Preparation of Hierarchically Porous Graphitic Carbon Spheres and Their Applications in Supercapacitors and Dye Adsorption



Abstract
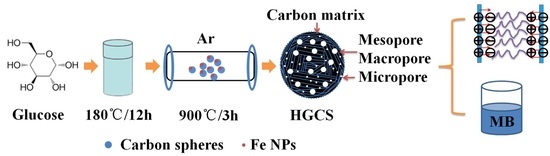
:
1. Introduction
2. Experimental Section
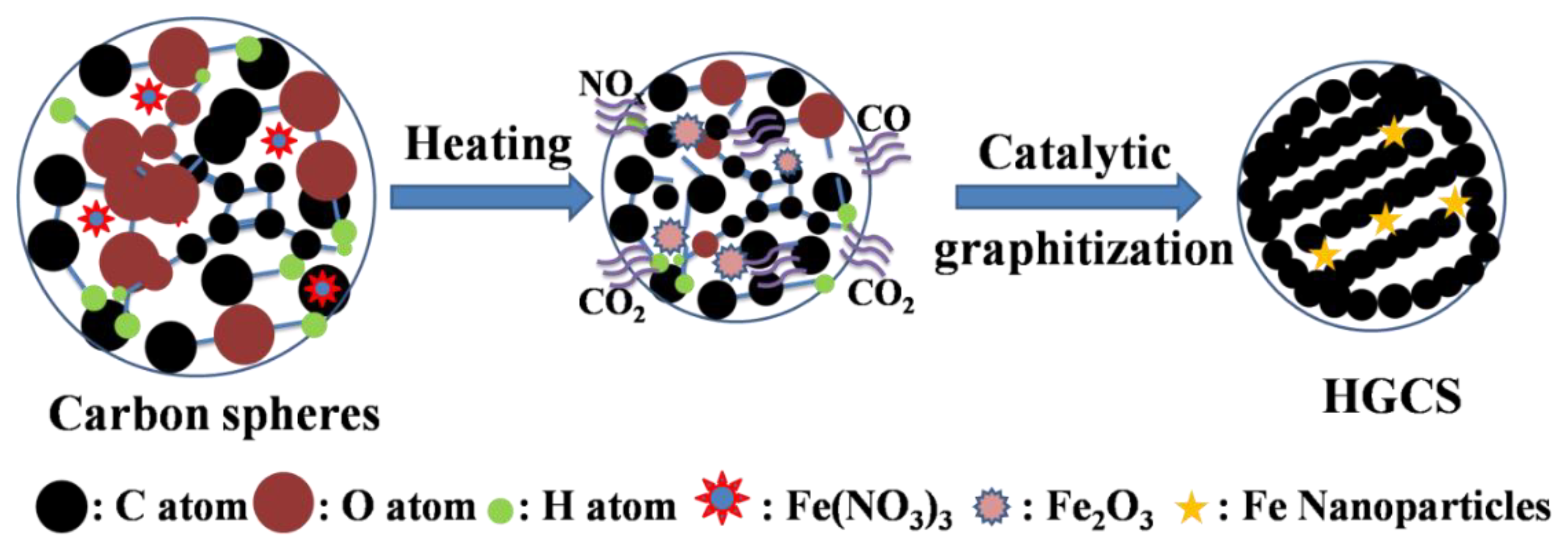
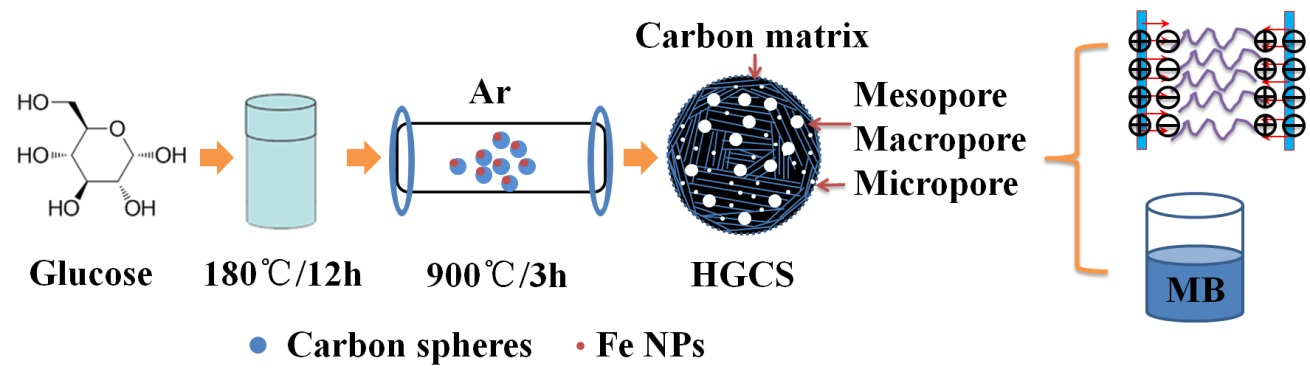
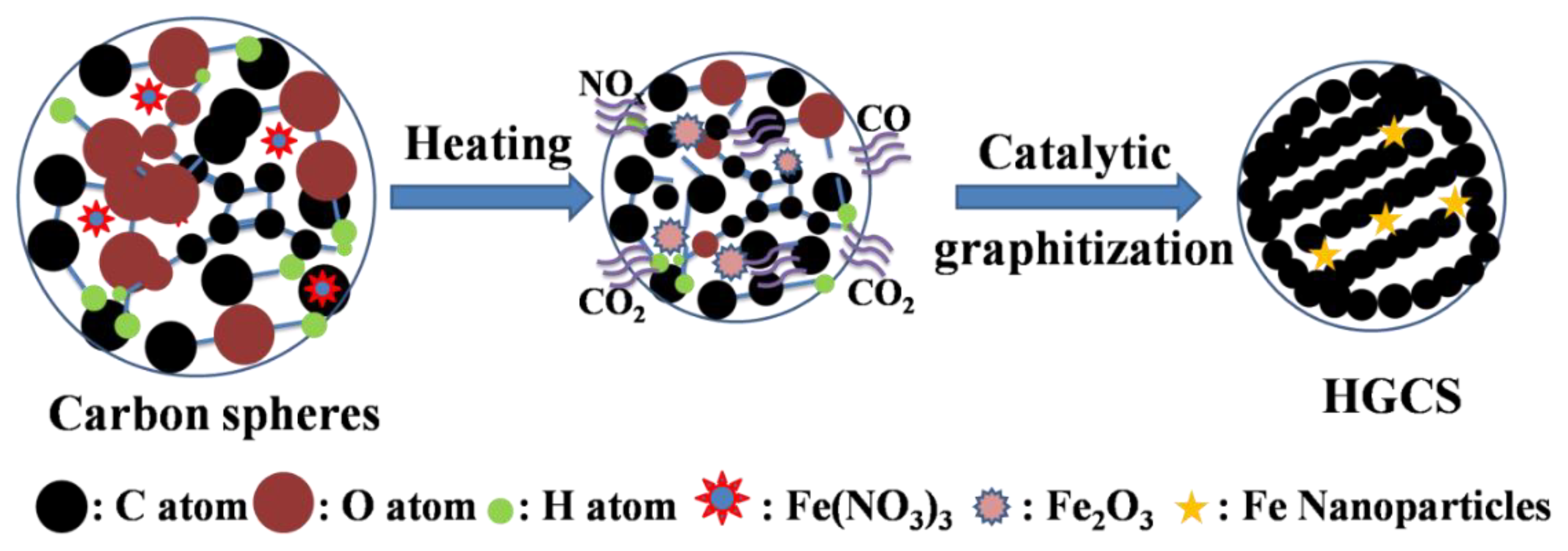
2.1. Preparation of HGCS
2.2. Sample Characterization
2.3. Electrochemical Measurement
2.4. Adsorption Test
3. Results and Discussion
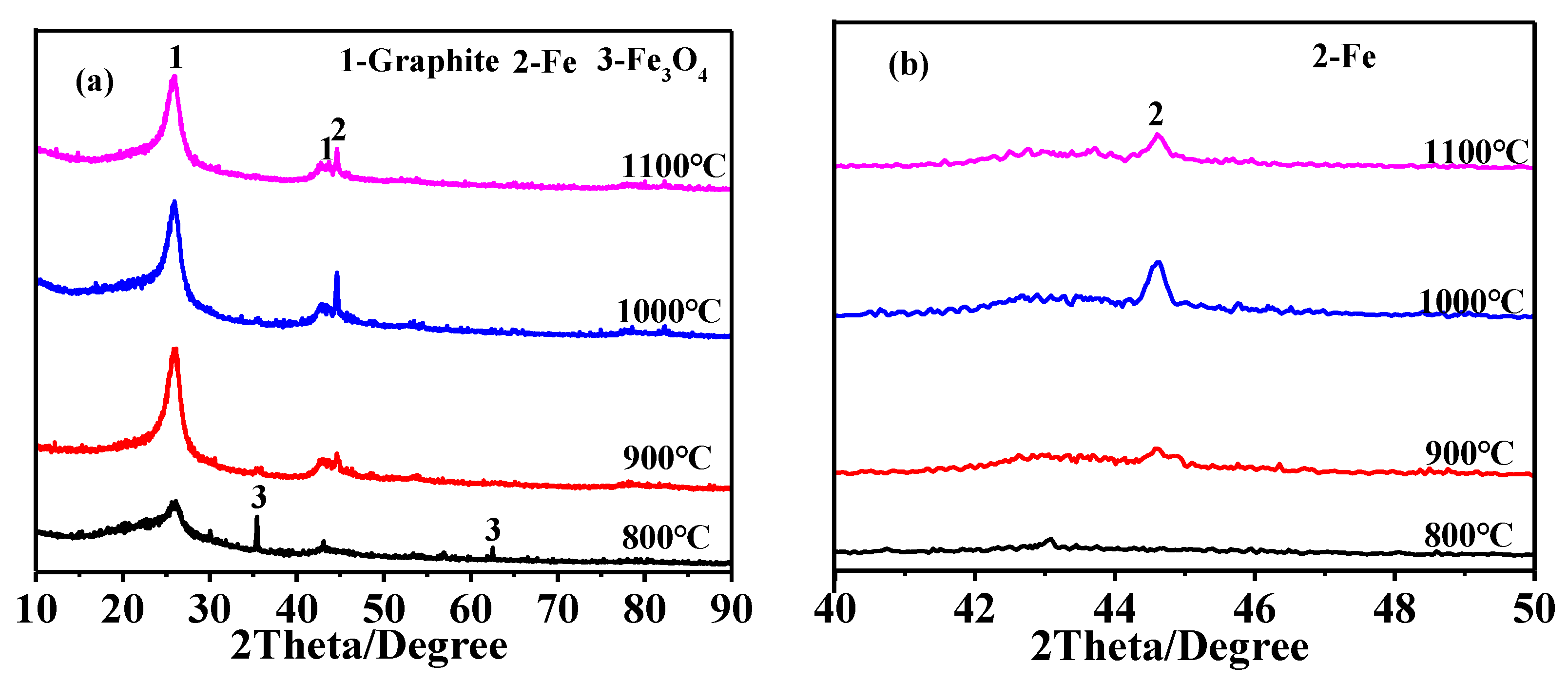
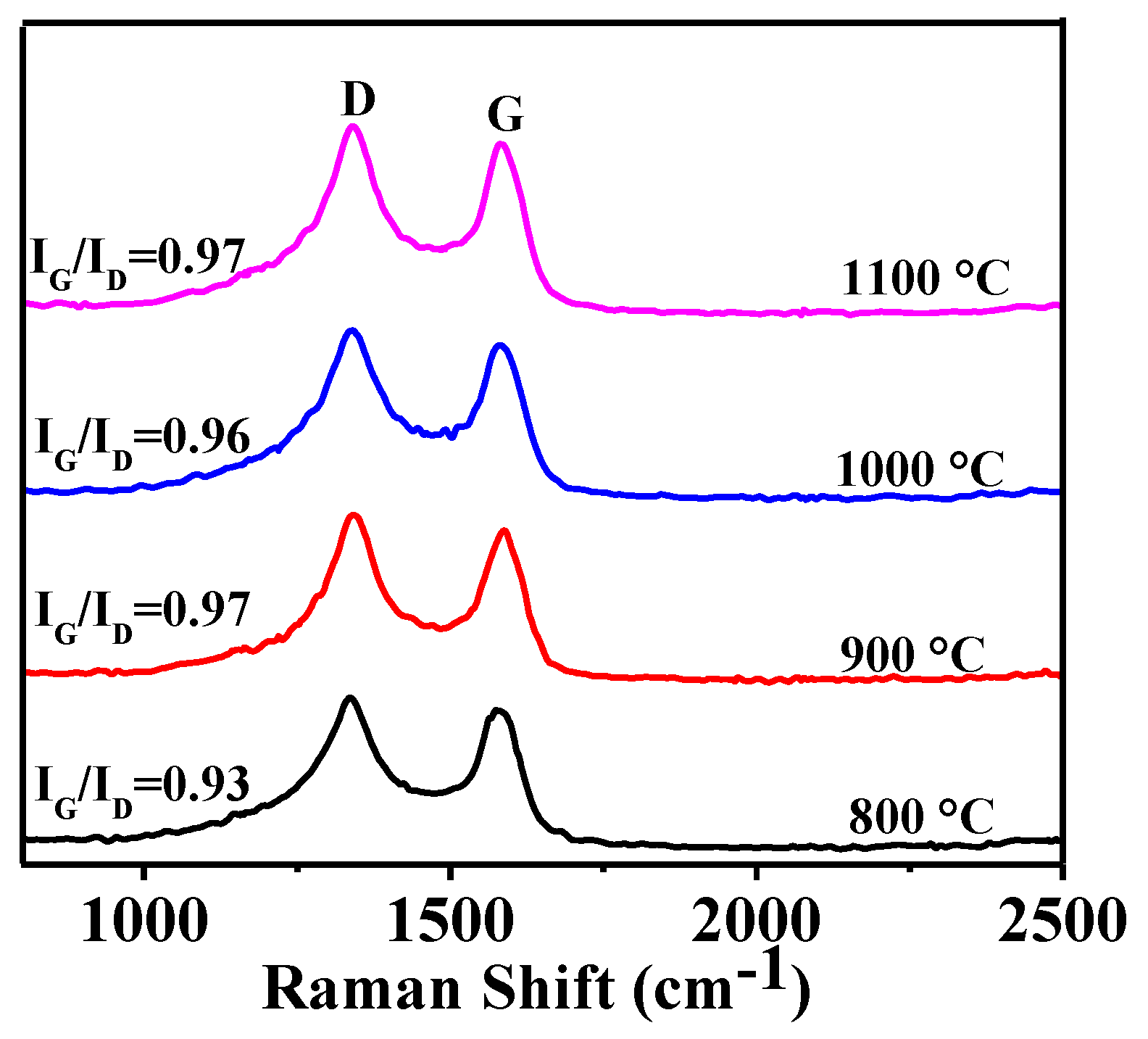
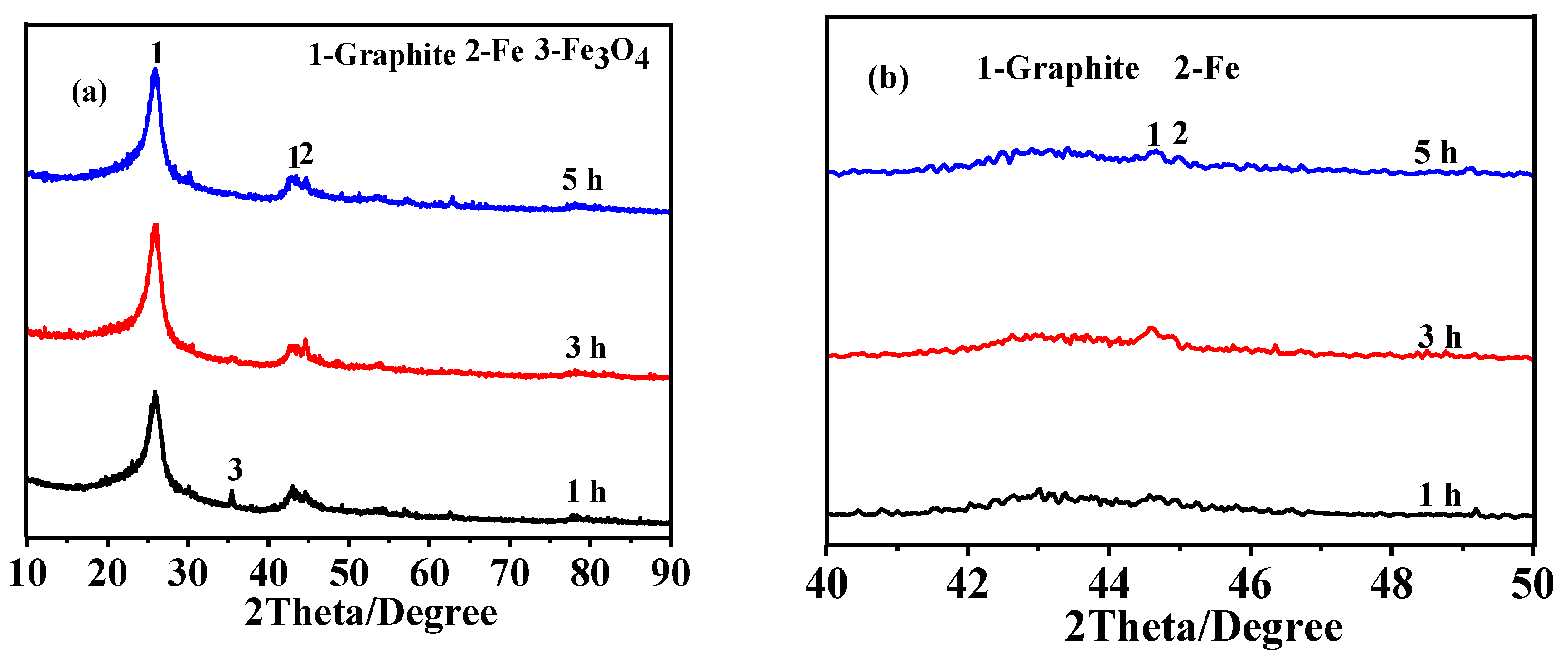
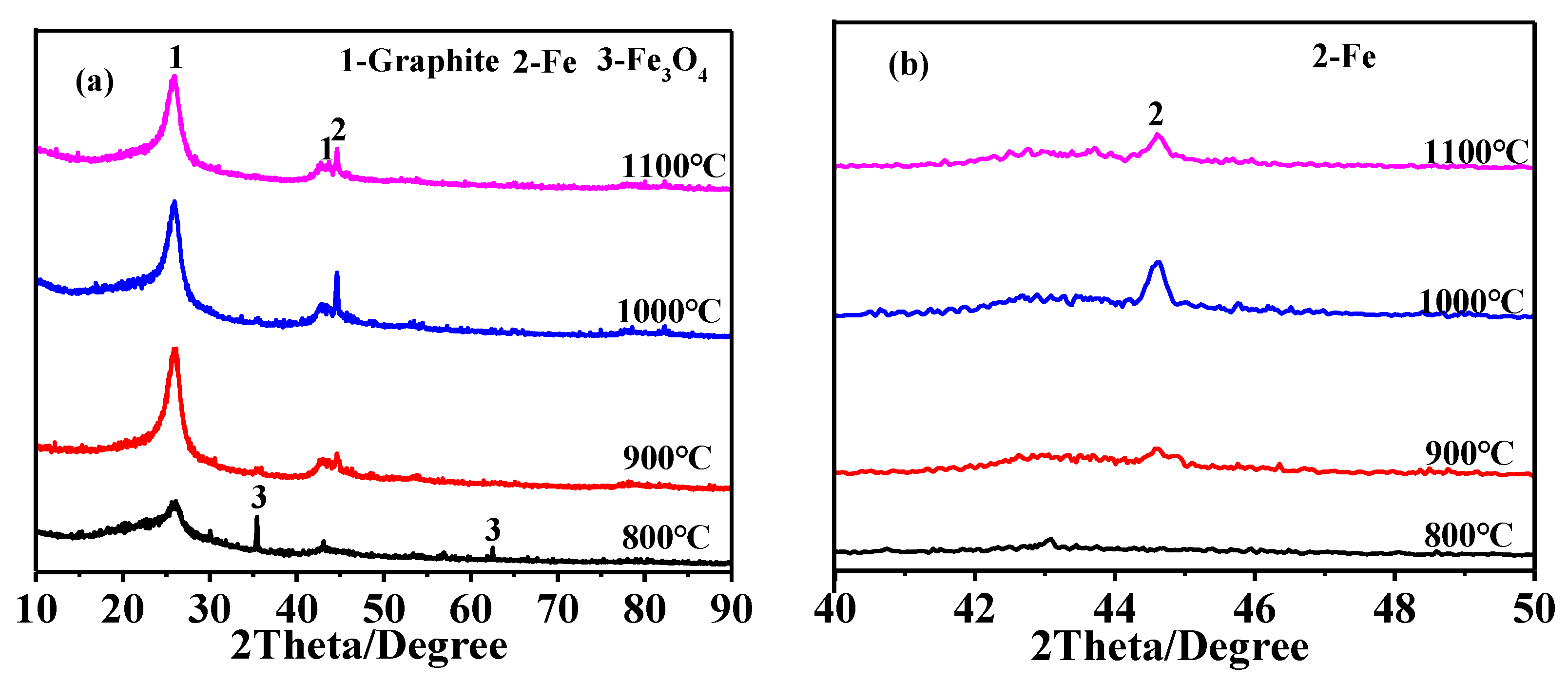
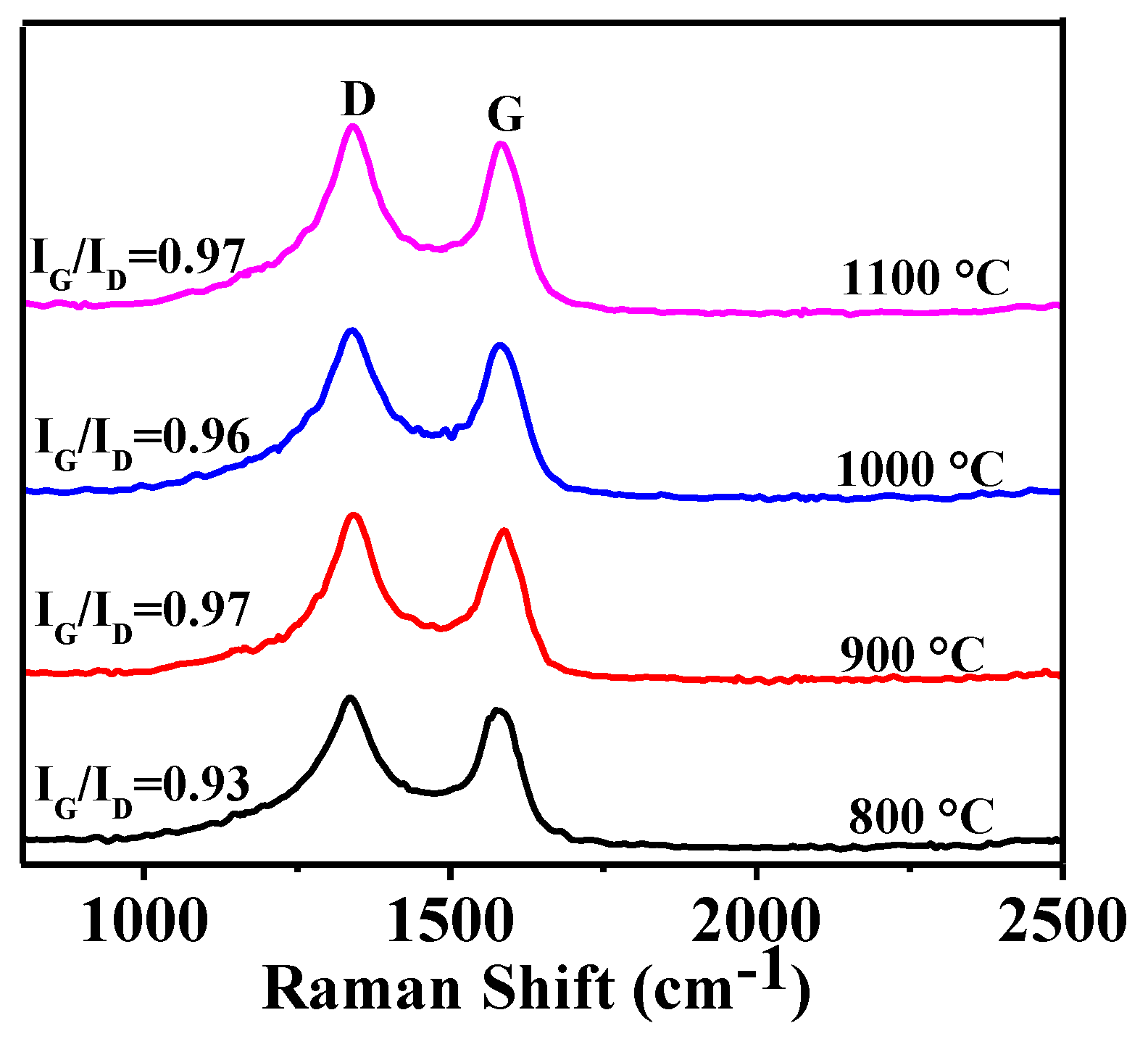
3.1. Effect of Heating Temperature on Graphitization
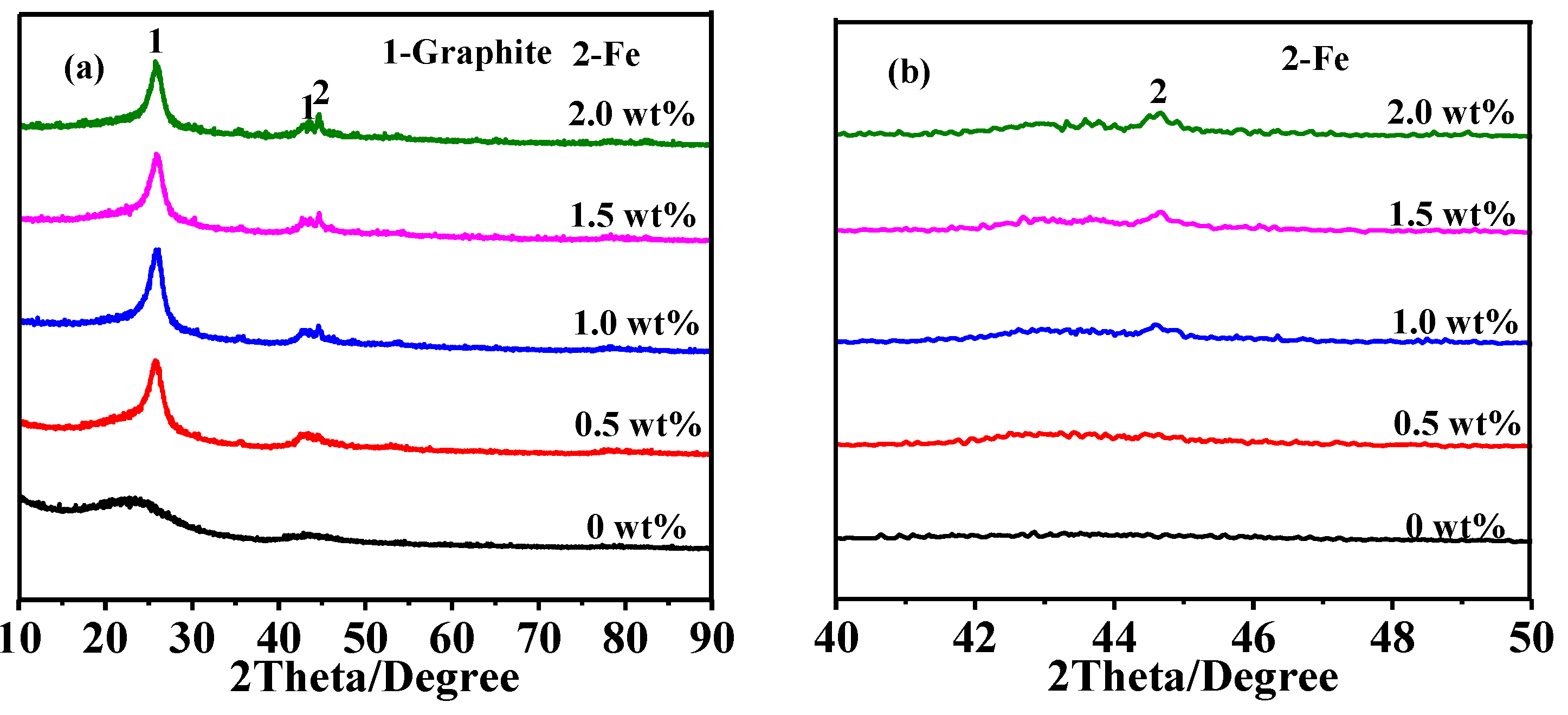
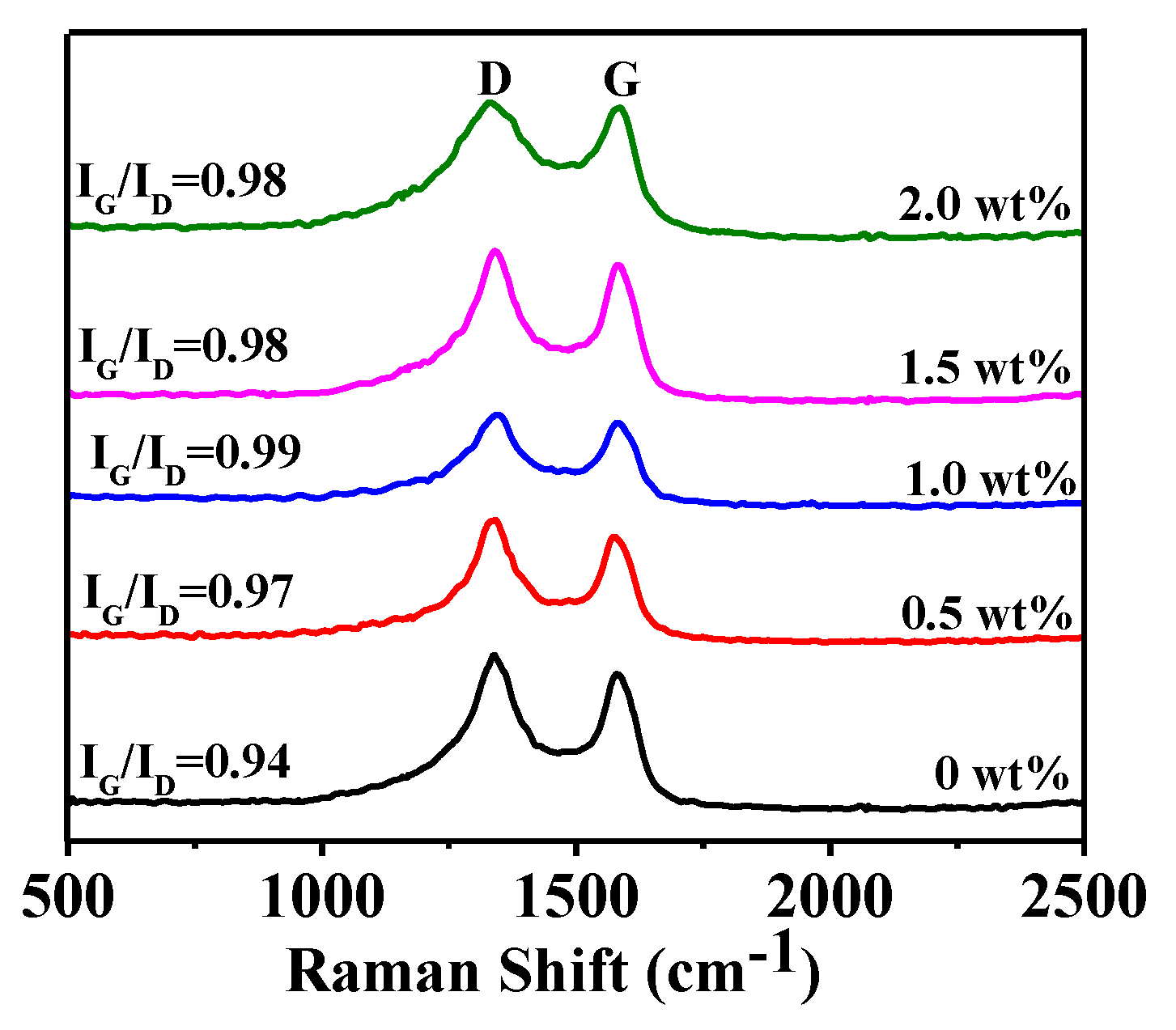
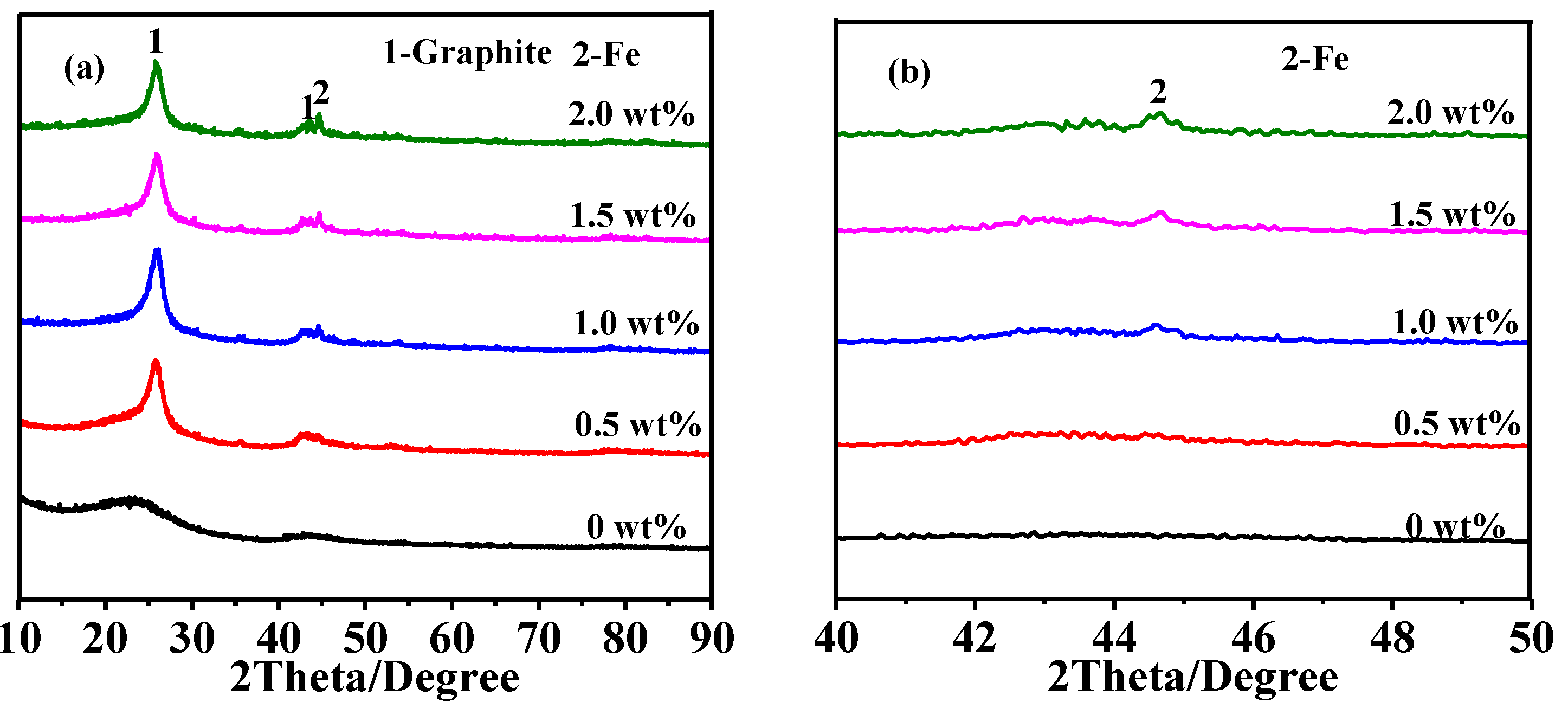
3.2. Effect of Catalyst Content on Graphitization
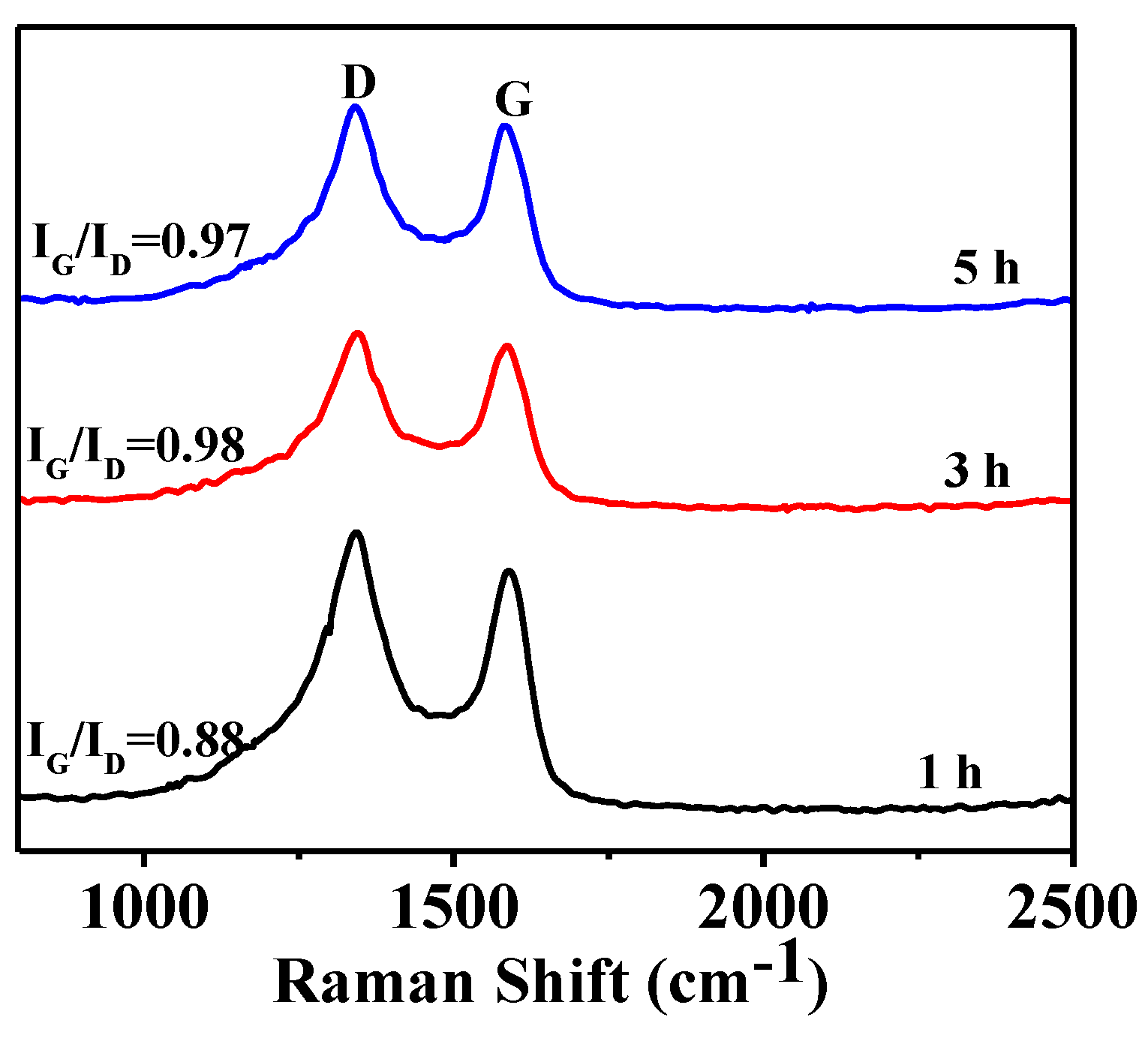
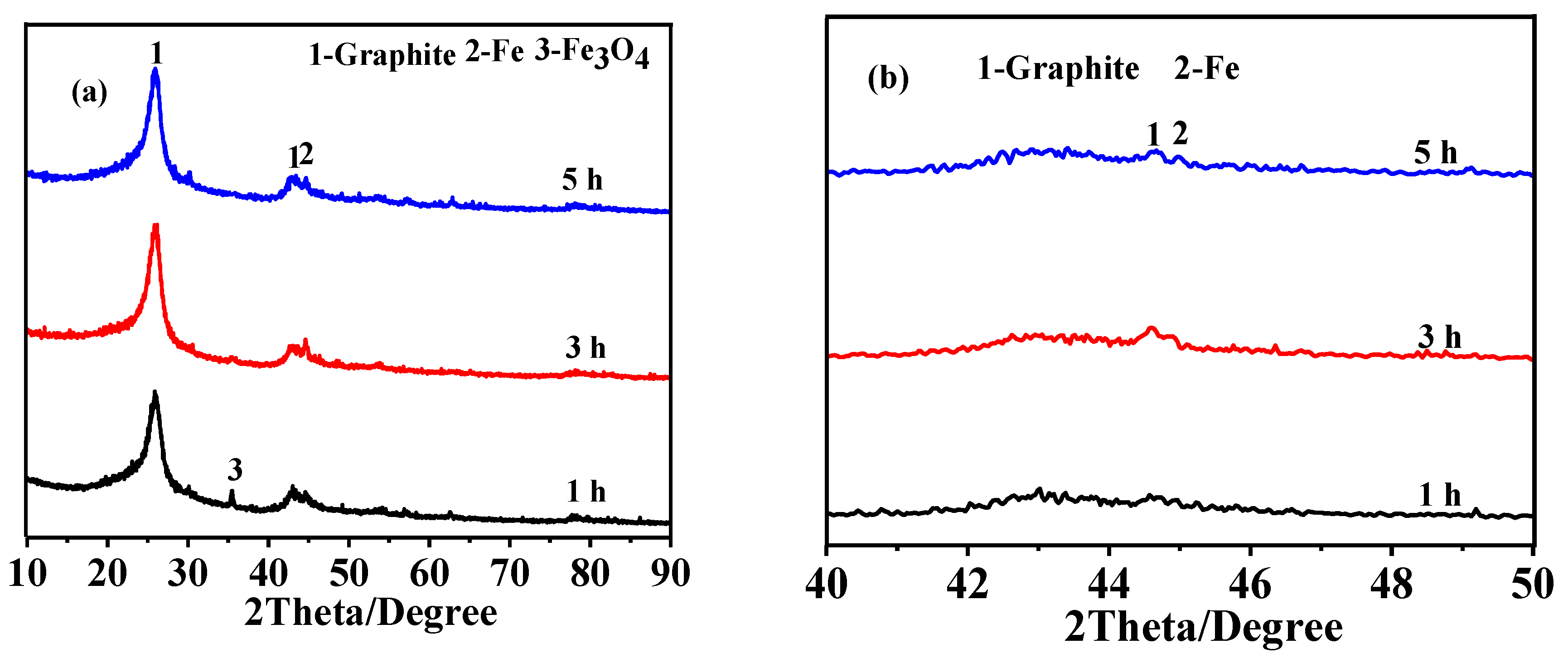
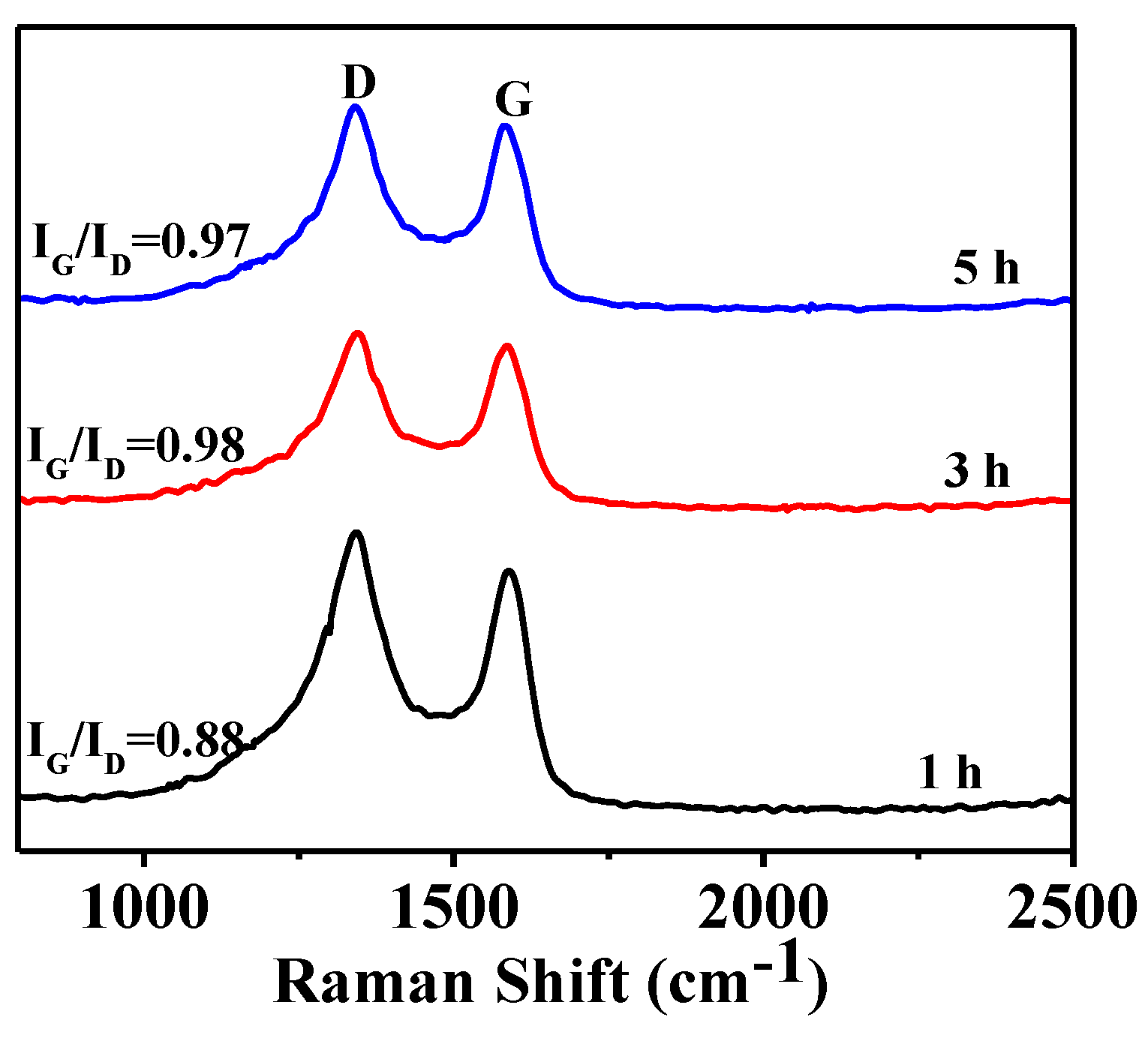
3.3. Effect of Soaking Time on Graphitization
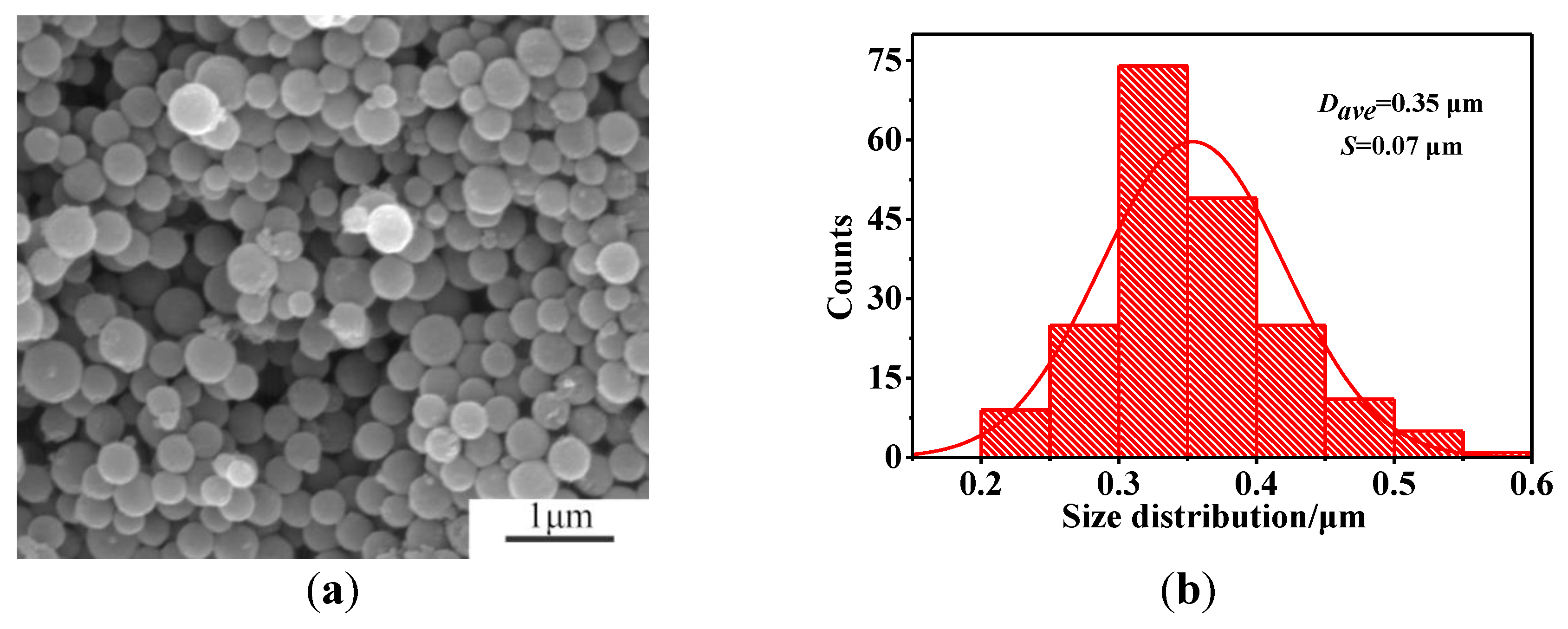
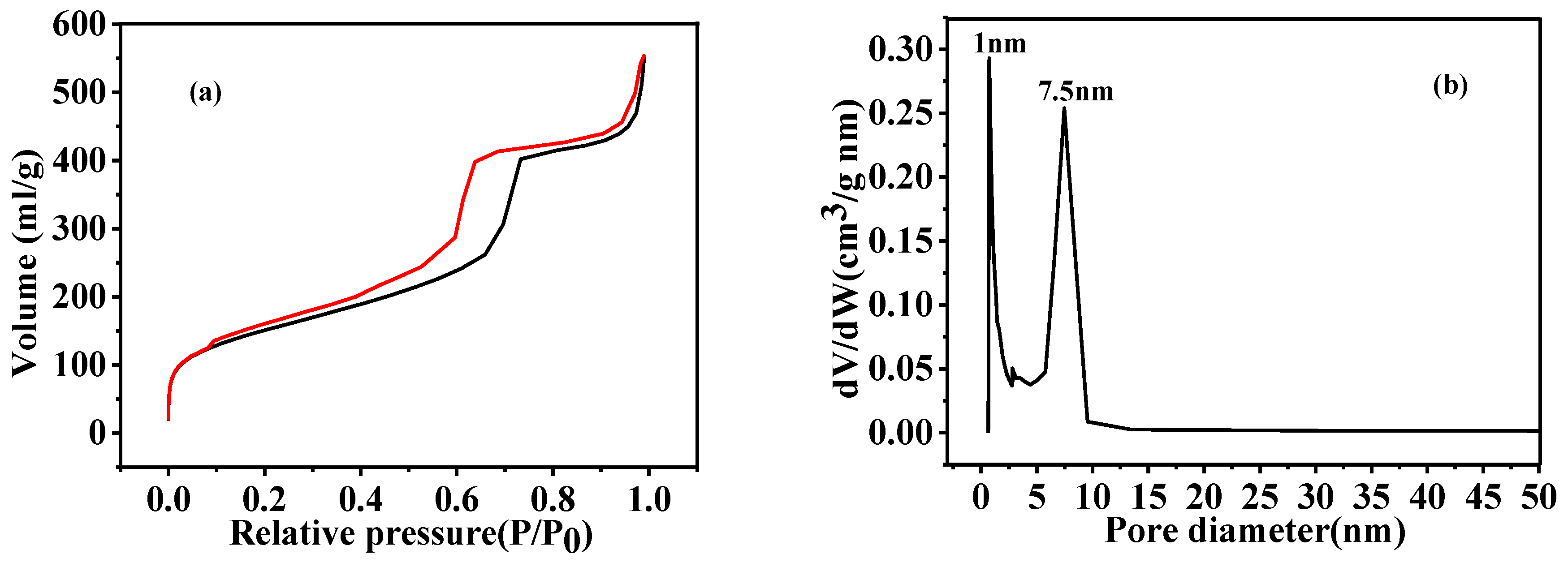
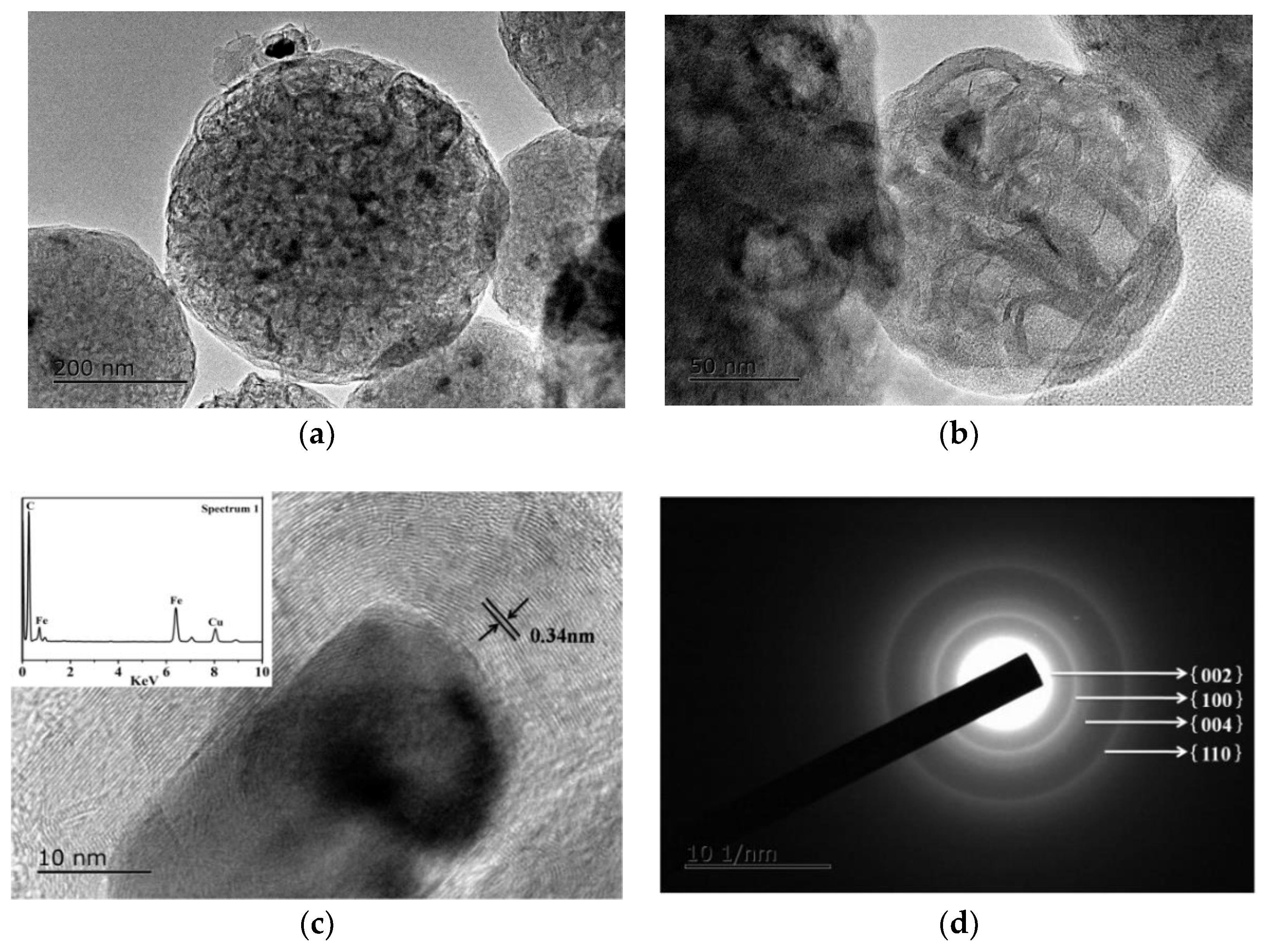
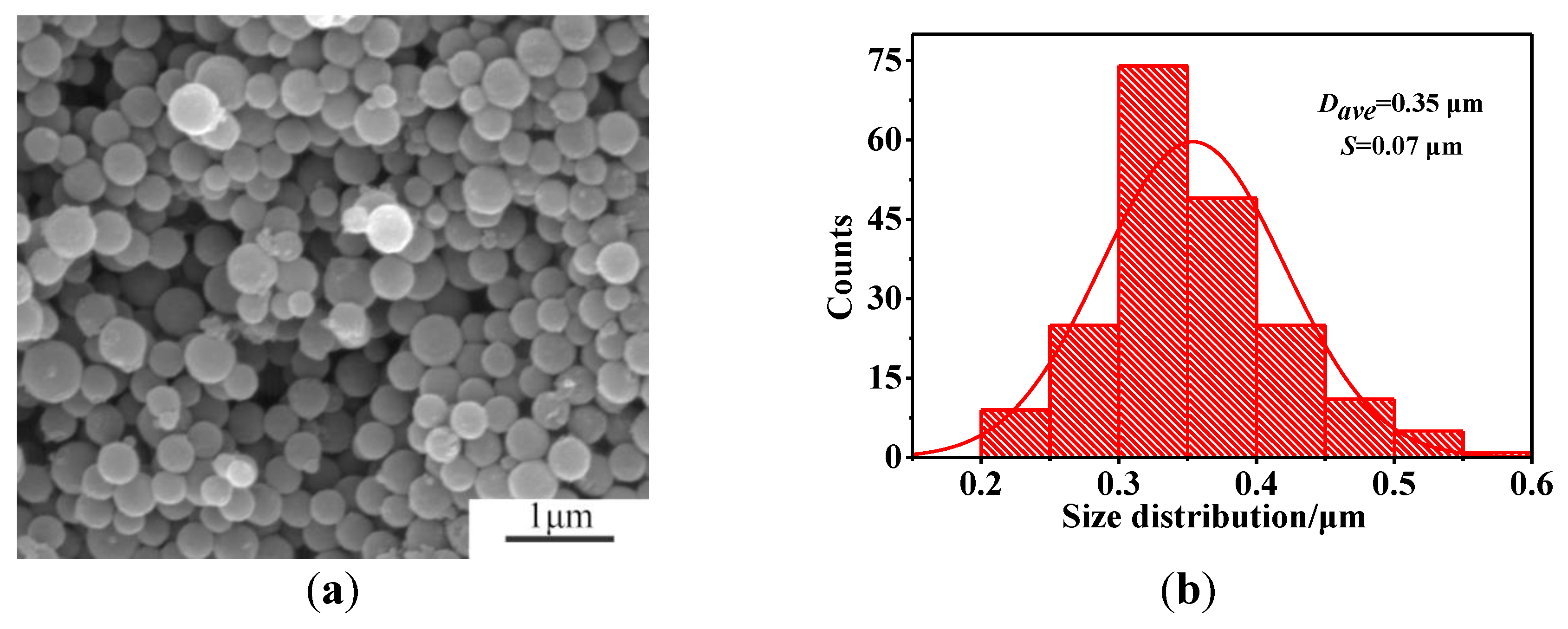
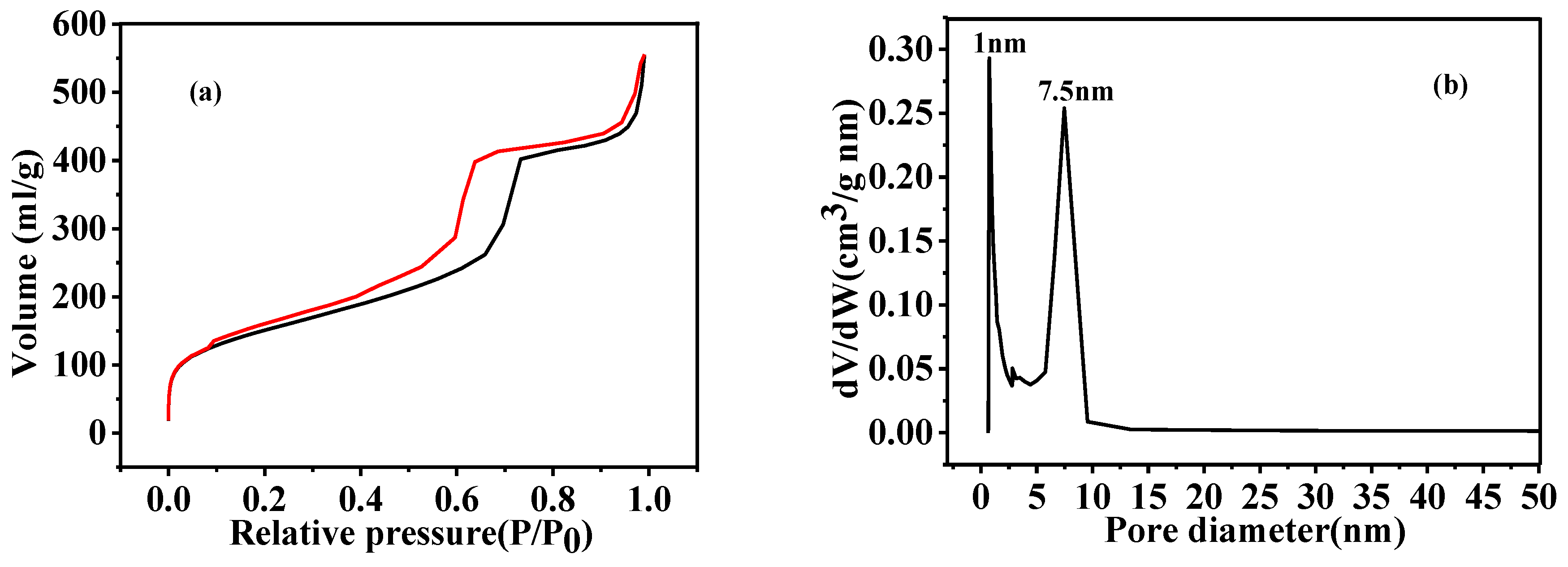
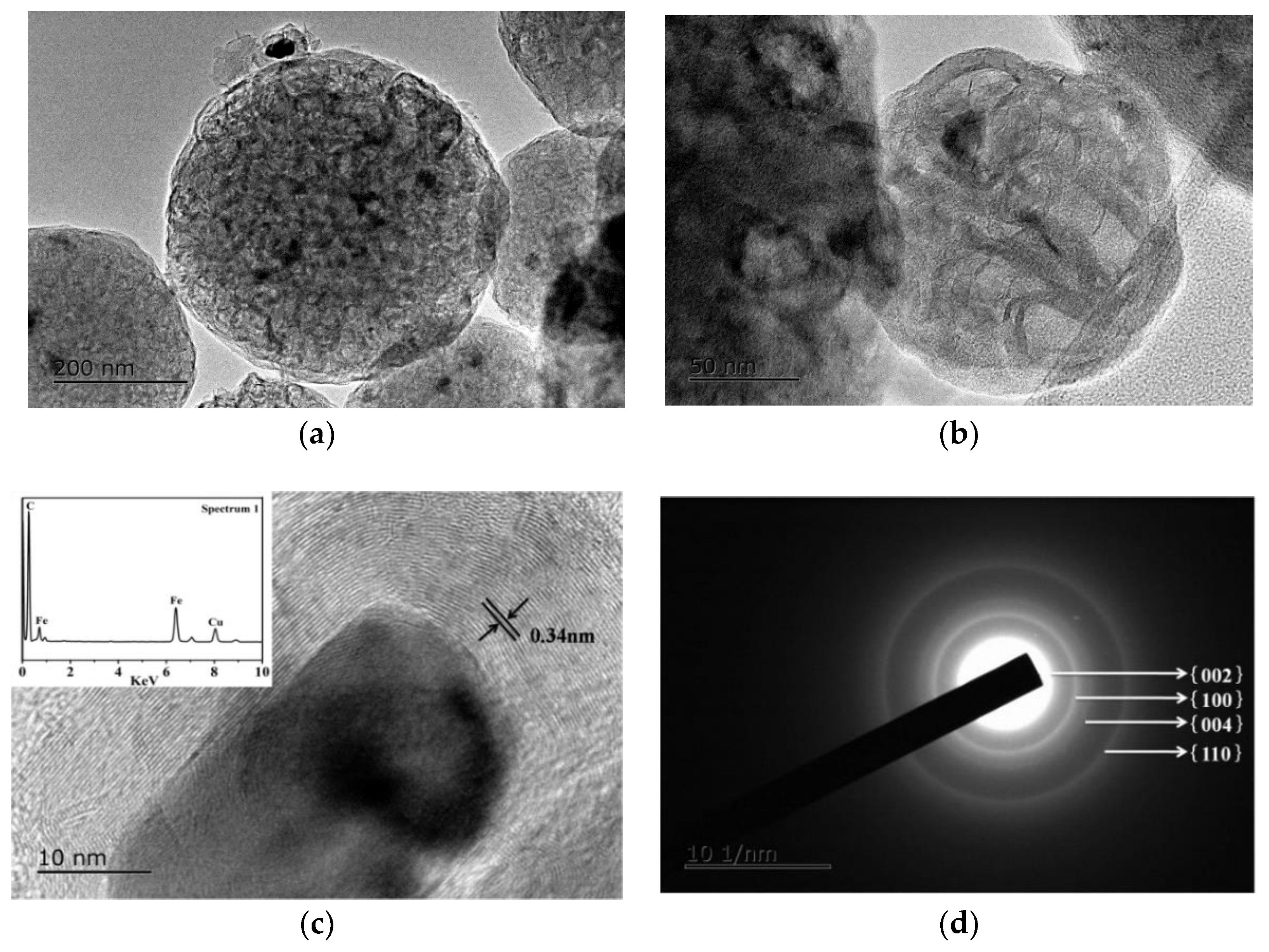
