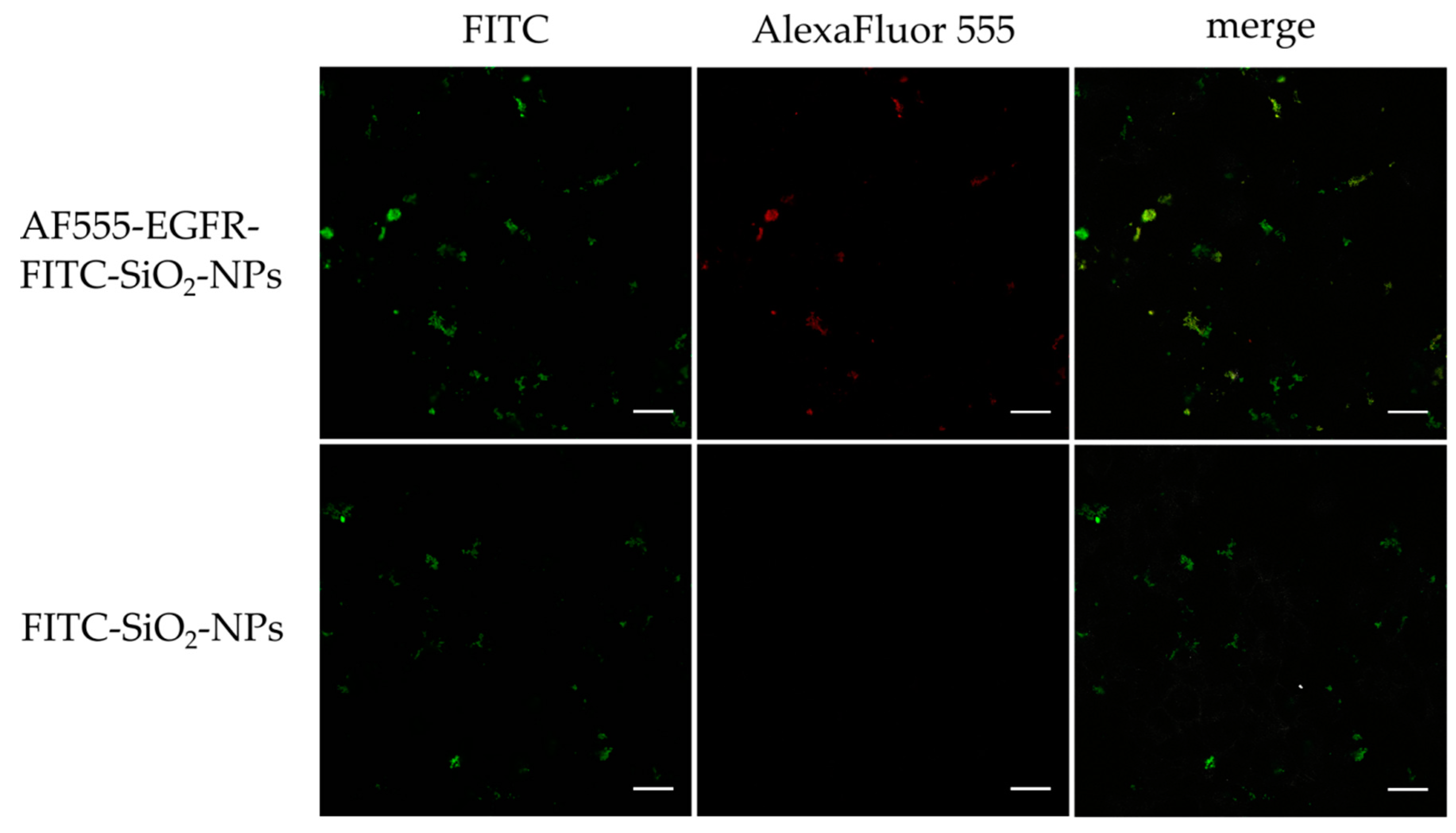
Confocal laser scanning microscopy of in vitro staining with AF555-EGFR-FITC-SiO2-NPs and FITC-SiO2-NPs: To show the binding of the AF555-EGFR-FITC-SiO2-NPs to the surface of EGF receptor expressing tumor cells, HNSCCUM-02T cells were seeded onto μ-slides 8-well ibidiTreat (ibidi GmbH, Munich, Germany) at 5000 cells per well. After adherence of the cells, 100 µg/mL AF555-EGFR-FITC-SiO2-NPs and FITC-SiO2-NPs were added for 30 min, respectively. Then, samples were washed with PBS (3 × 5 min), fixed with 4% paraformaldehyde (PFA) in PBS for 15 min and washed with PBS (3 × 5 min). Samples were fixed 4 min with ice-cold acetone, washed with PBS (3 × 5 min) and incubated with 1% bovine serum albumin (BSA) in PBS for 30 min. Cells were stained with AlexaFluor™ 350 Phalloidin (1:40, Invitrogen by Thermo Fisher Scientific, Waltham, MA, USA) in 1% BSA in PBS for 45 min, washed with PBS (3 × 5 min), rinsed briefly with double-distilled water and embedded in mounting medium (Vectashield, Vector Laboratories, Burlingame, CA, USA). All slides were examined using a Leica DMi8 confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany) at 630× magnification. Unfortunately, the Phalloidin staining was very weak and therefore not depicted in the results.
