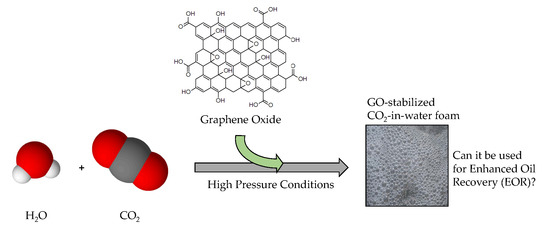
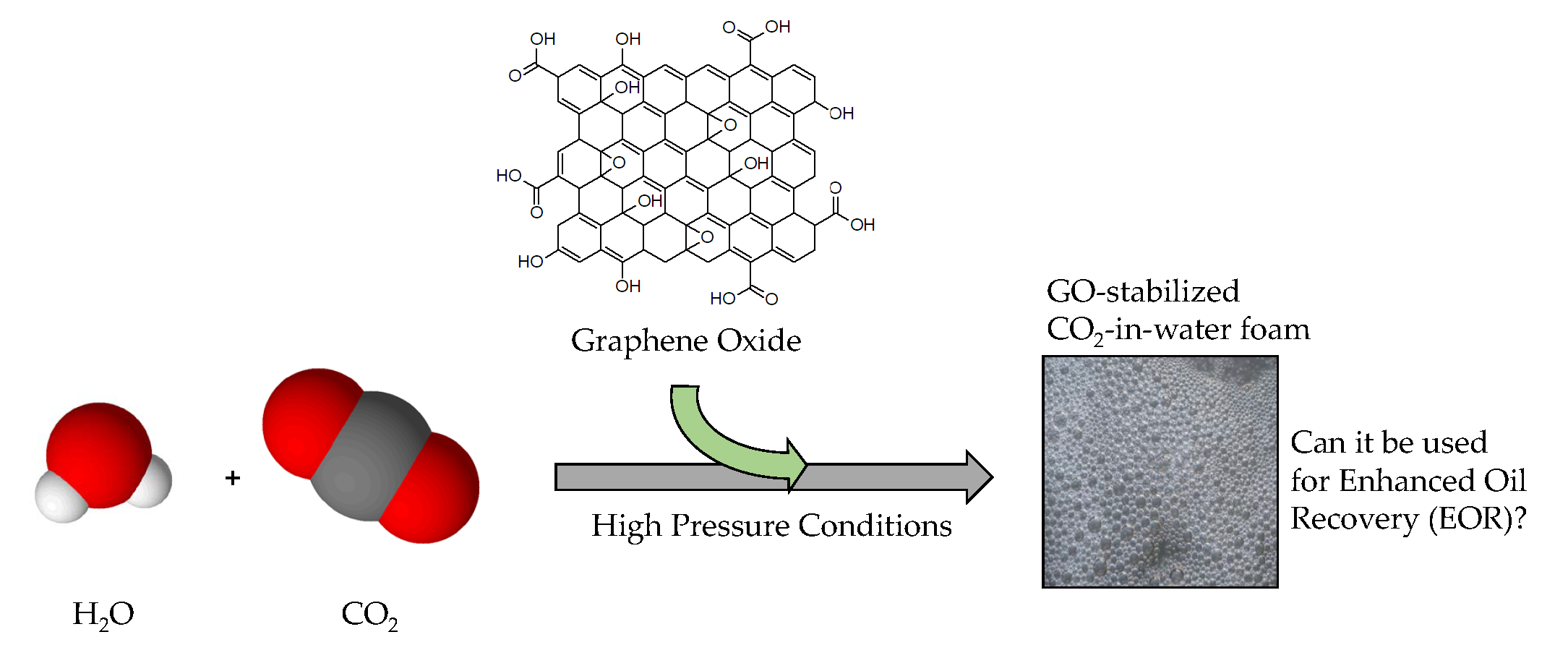
An Evaluation of Graphene Oxides as Possible Foam Stabilizing Agents for CO2 Based Enhanced Oil Recovery

Abstract
:
1. Introduction
2. Experimental
2.1. Materials
2.2. Methods
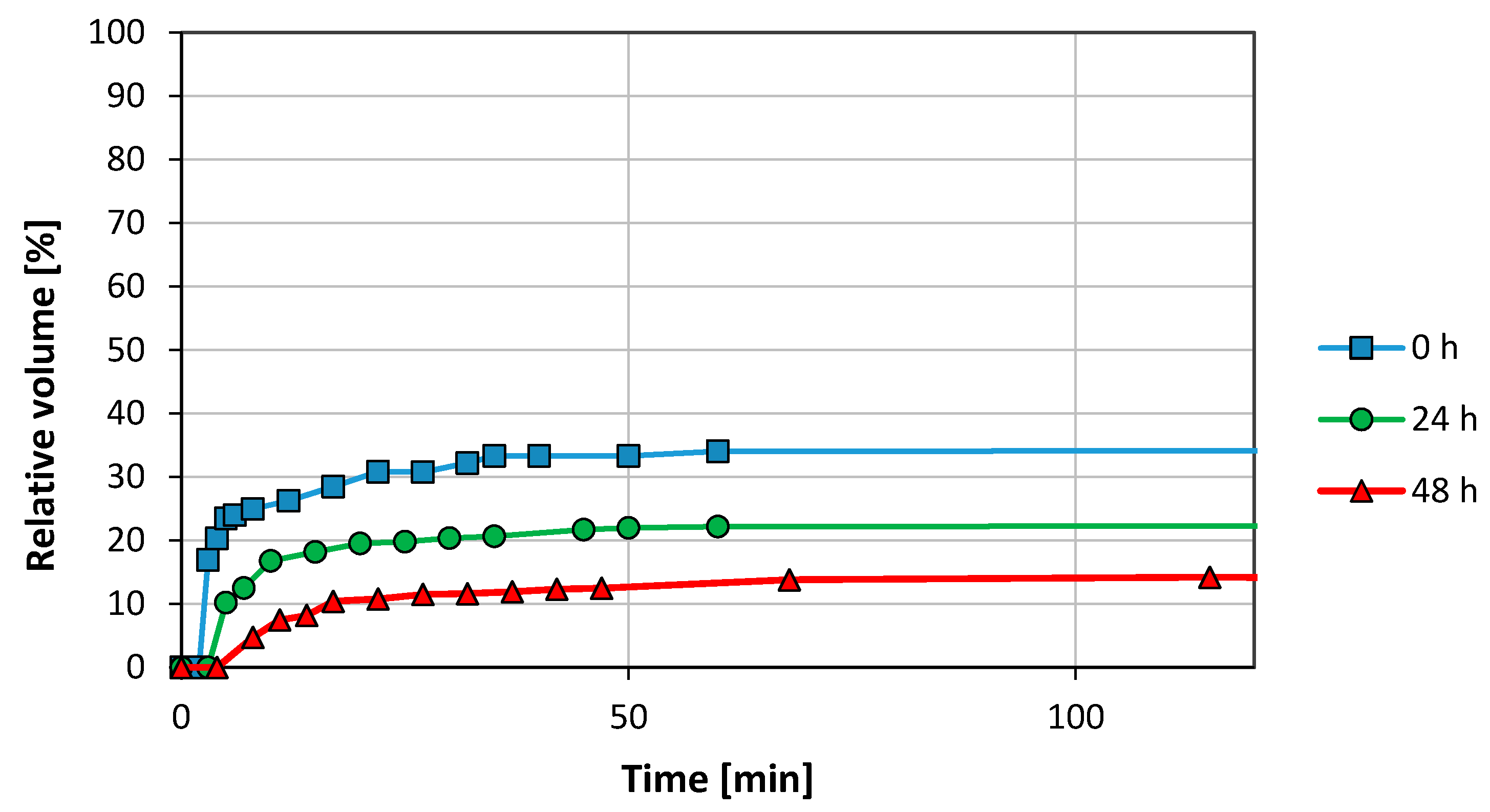
2.2.1. Bottle Test
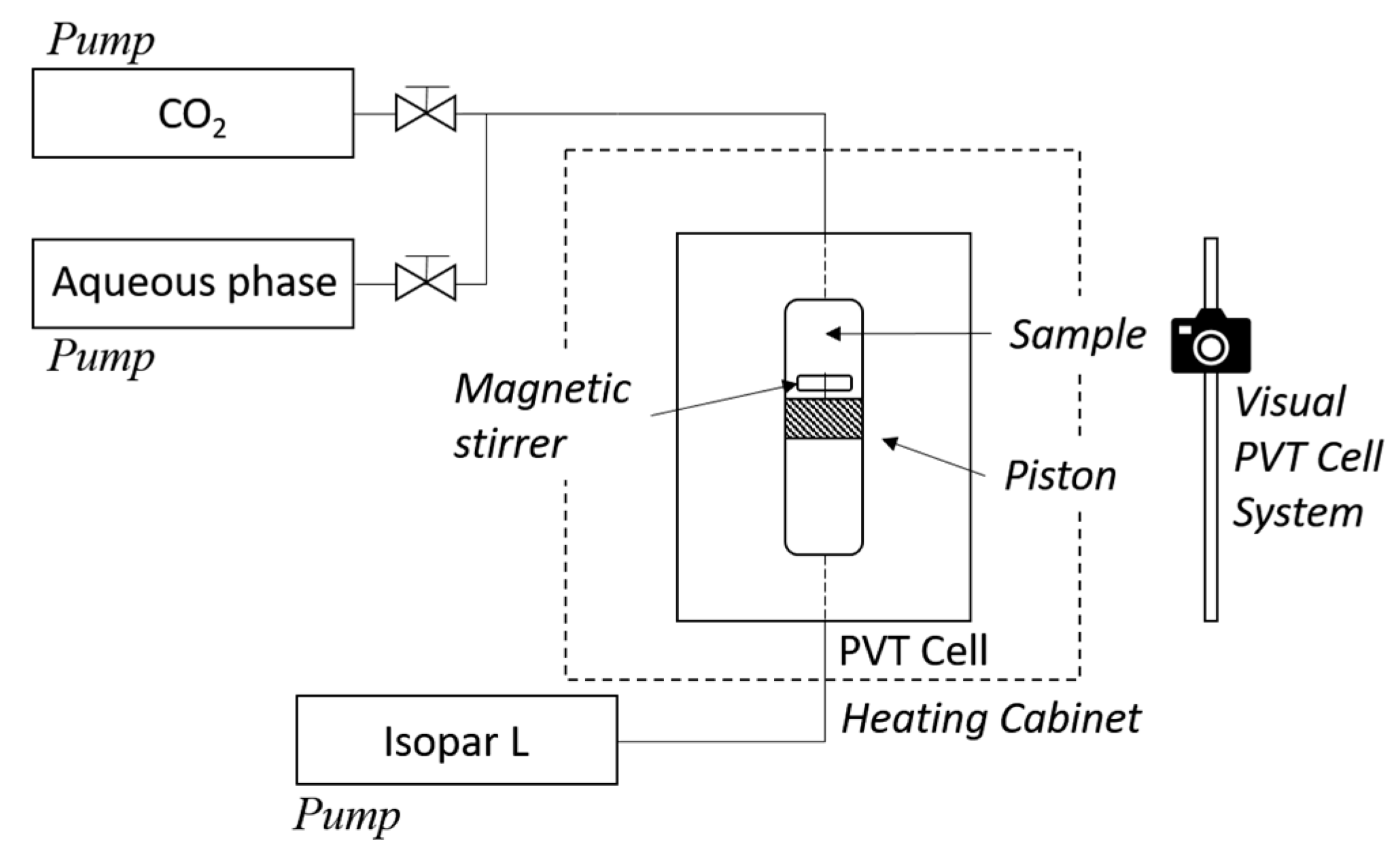
2.2.2. Phase Equilibria Studies
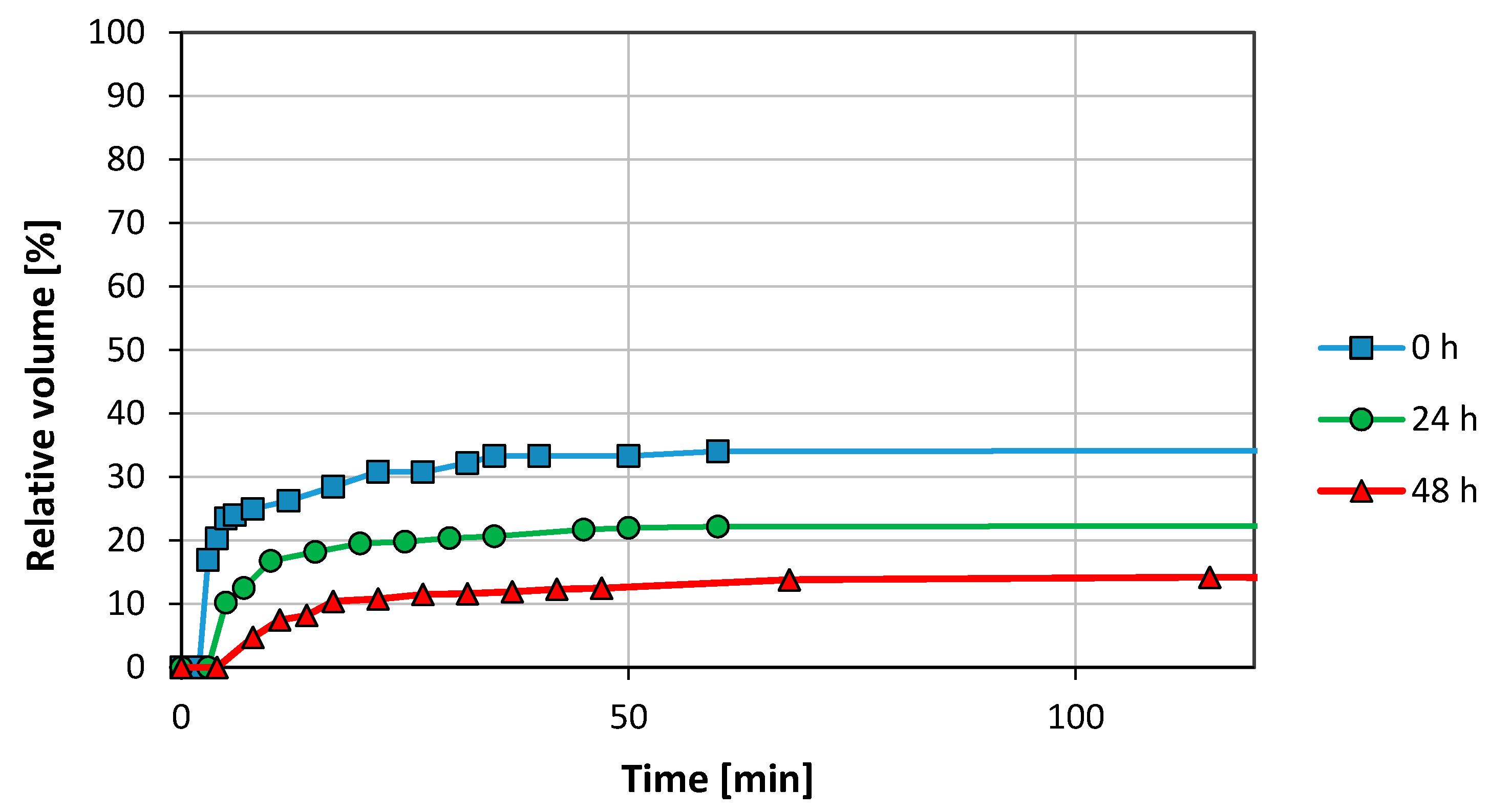
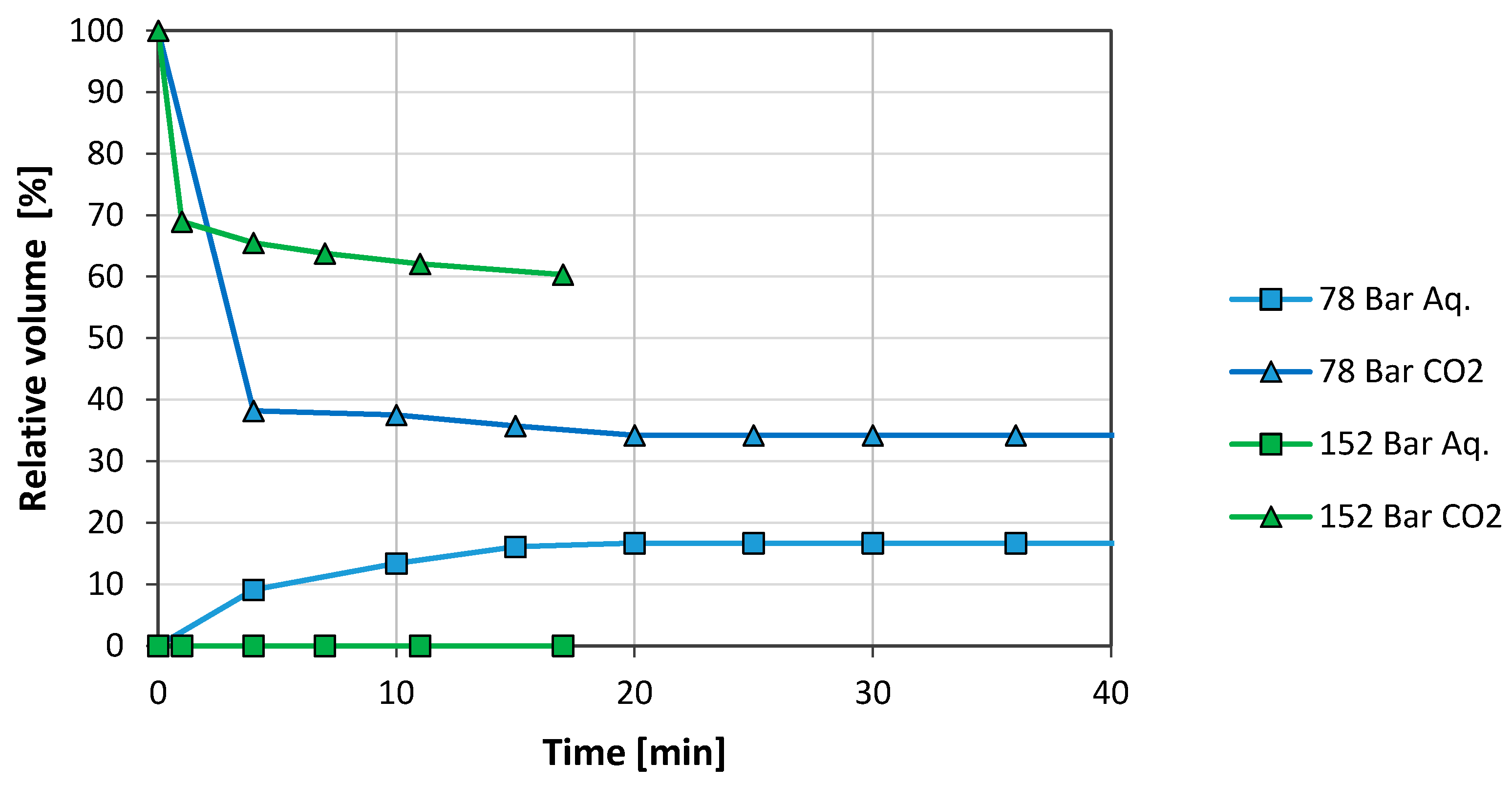
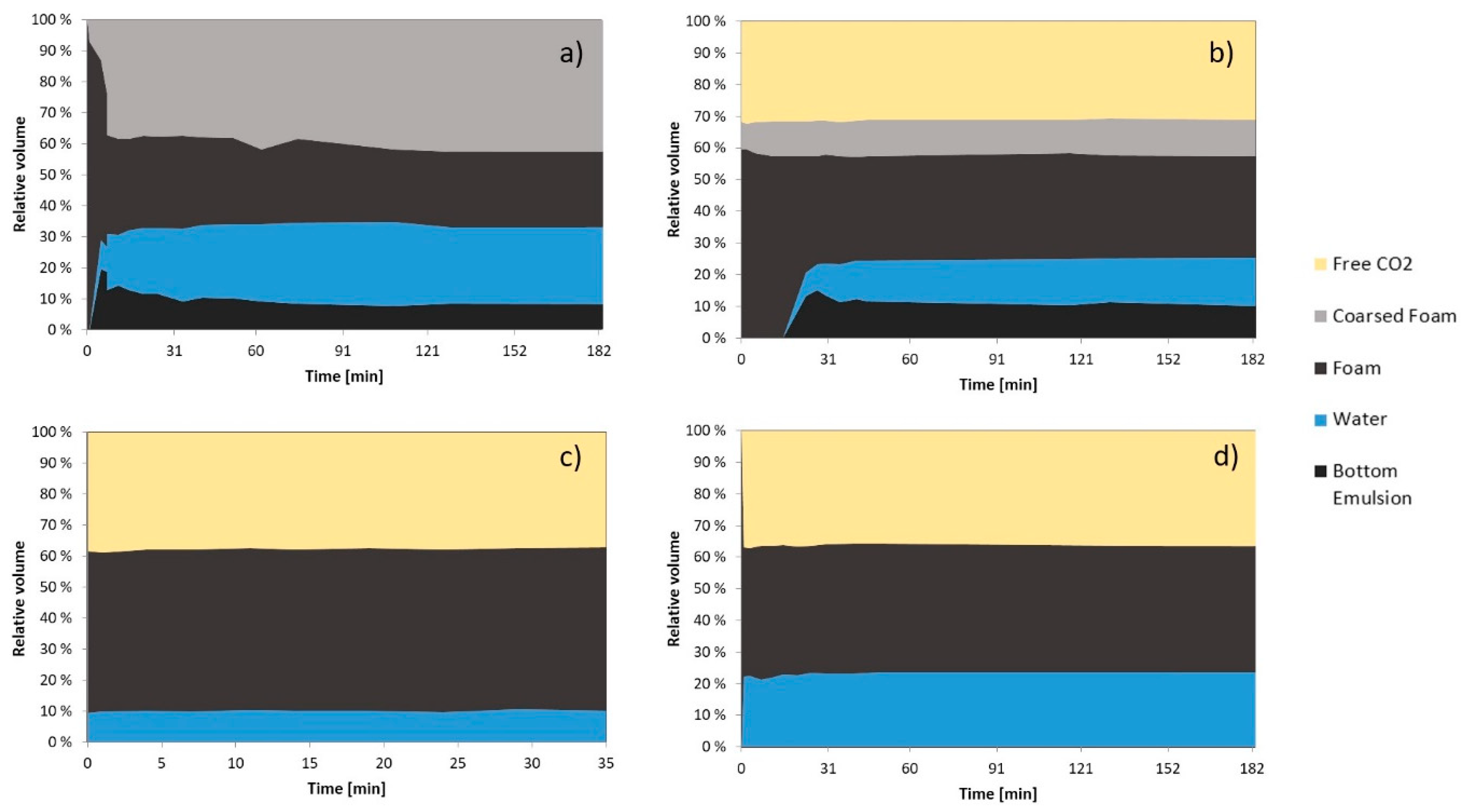
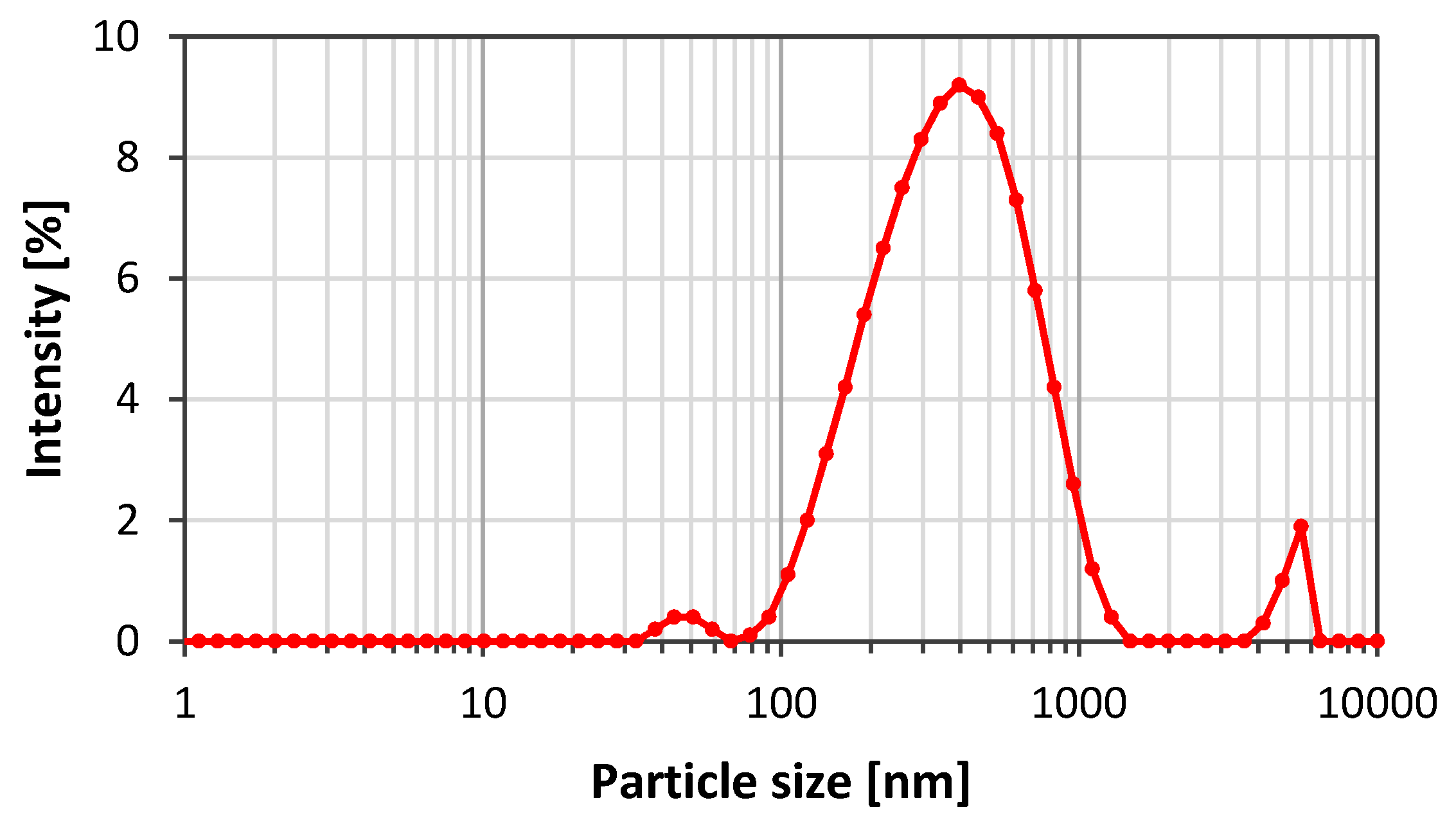
3. Results and Discussion
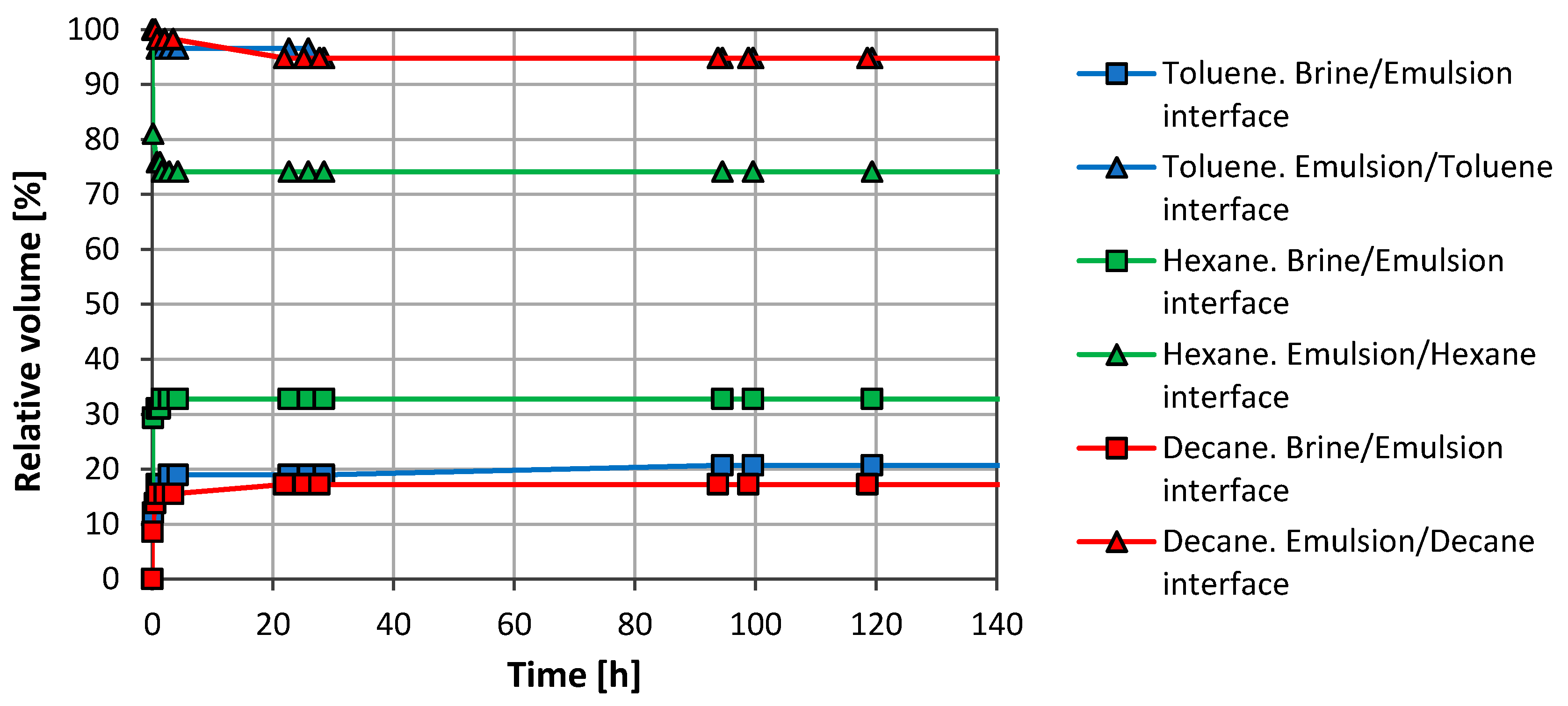
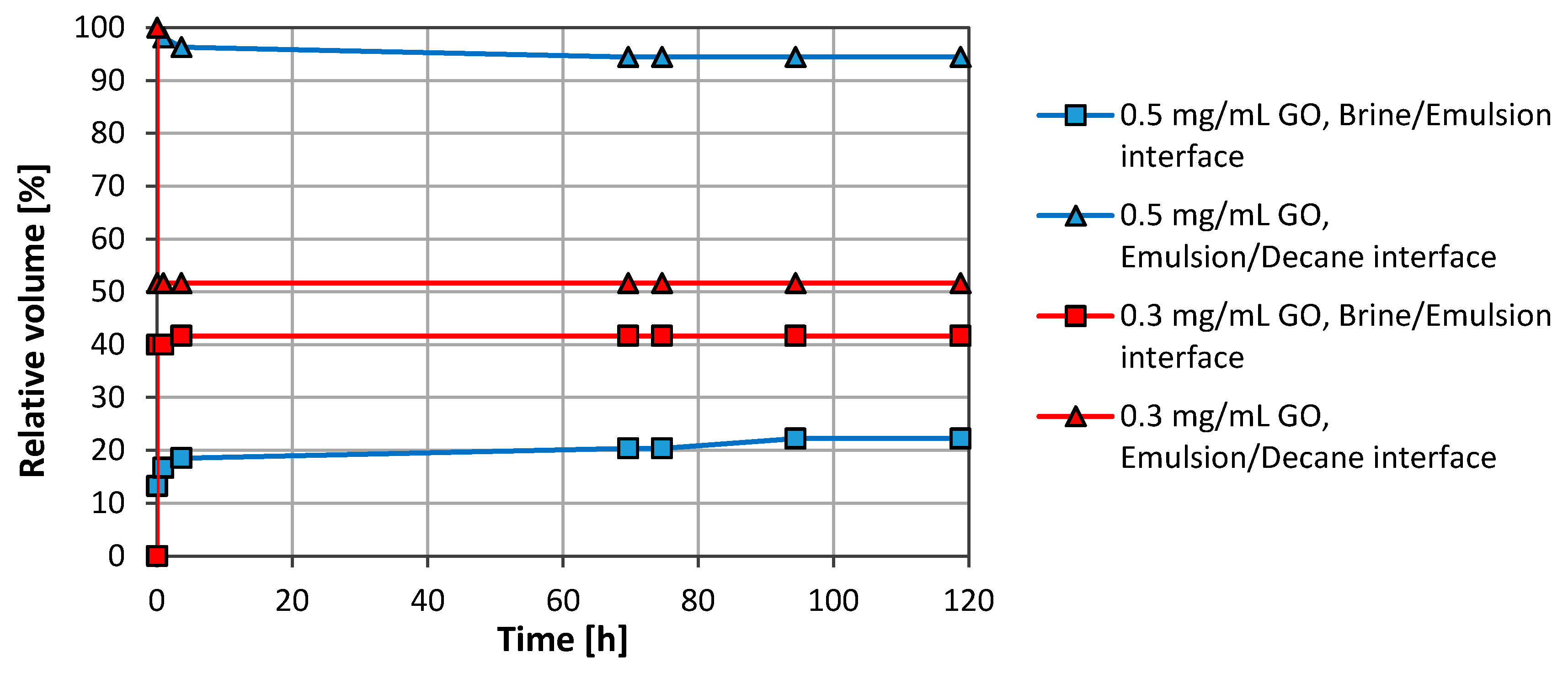
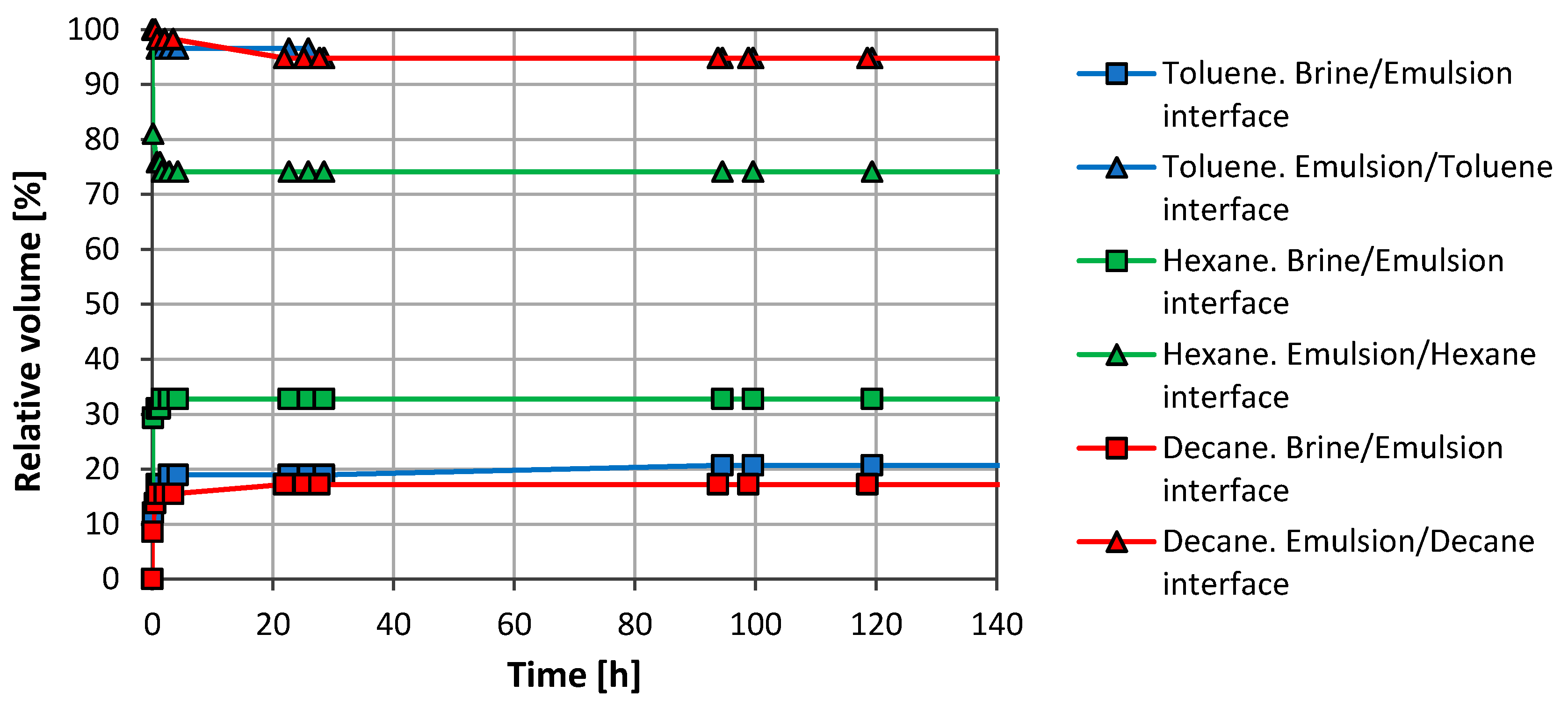