3.4. Microstructural Characterization of As-Prepared HGCS
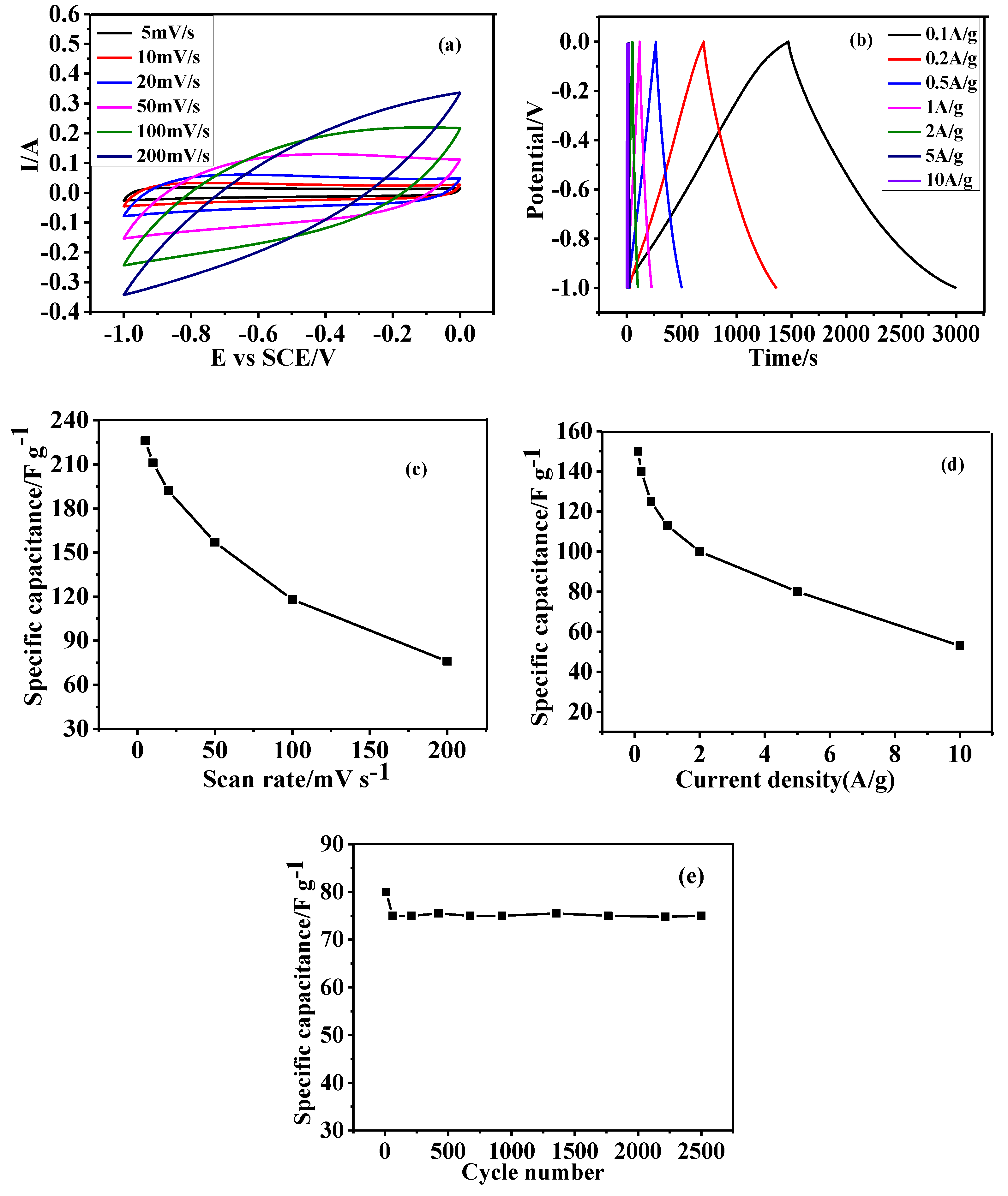
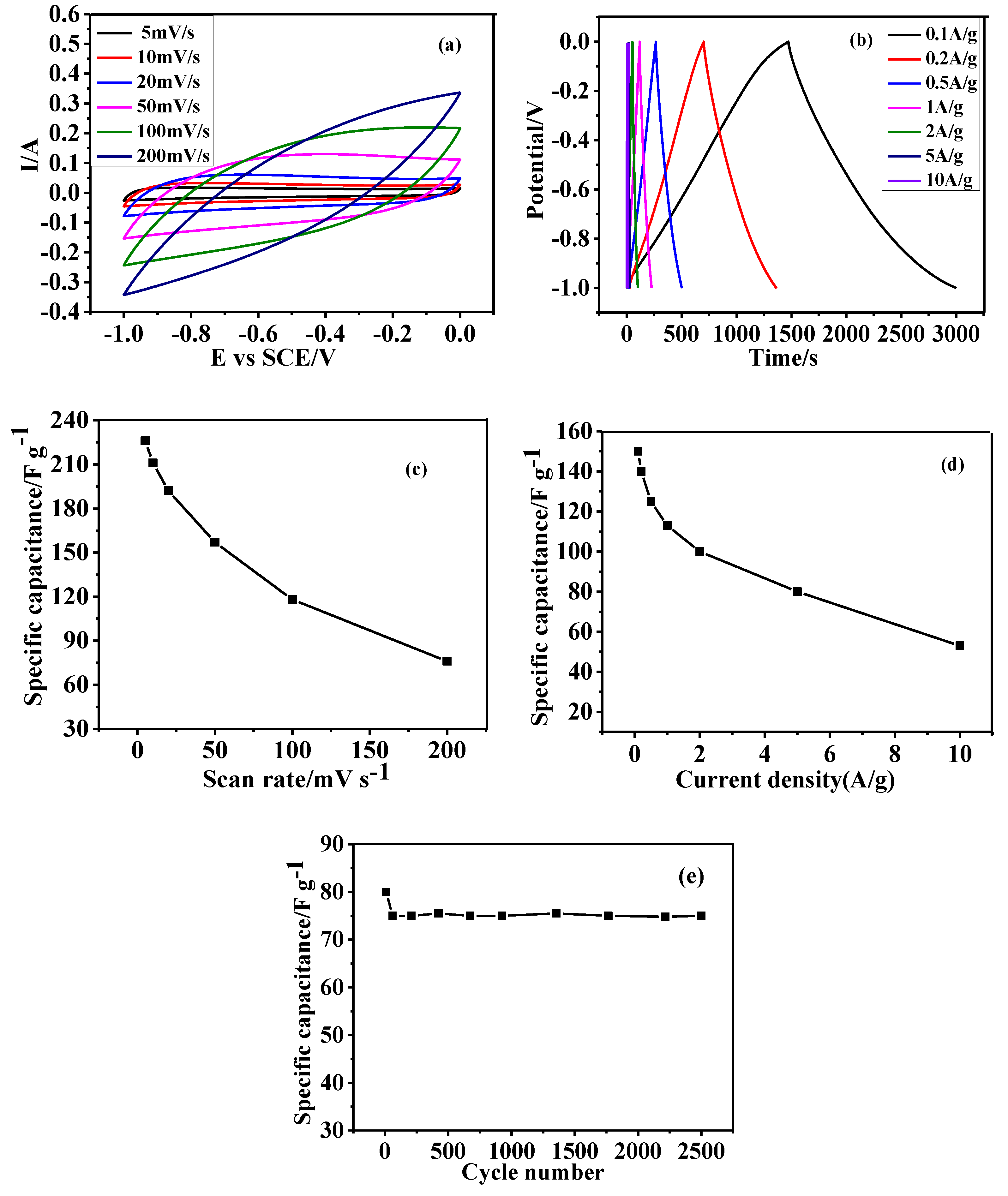
3.5. Electrochemical Performance of as Prepared HGCS
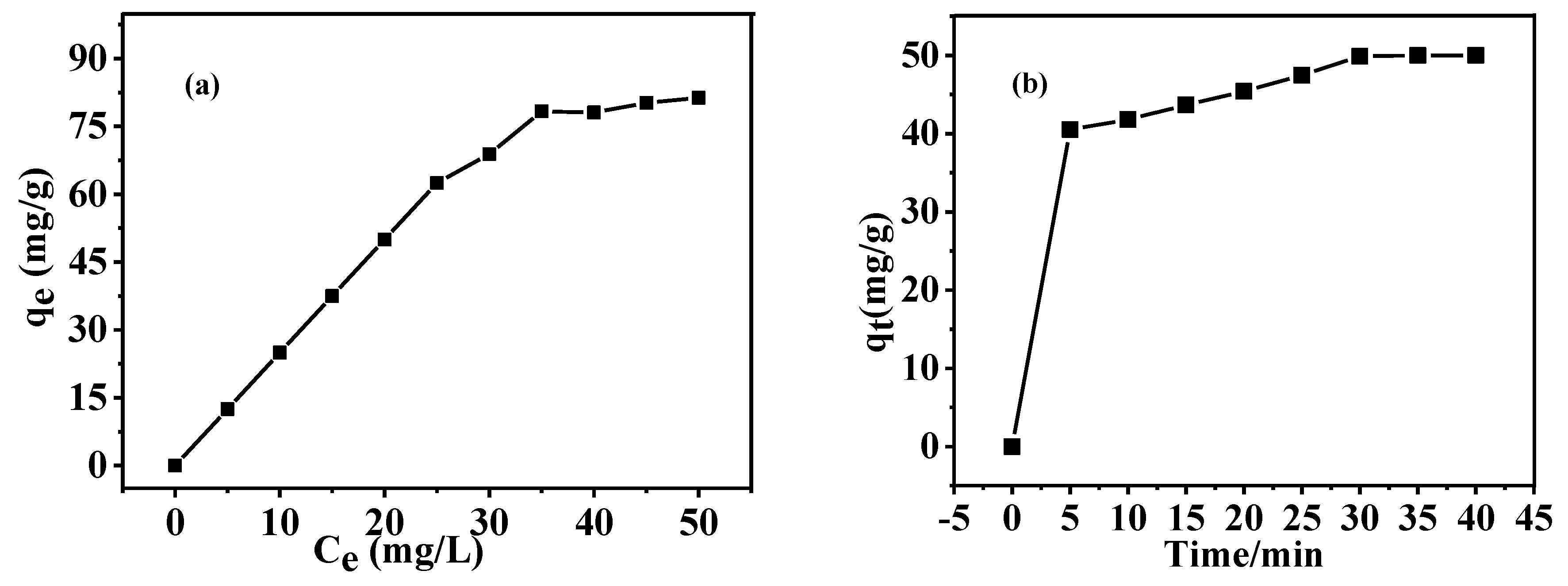
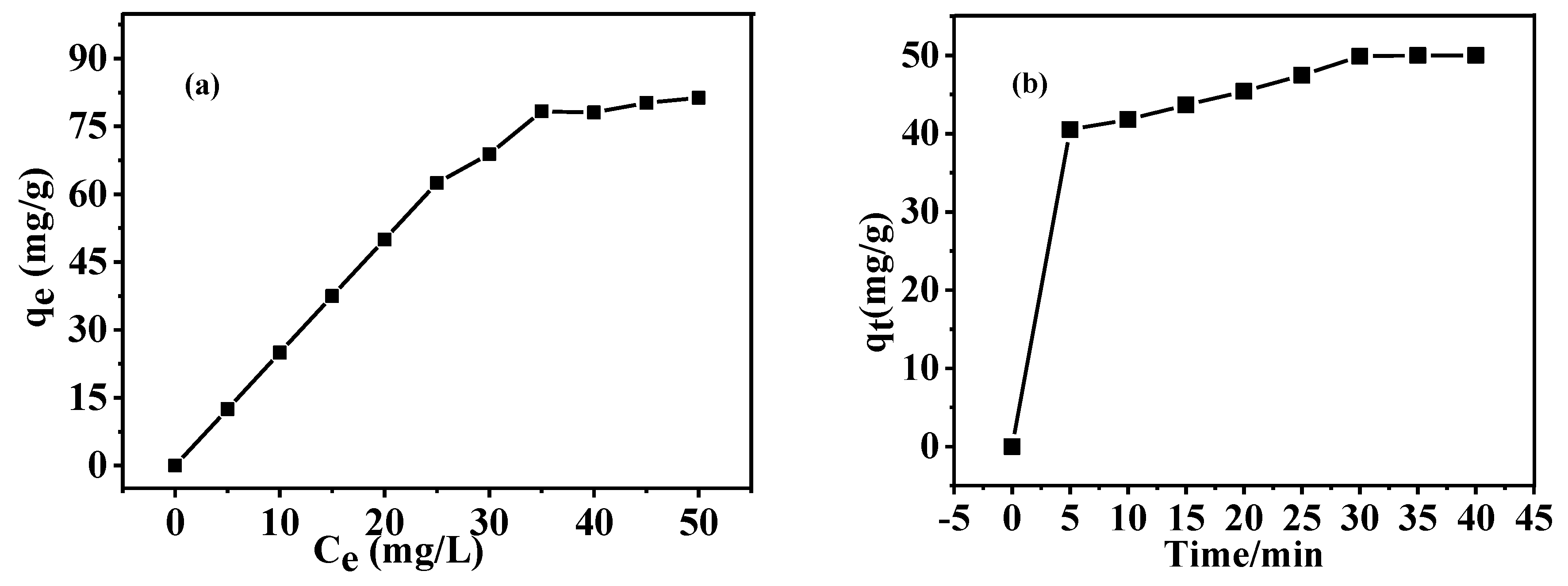
3.6. Adsorption Capacity of As-Prepared HGCS for MB
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pang, J.; Zhang, W.; Zhang, H.; Zhang, J.; Zhang, H.; Cao, G. Sustainable nitrogen-containing hierarchical porous carbon spheres derived from sodium lignosulfonate for high-performance supercapacitors. Carbon 2018, 132, 280–293. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, Y.; Zhang, B.; Xia, H.; Zhou, J.; Xie, W. Nano-micro carbon spheres anchored on porous carbon derived from dual-biomass as high rate performance supercapacitor electrodes. J. Power Sources 2018, 381, 116–126. [Google Scholar] [CrossRef]
- Yu, Z.; Tetard, L.; Zhai, L.; Thomas, J. Supercapacitor electrode materials: Nanostructures from 0 to 3 dimensions. Energy Environ. Sci. 2015, 8, 702–730. [Google Scholar] [CrossRef]
- Gu, W.; Yushin, G. Review of nanostructured carbon materials for electrochemical capacitor applications: Advantages and limitations of activated carbon, carbide-derived carbon, zeolite-templated carbon, carbon aerogels, carbon nanotubes, onion-like carbon, and grapheme. WIREs Energy Environ. 2014, 3, 424–473. [Google Scholar] [CrossRef]