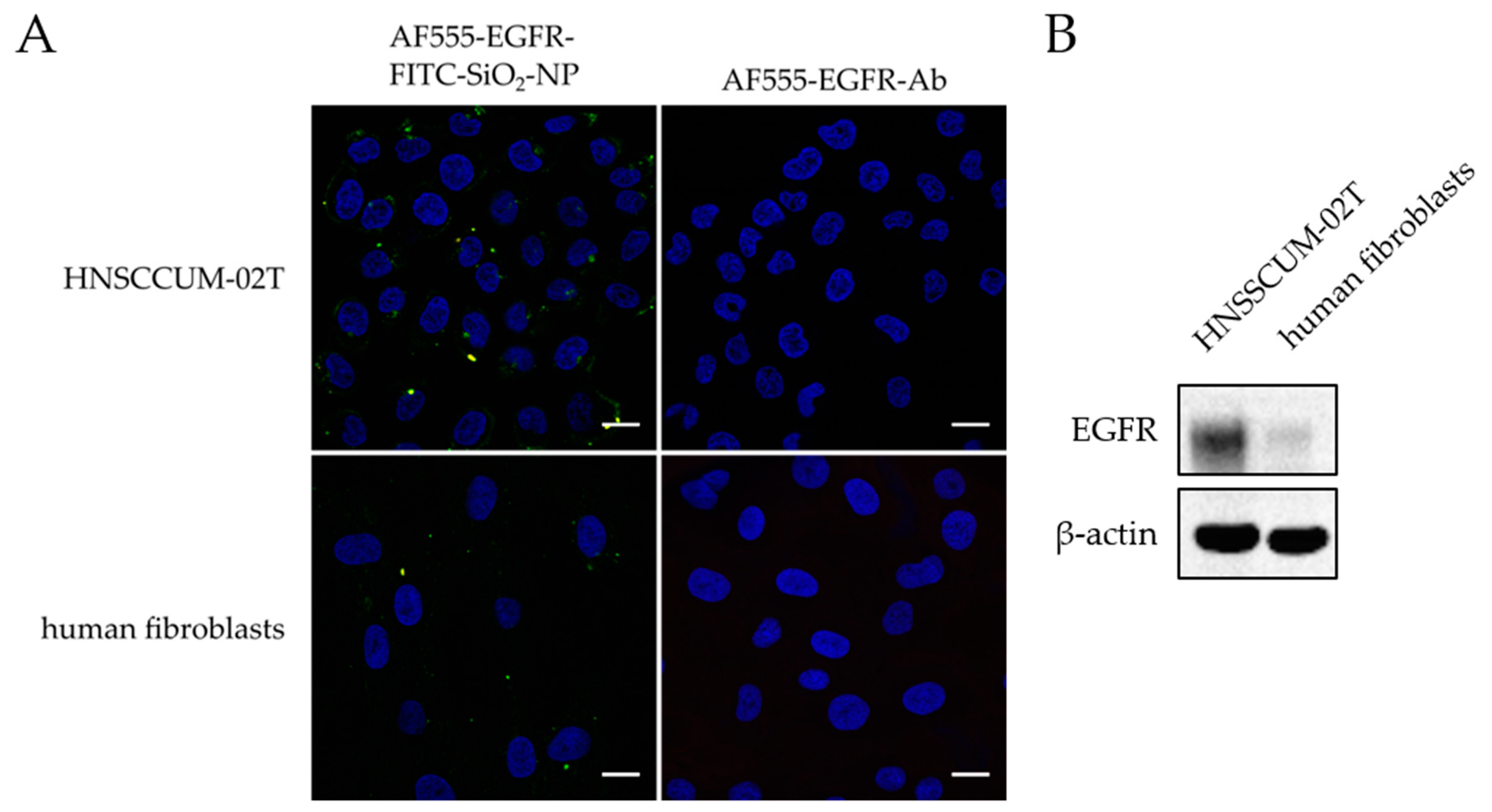
Confocal laser scanning microscopy of in vitro staining with AF555-EGFR-FITC-SiO2-NPs and AF555-EGFR-Ab: The contrast enhancement capabilities of AF555-EGFR-FITC-SiO2-NPs were compared to AF555-EGFR-Ab in HNSCCUM-02T cells and primary human fibroblasts. The cells were seeded in µ-slides 8-well ibidiTreat, let to adhere overnight, and pre-incubated with 1% BSA in PBS for 30 min. Then, the cells were treated for 30 min with 100 µg/mL AF555-EGFR-FITC-SiO2-NPs or the corresponding amount of AF555-EGFR-Ab in cell culture medium, respectively. The cells were washed and fixed as mentioned above and finally, the samples were embedded in VECTASHIELD® Hardset™ Antifade Mounting Medium with DAPI (Vectashield, Vector Laboratories, Burlingame, CA, USA). Images were obtained with Leica DMi8 confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany) at 630× magnification.