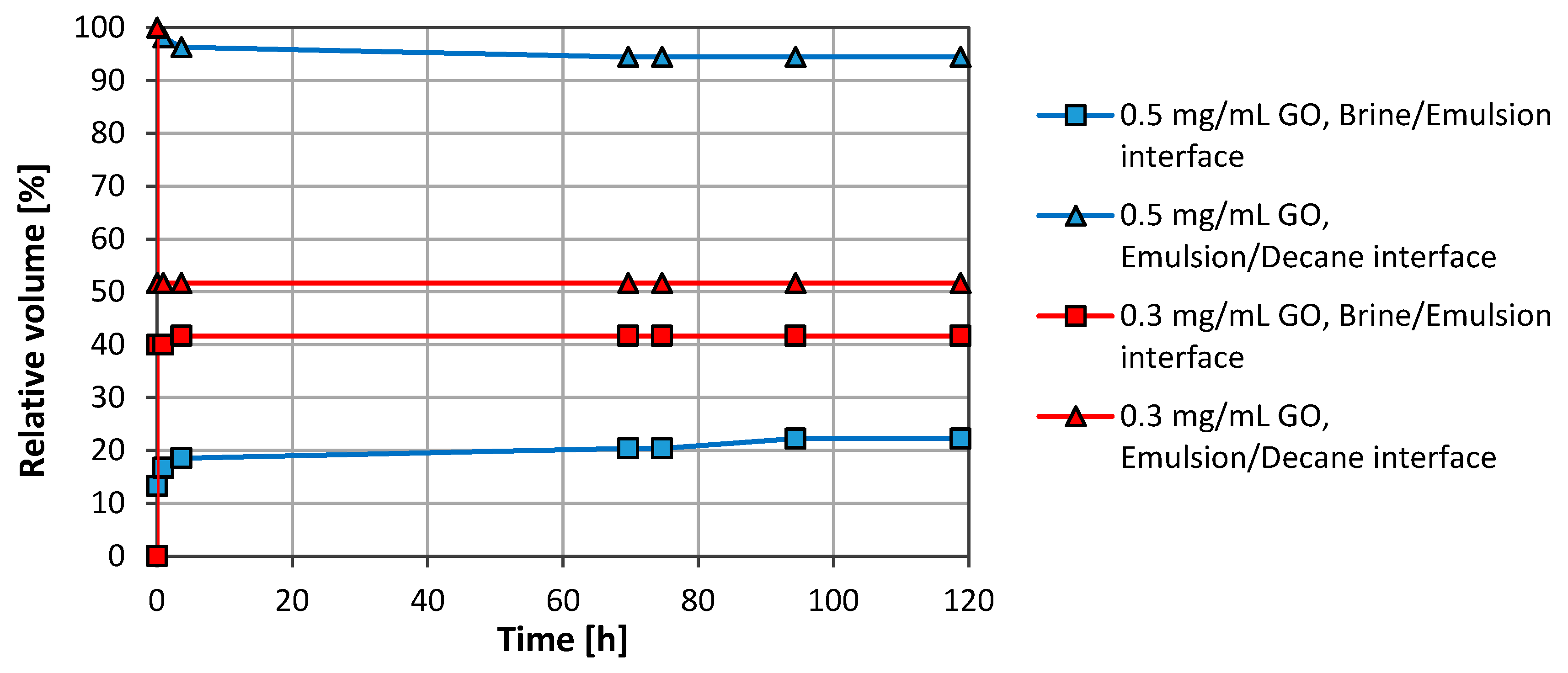
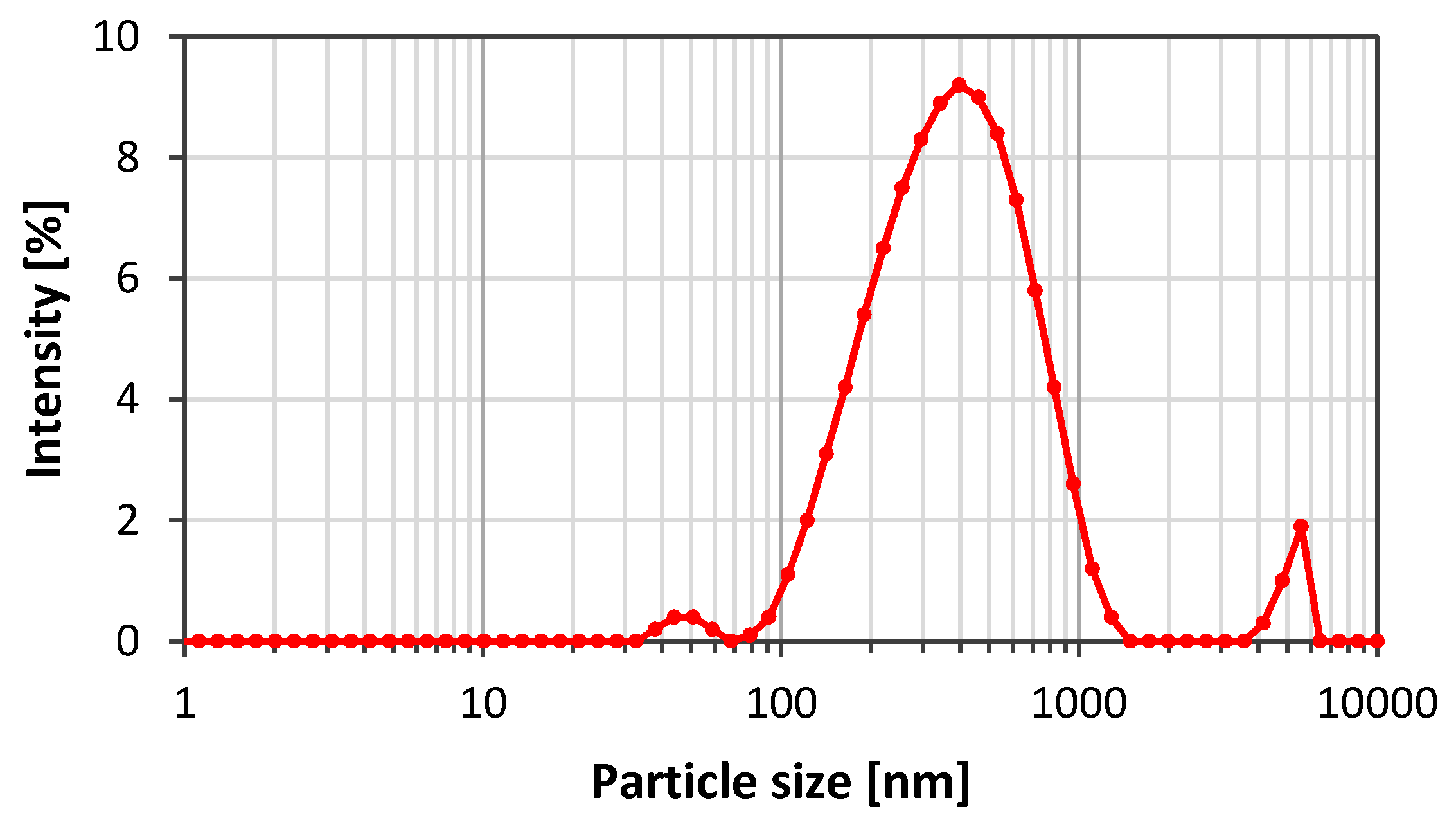
3.1. Emulsion Stability
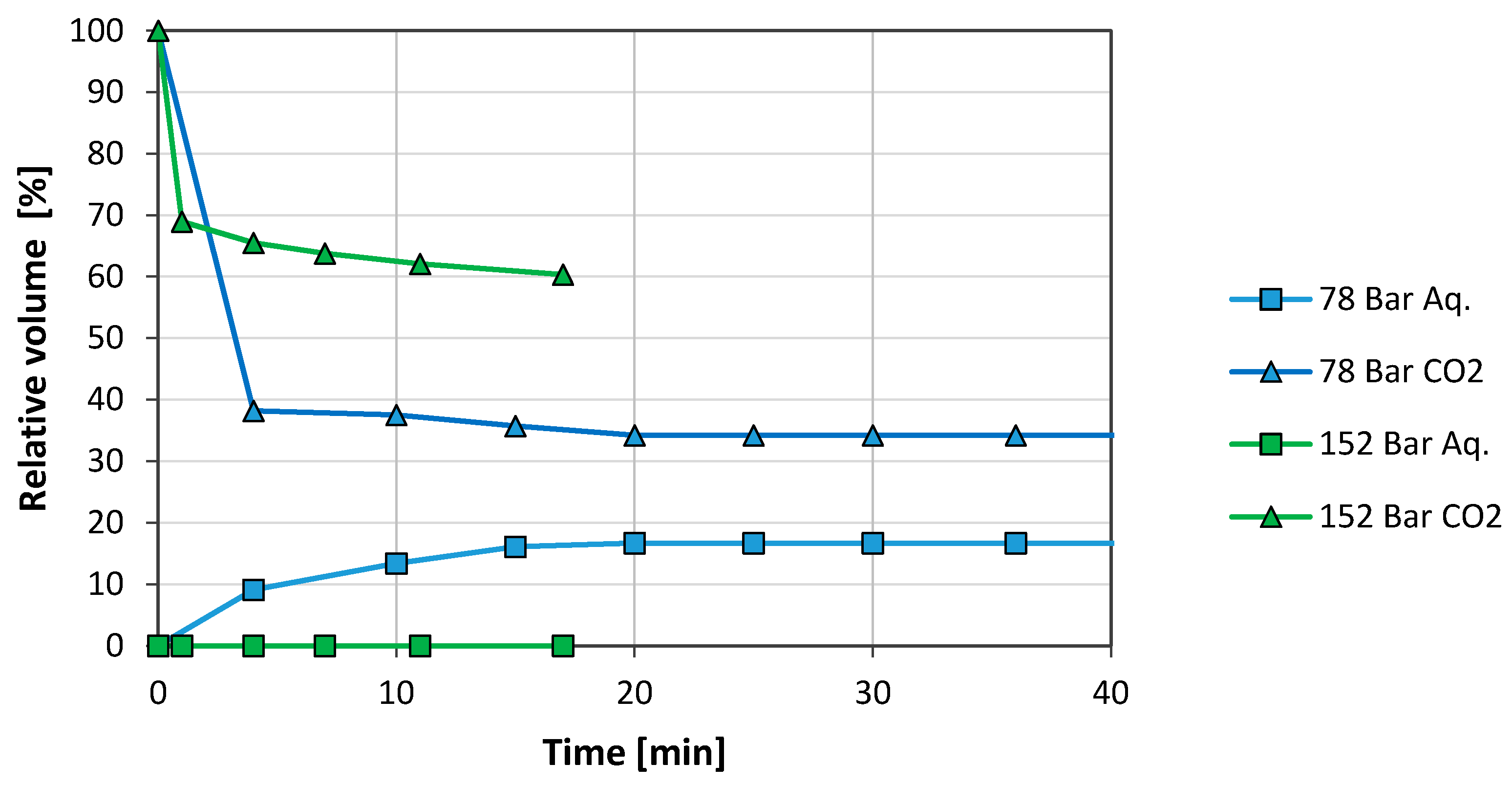
3.2. Brine/CO2 Systems
3.2.1. Graphene Oxide (GO)
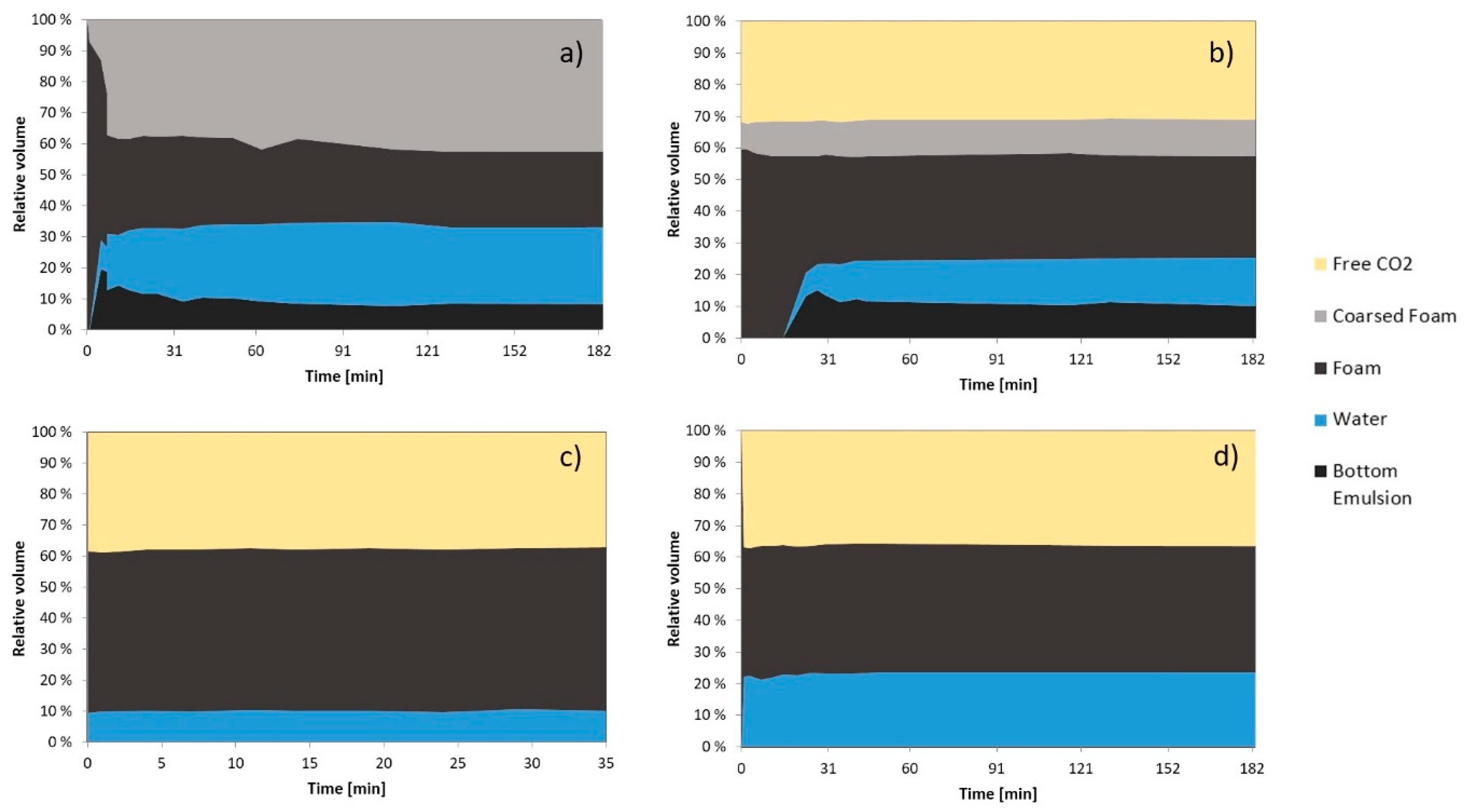
3.2.2. Foam Morphology
3.2.3. Partially Reduced Graphene Oxide (rGO)
3.2.4. Nanographene Oxide (nGO)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Enick, R.M.; Olsen, D.; Ammer, J.; Schuller, W. Mobility and conformance control for CO2 eor via thickeners, foams, and gels—A literature review. In Proceedings of the SPE Improved Oil Recovery Symposium, Tulsa, OK, USA, 14–18 April 2012. [Google Scholar]