- Simon, P.; Gogotsi, Y. Capacitive energy storage in nanostructured carbon-electrolyte systems. Acc. Chem. Res. 2013, 46, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Wang, K.P.; Teng, H. A simplified preparation of mesoporous carbon and the examination of the carbon accessibility for electric double layer formation. Carbon 2005, 43, 559–566. [Google Scholar] [CrossRef]
- Zhang, L.; Gu, H.; Sun, H.; Cao, F.; Chen, Y.; Chen, G. Molecular level one-step activation of agar to activated carbon for high performance supercapacitors. Carbon 2018, 132, 573–578. [Google Scholar] [CrossRef]
- Wang, L.; Mu, G.; Tian, C.; Sun, L.; Zhou, W.; Tan, T. In Situ intercalating expandable graphite for mesoporous carbon/graphite nanosheet composites as high-performance supercapacitor electrodes. ChemSusChem 2012, 5, 2442–2450. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Hu, X.; Yan, Z.; Lei, K.; Li, F.; Cheng, F. Template-free synthesis of porous graphitic carbon nitride/carbon composite spheres for electrocatalytic oxygen reduction reaction. Chem. Commun. 2016, 52, 1725–1728. [Google Scholar] [CrossRef] [PubMed]
- Estevez, L.; Prabhakaran, V.; Garcia, A.L.; Shin, Y.; Tao, J.; Schwarz, A.M. Hierarchically porous graphitic carbon with simultaneously high surface area and colossal pore volume engineered via ice templating. ACS Nano 2017, 11, 11047–11055. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, Q.; Jiang, D.; Huang, Y. Facile synthesis of B, N co-doped three-dimensional porous graphitic carbon toward oxygen reduction reaction and oxygen evolution reaction. Catal. Commun. 2017, 100, 89–92. [Google Scholar] [CrossRef]
- Dong, Y.; Lin, H.; Zhou, D.; Niu, H.; Jin, Q.; Qu, F. Synthesis of mesoporous graphitic carbon fibers with high performance for supercapacitor. Electrochim. Acta 2015, 159, 116–123. [Google Scholar] [CrossRef]
- Chen, Z.; Wen, J.; Yan, C.; Rice, L.; Sohn, H.; Shenl, M. High-performance supercapacitors based on hierarchically porous graphite particles. Adv. Energy Mater. 2011, 1, 551–556. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.; Hyeon, T. Recent progress in the synthesis of porous carbon material. Adv. Mater. 2006, 18, 2073–2094. [Google Scholar] [CrossRef]
- Xie, M.; Yang, J.; Liang, J.; Guo, X.; Ding, W. In situ hydrothermal deposition as an efficient catalyst supporting method towards low-temperature graphitization of amorphous carbon. Carbon 2014, 77, 215–225. [Google Scholar] [CrossRef]
- Jaroniec, M.; Choma, J.; Gorka, J.; Zawislak, A. Colloidal silica templating synthesis of carbonaceous monoliths assuring formation of uniform spherical mesopores and incorporation of inorganic nanoparticles. Chem. Mater. 2008, 20, 1069–1075. [Google Scholar] [CrossRef]