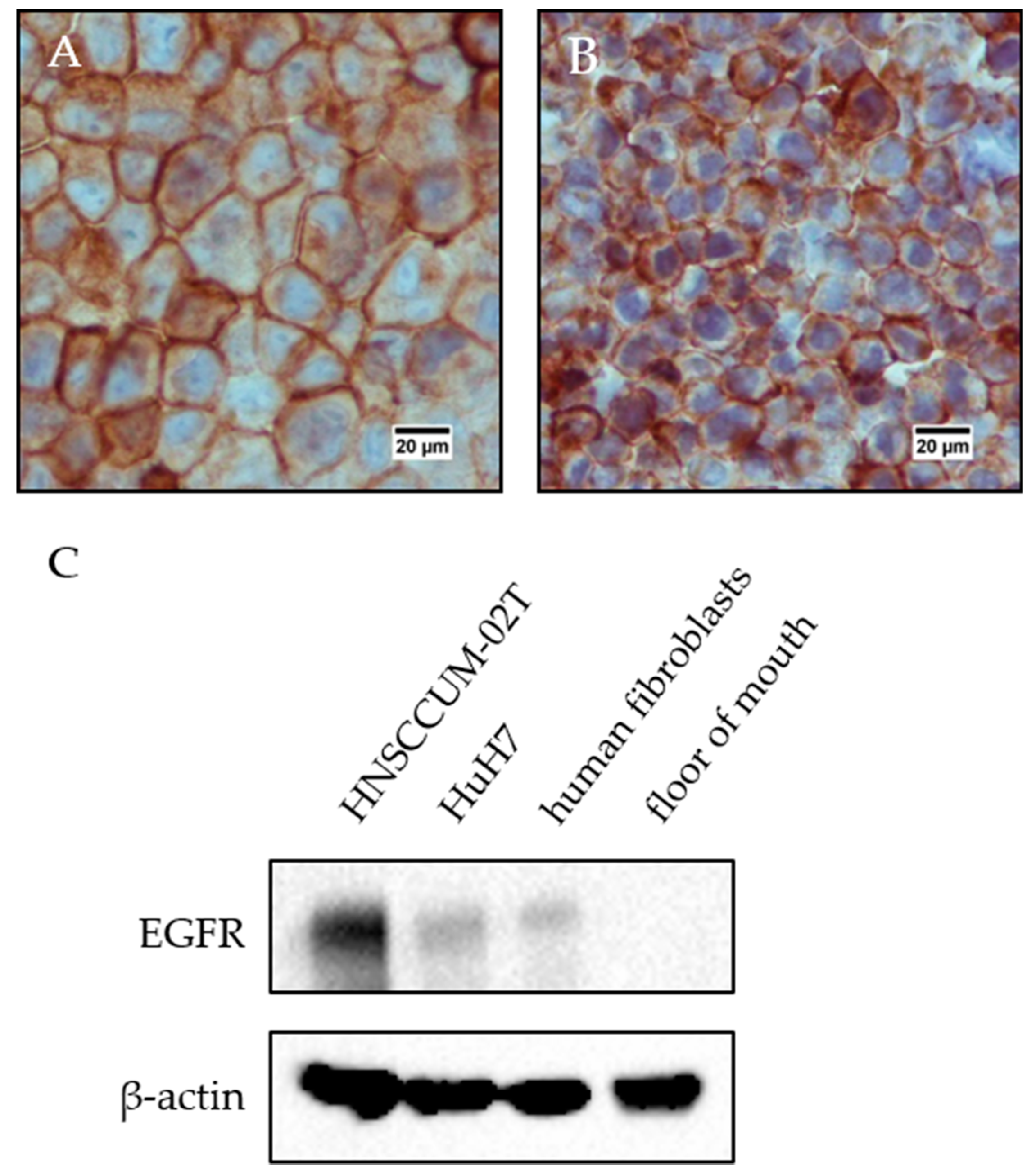
EGFR protein expression in cell lysates and floor of mouth lysate: HNSCCUM-02T cells, HuH7 cells, and human fibroblasts were lysed with RIPA lysis buffer [65 mM Tris, 154 mM NaCl (Carl Roth, Karlsruhe, Germany), 1% NP-40 (Hoffmann- La Roche AG, Basel, Switzerland), 0.025% sodium deoxycholate (Sigma-Aldrich, St. Louis, MO, USA), 1 mM EDTA (Carl Roth, Karlsruhe, Germany), pH 7.4 with protease inhibitor cocktail (Complete®, Hoffmann- La Roche AG, Basel, Switzerland) and phosphatase inhibitor cocktail (PhosSTOP, Hoffmann- La Roche AG, Basel, Switzerland)]. Samples were sonicated for 30 s, incubated on ice for 20 min and sonicated twice for 30 s. In the next step, the lysates were centrifuged (13,000 rpm, 20 min, 12 °C, BioFuge Fresco, Heraeus, Kendro Laboratory Products, Hanau, Germany), and the supernatants were transferred to new centrifuge tubes. The floor of mouth tissue was weighed and cut into small pieces, liquid nitrogen was added, and the tissue sample was further ground in a mortar. This was repeated two to three times. Then, the sample was lysed with RIPA lysis buffer (volume dependent on the amount of sample) and transferred to a centrifuge tube. The lysate was sonicated twice for 30 s (75%), incubated on ice for 20 min and sonicated twice again. Finally, the sample was centrifuged (13,000 rpm, 20 min, 12 °C, BioFuge Fresco, Heraeus) and transferred to a fresh centrifuge tube. The protein concentration was determined with DC™ Protein Assay (Bio-Rad, Munich, Germany) in accordance with the manufacturer’s instructions. Samples were subjected to discontinuous polyacrylamide gel electrophoresis. 