- Solbakken, J.S.; Skauge, A.; Aarra, M.G. Supercritical CO2 foam—The importance of CO2 density on foams performance. In Proceedings of the SPE Enhanced Oil Recovery, Society of Petroleum Engineers, Kuala Lumpur, Malaysia, 2013. [Google Scholar]
- Tsau, J.-S.; Grigg, R.B. Assessment of foam properties and effectiveness in mobility reduction for CO2-foam floods. In Proceedings of the SPE International Symposium on Oilfield Chemistry, Houston, TX, USA, 18–21 February 1997. [Google Scholar]
- Bernard, G.C.; Holm, L.W.; Harvey, C.P. Use of surfactant to reduce CO2 mobility in oil displacement. Soc. Pet. Eng. J. 1980, 20, 281–292. [Google Scholar] [CrossRef]
- Wasan, D.T.; Koczo, K.; Nikolov, A.D. Mechanisms of aqueous foam stability and antifoaming action with and without oil. In Foams: Fundamentals and Applications in the Petroleum Industry; American Chemical Society: Washington, DC, USA, 1994; Volume 242, pp. 47–114. [Google Scholar]
- Schramm, L.L.; Novosad, J.J. Micro-visualization of foam interactions with a crude oil. Colloids Surf. 1990, 46, 21–43. [Google Scholar] [CrossRef]
- Manlowe, D.J.; Radke, C.J. A pore-level investigation of foam/oil interactions in porous media. SPE Reserv. Eng. 1990, 5, 495–502. [Google Scholar] [CrossRef]
- Vassenden, F.; Holt, T.; Moen, A.; Ghaderi, A. Foam propagation in the absence and presence of oil. In Proceedings of the SPE/DOE Improved Oil Recovery Symposium, Tulsa, OK, USA, 3–5 April 2000. [Google Scholar]