- Yang, G.; Han, H.; Li, T.; Du, C. Synthesis of nitrogen-doped porous graphitic carbons using nano-CaCO3 as template, graphitization catalyst, and activating agent. Carbon 2012, 50, 3753–3765. [Google Scholar] [CrossRef]
- Ferrero, G.A.; Fuertes, A.B.; Sevilla, M. N-doped microporous carbon microspheres for high volumetric performance supercapacitors. Electrochim. Acta 2015, 168, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Xie, L.; Li, H.; Wang, Y. Sustainable and scalable production of monodisperse and highly uniform colloidal carbonaceous spheres using sodium polyacrylate as the dispersant. Chem. Commun. 2014, 50, 12633–12636. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liang, F.; Wang, J.; Zhang, H.; Zhang, S. Preparation of mono-dispersed carbonaceous spheres via hydrothermal process. Adv. Powder Technol. 2017, 28, 2648–2657. [Google Scholar] [CrossRef]
- Sevilla, M.; Fuertes, A.B. Graphitic carbon nanostructures from cellulose. Chem. Phys. Lett. 2010, 490, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Huang, R.; Mu, D.; Wu, B.; Chen, Y. Controlled synthesis of graphitic carbon-encapsulated α-Fe2O3 nanocomposite via low-temperature catalytic graphitization of biomass and its lithium storage property. Electrochim. Acta 2016, 187, 508–516. [Google Scholar] [CrossRef]