20 µg proteins per sample were separated in a 7.5% stain-free polyacrylamide gel (prepared according to the instructions by Bio-Rad) with 10 mA for 2.5 h. The stain-free gel was activated, and protein bands were transferred to a methanol-activated PVDF membrane with Biometra tank blot (20 V, overnight). The blotting buffer contained 48 mM Tris, 39 mM Glycine (Carl Roth, Karlsruhe, Germany) and 20% (V/V) methanol (Honeywell Riedel- de Haën®, Seelze, Germany) in desalted water. The membrane was briefly washed in 20 mM Tris-buffered saline (137 mM NaCl) containing 0.1% Tween20 (TBS-T20) and all protein bands were detected at ChemiDoc (Bio-Rad, Munich, Germany). After blocking with 5% dry-milk in TBS-T20 for 1 h at room temperature, the membrane was washed with TBS-T20 (3 × 5 min) and incubated overnight at 4 °C with EGFR-Ab 1:1000 in 5% BSA in TBS-T20 under agitation. The next day, the membrane was washed with TBS-T20 (3 × 5 min) and incubated with anti-rabbit IgG, HRP-linked antibody (Cell Signaling Technologies) 1:5000 in 5% dry-milk in TBS-T20 for 1 h at RT. The protein bands were detected at ChemiDoc™ MP Imaging System (Bio-Rad) after 2 min incubation with Western Lightning® Plus ECL (PerkinElmer, Waltham, MA, USA) reagent. Then, the membrane was stripped at RT: 30 min with 25 mM glycine pH 2.0 and 30 min with 1% (m/V) SDS in desalted water. For the detection of β-actin, the membrane was blocked with 5% dry-milk in TBS-T20 (1 h at RT), washed with TBS-T20 (3 × 5 min), incubated with anti-β-actin monoclonal antibody, clone AC-15 (Sigma-Aldrich, St. Louis, MO, USA) 1:10,000 in 5% BSA in TBS-T20 overnight at 4 °C, washed again thrice, incubated with anti-mouse IgG, HRP-linked antibody (Cell Signaling Technologies Inc, Danvers, MA, USA) 1:5000 in 5% dry-milk in TBS-T20 (1 h, RT) and visualized with Western Lightning® Plus ECL reagent at ChemiDoc™MP Imaging System.