- Espinosa, D.; Caldelas, F.; Johnston, K.; Bryant, S.L.; Huh, C. Nanoparticle-stabilized supercritical CO2 foams for potential mobility control applications. In Proceedings of the SPE Improved Oil Recovery Symposium, Tulsa, OK, USA, 24–28 April 2010. [Google Scholar]
- Yu, J.; Liu, N.; Li, L.; Lee, R. Generation of nanoparticle-stabilized supercritical CO2 foams. In Proceedings of the Carbon Management Technology Conference, Orlando, FL, USA, 7–9 February 2012. [Google Scholar]
- San, J.; Wang, S.; Yu, J.; Lee, R.; Liu, N. Nanoparticle stabilized CO2 foam: Effect of different ions. In Proceedings of the SPE Improved Oil Recovery Conference, Tulsa, OK, USA, 11–13 April 2016. [Google Scholar]
- Binks, B.P. Particles as surfactants—Similarities and differences. Curr. Opin. Colloid Interface Sci. 2002, 7, 21–41. [Google Scholar] [CrossRef]
- Singh, R.; Gupta, A.; Mohanty, K.K.; Huh, C.; Lee, D.; Cho, H. Fly ash nanoparticle-stabilized CO2-in-water foams for gas mobility control applications. In Proceedings of the SPE Annual Technical Conference and Exhibition, Houston, TX, USA, 28–30 September 2015. [Google Scholar]
- Manan, M.A.; Farad, S.; Piroozian, A.; Esmail, M.J.A. Effects of nanoparticle types on carbon dioxide foam flooding in enhanced oil recovery. Pet. Sci. Technol. 2015, 33, 1286–1294. [Google Scholar] [CrossRef]