- Wang, J.; Deng, X.; Zhang, H. Synthesis of carbon nanotubes via Fe-catalyzed pyrolysis of phenolic resin. Phys. E Low-Dimens. Syst. Nanostruct. 2017, 86, 24–35. [Google Scholar] [CrossRef]
- Gomez-Martin, A.; Martinez-Fernandez, J.; Ruttert, M.; Heckmann, A.; Winter, M.; Placke, T.; Ramirez-Rico, J. Iron-Catalyzed Graphitic Carbon Materials from Biomass Resources as Anodes for Lithium-Ion Batteries. ChemSusChem 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Deng, X.; Zhang, H. Synthesis of carbon nanotubes from phenolic resin using nickel nitrate as a catalyst precursor. Key Eng. Mater. 2016, 697, 691–694. [Google Scholar] [CrossRef]
- Ma, C.; Chen, X.; Long, D. High-surface-area and high-nitrogen-content carbon microspheres prepared by a pre-oxidation and mild KOH activation for superior supercapacitor. Carbon 2017, 118, 699–708. [Google Scholar] [CrossRef]
- Sevilla, M.; Ferrero, G.A.; Fuertes, A.B. One-Pot Synthesis of Biomass-Based Hierarchical Porous Carbons with a Large Porosity Development. Chem. Mater. 2017, 29, 6900–6907. [Google Scholar] [CrossRef]
- Li, S.; Liu, J.; Wang, J.; Zhu, Q.; Zhao, X.; Zhang, H. Fabrication of graphitic carbon spheres and their application in Al2O3-SiC-C refractory castables. Int. J. Appl. Ceram. Technol. 2018, 15, 1166–1181. [Google Scholar] [CrossRef]