3D cell culture: 3D solid tumors were prepared with the cell line HuH7 and 5,000,000 cells were used for one tumor, which was applied to the chorioallantoic membrane (CAM) assay. First, the cells were transferred to a 1.5 mL centrifuge tube and generated a cellular pellet by centrifugation (1400 rpm, 10 min, 20 °C, Hettich Universal 16R, Hettich Zentrifugen, Tuttlingen, Germany). The supernatants were carefully removed, the cells were quickly suspended in 25 µL Matrigel® Basement Membrane Matrix (Corning®, Corning Inc, NY, USA) and pipetted into six-well plates. After 30 min incubation in the incubator (37 °C, 5% CO2 (V/V)), the tumor model stiffed, medium was added to the wells and the 3D-cultures were incubated overnight until use in the CAM assay.
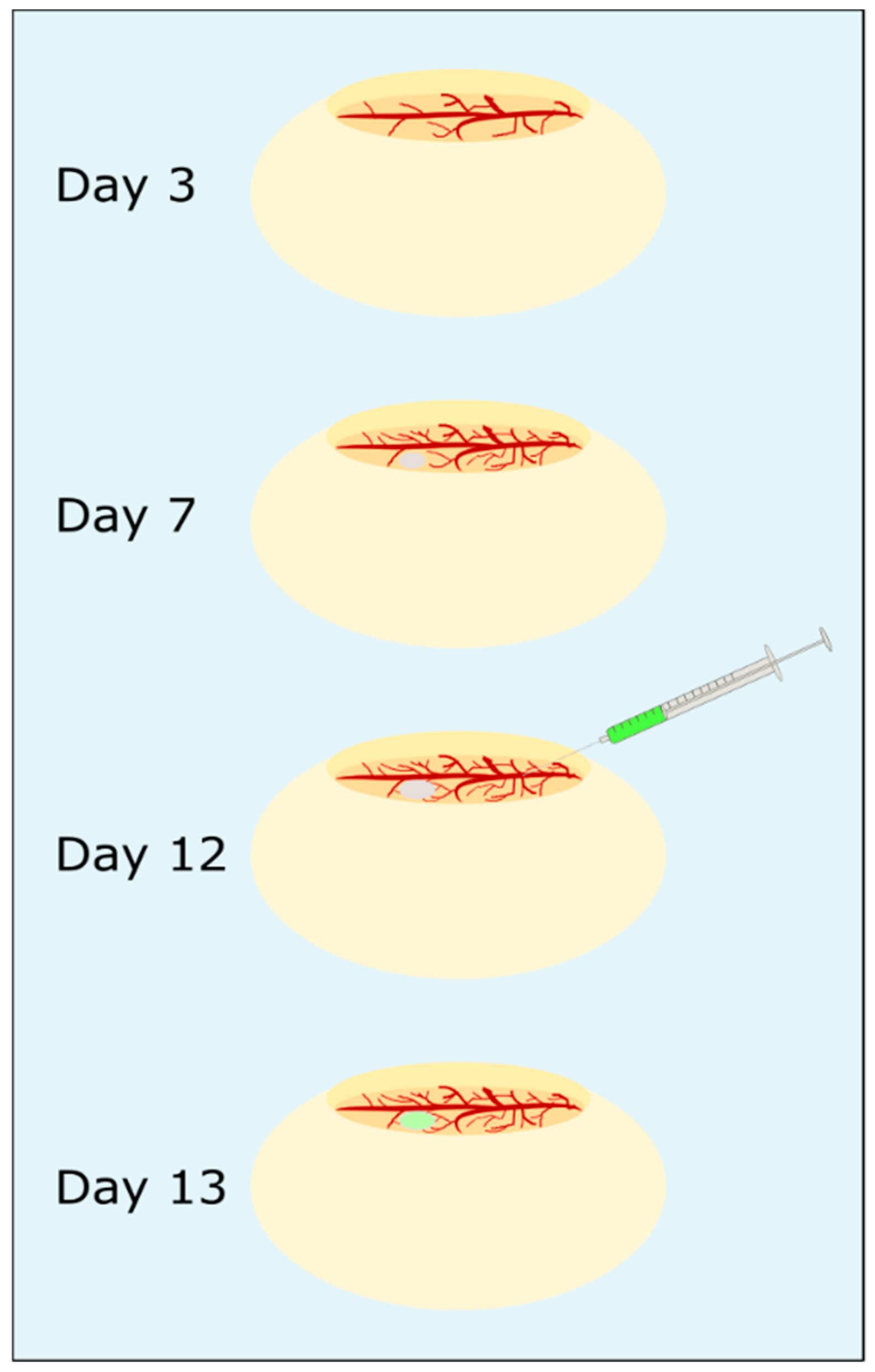
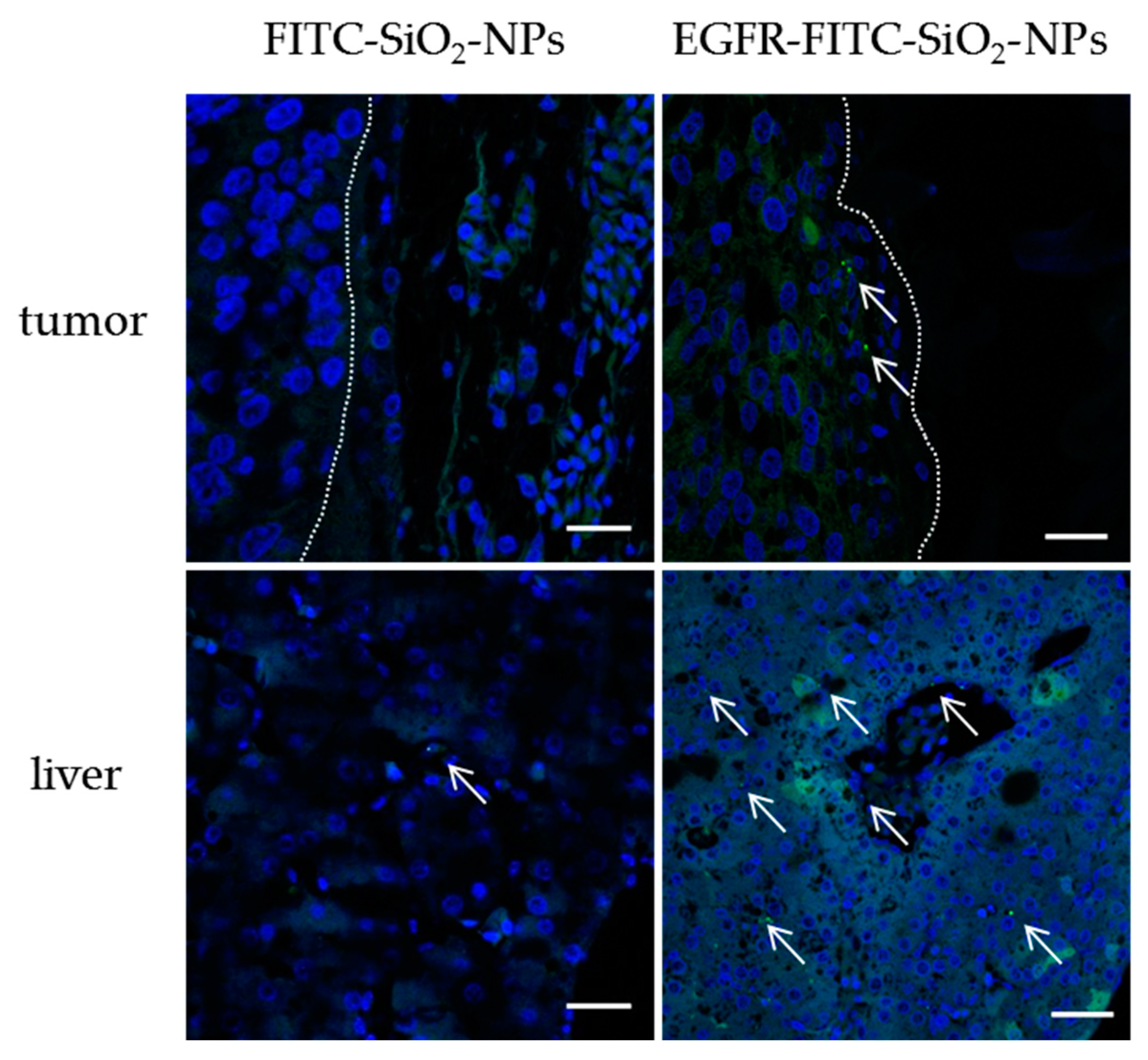
Chorioallantoic membrane assay: The steps of the CAM assay are presented in
Figure 3. Fertilized white leghorn eggs (LSL, Dieburg, Germany) were cleaned with sterilized water, screened for any damage and intact eggs were horizontally placed in a freshly cleaned incubator at 37.5 °C. After three days incubation, about 6 mL albumen were removed from each egg with a syringe and the hole was sealed with sticky tape. Then, a micropore bandage was attached horizontally to the top of the egg to prevent eggshells falling into the egg while an oval hole was cut out. The egg was checked for fertilization, sealed with Parafilm “M”™, and was further incubated. The chicken embryos (CEs) were monitored daily for viability. Viability was characterized by steady blood flow and visible heartbeat. On the seventh day of incubation, a blood vessel was carefully cut with a scalpel, a 3D-culture of HuH7 cells (preparation described above) was directly placed on the wound and 20 µL Matrigel
® Basement Membrane Matrix (Corning
®, Corning Inc, Corning, NY, USA) were added to the cells. Then, the CEs were incubated further with minimal movement to enhance tumor growth. After the tumor grew for five days, 50 µL of 0.9% (m/V) NaCl solution, 1.0 mg/mL EGFR-FITC-SiO
2-NPs in 20% ethanol/80% 0.9% NaCl (
V/
V) or FITC-SiO