- Ran, F.; Shen, K.; Tan, Y.; Peng, B.; Chen, S.; Zhang, W. Activated hierarchical porous carbon as electrode membrane accommodated with triblock copolymer for supercapacitors. J. Membr. Sci. 2016, 514, 366–375. [Google Scholar] [CrossRef]
- Hulicova-Jurcakova, D.; Kodama, M.; Shiraishi, S.; Hatori, H.; Zhu, Z.H.; Lu, G.Q. Nitrogen-enriched nonporous carbon electrodes with extraordinary supercapacitance. Adv. Funct. Mater. 2010, 19, 1800–1809. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, D.; Lin, H.; Lu, H.; Xu, J.; Liu, D. On the cycling stability of the supercapacitive performance of activated carbon in KOH and H2SO4 electrolytes. Colloids Surf. A 2016, 511, 294–302. [Google Scholar] [CrossRef]
- Wilgosz, K.; Chen, X.; Kierzek, K.; Machnikowski, J.; Kalenczuk, R.J.; Mijowska, E. Template method synthesis of mesoporous carbon spheres and its applications as supercapacitors. Nanoscale Res. Lett. 2012, 7, 269. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; He, B.; Xu, F.; Chen, X. Equilibrium and kinetic studies of methyl orange adsorption on multiwalled carbon nanotubes. Chem. Eng. J. 2011, 170, 82–89. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Langmuir | Freundlich | Pseudo-First-Order | Pseudo-Second-Order | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Q0 | B | R2 | KF | N | R2 | Qe | K1 | R2 | Qe | K2 | R2 | |
| CS | 15.4 | 0.024 | 0.95 | 0.38 | 1.21 | 0.95 | 47.4 | 0.336 | 0.969 | 5.05 | 0.283 | 0.999 |
| HGCS | 182.8 | 0.019 | 0.98 | 3.6 | 1.19 | 0.97 | 47.4 | 0.337 | 0.968 | 51.6 | 0.0125 | 0.995 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Li, F.; Wang, J.; Tian, L.; Zhang, H.; Zhang, S. Preparation of Hierarchically Porous Graphitic Carbon Spheres and Their Applications in Supercapacitors and Dye Adsorption. Nanomaterials 2018, 8, 625. https://doi.org/10.3390/nano8080625
Li S, Li F, Wang J, Tian L, Zhang H, Zhang S. Preparation of Hierarchically Porous Graphitic Carbon Spheres and Their Applications in Supercapacitors and Dye Adsorption. Nanomaterials. 2018; 8(8):625. https://doi.org/10.3390/nano8080625
Chicago/Turabian StyleLi, Saisai, Faliang Li, Junkai Wang, Liang Tian, Haijun Zhang, and Shaowei Zhang. 2018. "Preparation of Hierarchically Porous Graphitic Carbon Spheres and Their Applications in Supercapacitors and Dye Adsorption" Nanomaterials 8, no. 8: 625. https://doi.org/10.3390/nano8080625
APA StyleLi, S., Li, F., Wang, J., Tian, L., Zhang, H., & Zhang, S. (2018). Preparation of Hierarchically Porous Graphitic Carbon Spheres and Their Applications in Supercapacitors and Dye Adsorption. Nanomaterials, 8(8), 625. https://doi.org/10.3390/nano8080625