2-NPs in 0.9% NaCl were injected into a blood vessel and the wound was sealed immediately with a silver nitrate stick. The CEs were incubated further for 24 h and from two embryos of each treatment group the CAM with tumor and liver were removed, transferred to immunohistochemistry cassettes and fixed in 4% PFA overnight. Tissue samples were stored in 1% PFA until further processing.
Fluorescence staining of chicken embryonic tissues: The washing and dehydration of the tissues were performed at RT and under light protection. First, the fixed tissues were washed three times with deionized water (20 min each) and then incubated in 70%, 80% and 90% (V/V) isopropanol (Hedinger, Stuttgart, Germany) in deionized water for one hour each. Next, the samples were incubated in 100% isopropanol for 1.5 h, afterwards for two hours in xylene (PanReac AppliChem, Darmstadt, Germany) and finally overnight in fresh xylene. The cassettes were transferred to liquid paraffin, incubated for four hours at 60 °C, transferred to freshly prepared liquid paraffin and incubated overnight at 60 °C. The embedding of the tissues was conducted with Leica EG 1140H tissue embedding center. The organs were placed in a small form, liquid paraffin was added, and the form was placed on a cooling plate. Paraffin tissue sections were cut with a Leica RM2165 rotary microtome to 3–5 µm sections and placed on microscope slides. Next, the tissue sections were deparaffinated as follows: Incubation in xylene for 5 min (twice), incubation in 100%, 90%, 80%, 70% (V/V) isopropanol in desalted water for 5 min each and 5 min of incubation in deionized water. Finally, the samples were embedded in Vectashield® Hardset™ Antifade Mounting Medium with DAPI (Vectashield) and solidified for at least 24 h before microscopic analysis. The stained samples were analyzed with a Leica DMi8 confocal laser scanning microscope with 400-fold magnification.
Immunohistochemistry staining of HNSCCUM-02T cells and HuH7 cells: HNSCCUM-02T and HuH7 cells were detached with trypsin/EDTA, transferred to 1.5 mL centrifuge tubes, centrifuged (350 g, 5 min, RT) and washed with PBS three times. The PBS was removed completely after the final washing step, and 4% PFA in PBS were added. The samples were stored at 4 °C. Then, cells were washed three times with deionized water and dehydrated in 70%, 80%, 90% and 100% (V/V) isopropanol in deionized water for 30 min each. Next, the cellular pellets were incubated twice in xylene and twice in liquid paraffin for 30 min each. The cell pellets were generated by centrifugation (4000 rpm, 5 min, RT, Universal 16 R, Hettich Zentrifugen, Tuttlingen, Germany) after each incubation step. After the final paraffin incubation step, the paraffin containing cell pellet was solidified briefly, transferred carefully to an immunohistochemistry cassette and embedded in paraffin with a Leica EG 1140H tissue embedding center (Leica Biosystems, Wetzlar, Germany). The samples were cut to 3–5 µm paraffin sections with a Leica RM2165 rotary microtome (Leica Biosystems, Wetzlar, Germany) and placed on microscope slides. Then, cellular samples were deparaffinated as described above, washed once with TBS-T20 (5 min) and antigen demasking was performed with 10 mM Tris/1 mM EDTA (pH 9.00) in desalted water for 30 min in a steamer (Steam Cuisine High Speed). After rinsing the sample briefly with deionized water, it was washed for 5 min with TBS-T20, blocked for 45 min with goat serum (normal) (DAKO Agilent, Santa Clara, CA, USA) 1:10 in 1% BSA in PBS and was incubated with EGFR-Ab 1:50 in 1% BSA in PBS for one hour at RT. The samples were washed twice with TBS-T20 for 5 min each, incubated with DEnVision™+Dual Link System-HRP (DAKO Agilent, Santa Clara, CA, USA) for 30 min and washed again twice with TBS-T20 for 5 min. For visualization the samples were incubated with DakoCytomation Liquid DAB+ Substrate Chromogen System (DAKO Agilent, Santa Clara, CA, USA) and hemalum solution acid according to Mayer ready-to-use (Carl Roth, Karlsruhe, Germany) for 5 min each and washed for 5 min with tap water. Finally, the samples were dehydrated by incubating in 70%, 80%, 90% and 100% isopropanol in desalted water and twice with xylene for 5 min each and embedded in Eukitt® Quick-hardening mounting medium (Sigma-Aldrich, St. Louis, MO, USA). Images were taken at a Nikon Eclipse E200 microscope with 400-fold magnification.
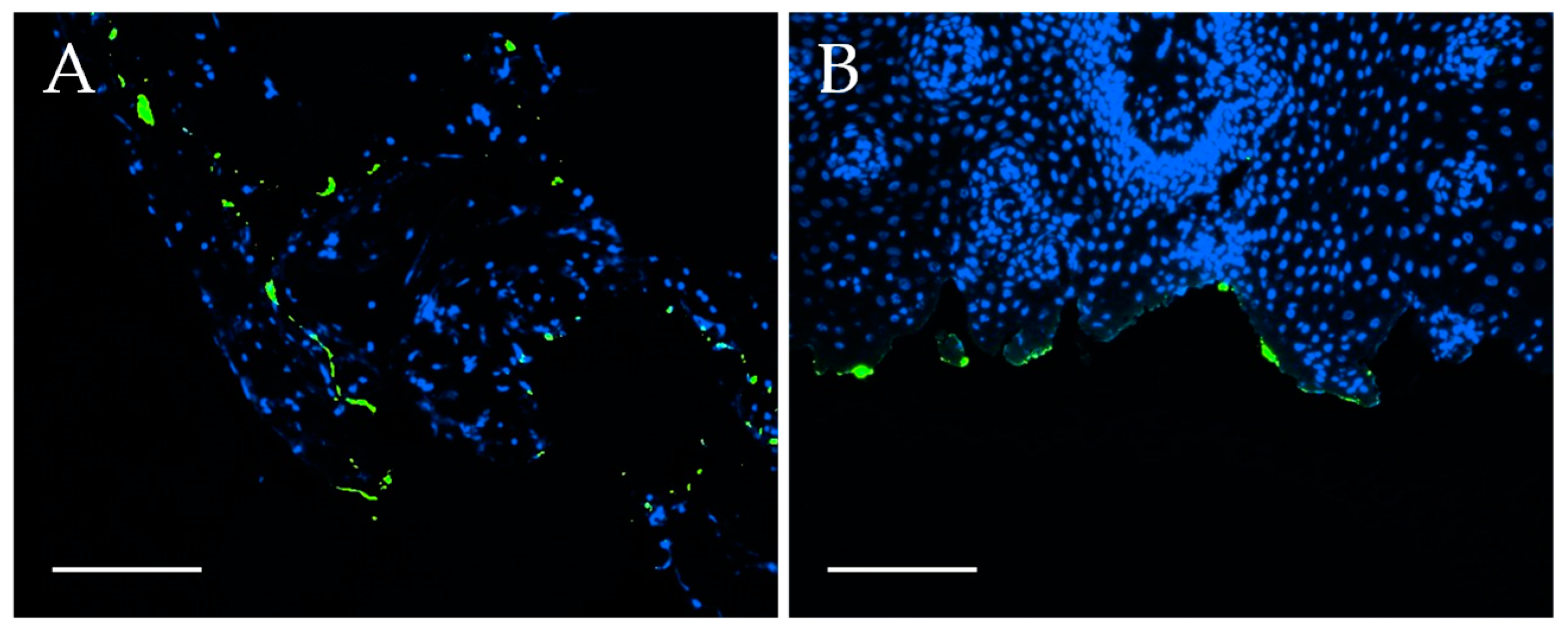
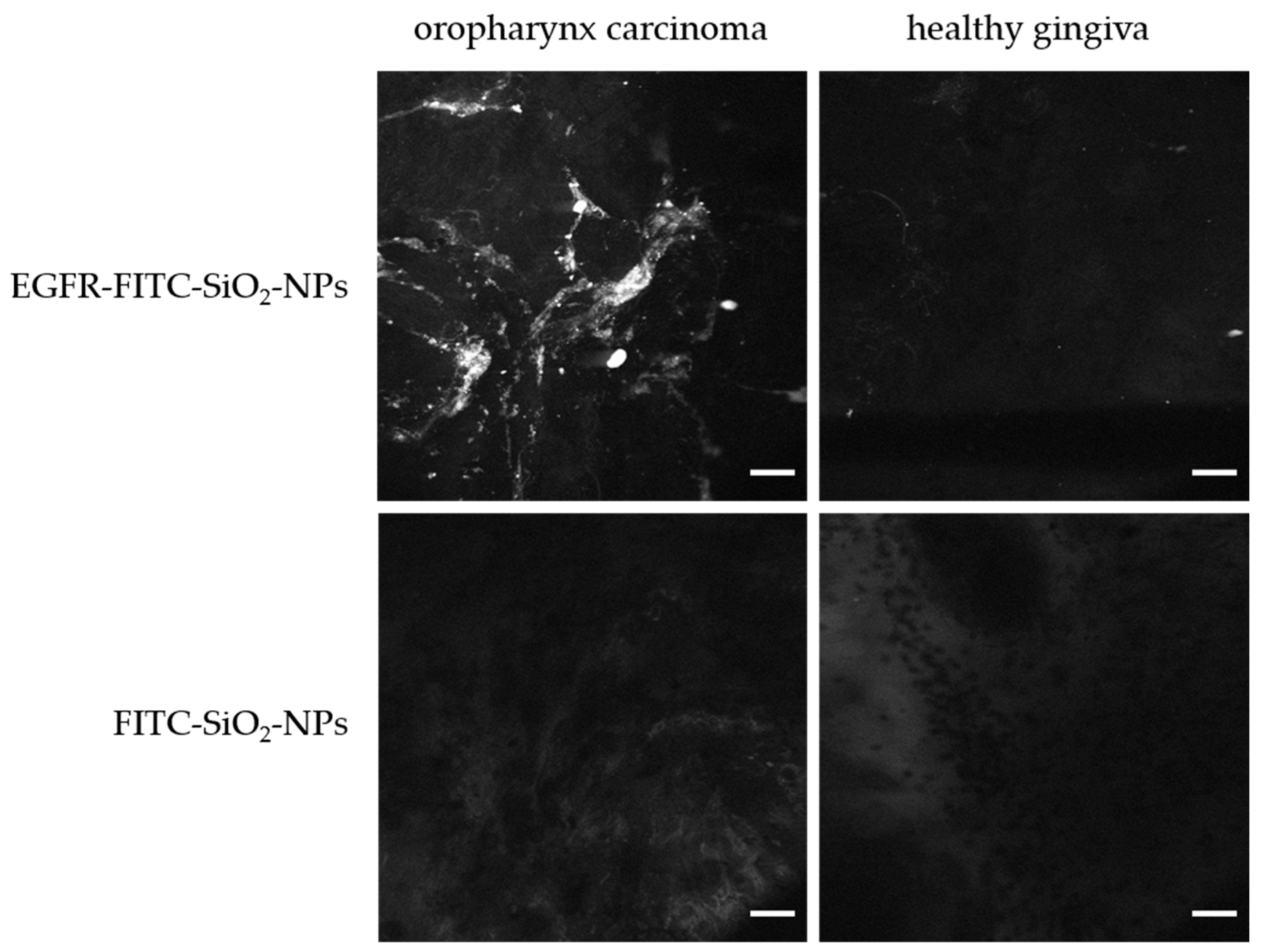
Confocal endomicroscopy: Tumor samples (oropharynxcarcinoma) and healthy tissue (gingiva samples) were incubated briefly in 100 µg/mL EGFR-FITC-SiO
2-NP or FITC-SiO
2-NP dispersions, respectively, and rinsed with deionized water. A miniaturized confocal microscope was integrated into the distal tip of a conventional video endoscope with an outer diameter of 13.2 mm (Pentax, Tokyo, Japan, EC 3830FK) and used for visualization. A single optical fiber was operated as the illumination point source and the detection pinhole. A solid-state laser delivered an excitation wavelength of 488 nm at a maximum laser power output of 1 mW or lower at the tissue surface. Confocal image data were collected with 1100-fold magnification at a scan rate of 0.8 frames/s (1024 × 1024 pixels) and an image size of 215.9 × 215.9 µm [
1].
Fluorescence staining of tumor and healthy tissue samples: After confocal endomicroscopic analysis, the tumor samples and healthy mucosa (gingiva) were fixed in 4% PFA. The samples were dehydrated and embedded in paraffin as described above. Paraffin sections with a thickness of 3–5 µm were prepared and deparaffinated as mentioned before. Next, the samples were embedded in Vectashield® Hardset™ Antifade Mounting Medium with DAPI and solidified for at least 24 h before microscopic analysis with an Eclipse TE2000-U fluorescence microscope (Nikon GmbH, Dusseldorf, Germany).

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