- Patel, A.; Nihalani, D.; Mankad, D.; Patel, D.; Chaudhari, R.; Dhameliya, M.; Tripathi, D.; Bhui, U.K. Evaluating feasibility of hydrophilic silica nanoparticles for in-situ emulsion formation in presence of co-surfactant: An experimental study. In Proceedings of the SPE Kingdom of Saudi Arabia Annual Technical Symposium and Exhibition, Dammam, Saudi Arabia, 24–27 April 2017. [Google Scholar]
- Pichot, R.; Spyropoulos, F.; Norton, I.T. Competitive adsorption of surfactants and hydrophilic silica particles at the oil–water interface: Interfacial tension and contact angle studies. J. Colloid Interface Sci. 2012, 377, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Loh, K.P. Carbocatalysts: Graphene oxide and its derivatives. Acc. Chem. Res. 2012, 46, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Kim, Y.-K.; Shin, D.; Ryoo, S.-R.; Hong, B.H.; Min, D.-H. Biomedical applications of graphene and graphene oxide. Acc. Chem. Res. 2013, 46, 2211–2224. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, V.; Tiwari, J.N.; Kemp, K.C.; Perman, J.A.; Bourlinos, A.B.; Kim, K.S.; Zboril, R. Noncovalent functionalization of graphene and graphene oxide for energy materials, biosensing, catalytic, and biomedical applications. Chem. Rev. 2016, 116, 5464–5519. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cote, L.J.; Kim, F.; Yuan, W.; Shull, K.R.; Huang, J. Graphene oxide sheets at interfaces. J. Am. Chem. Soc. 2010, 132, 8180–8186. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Cote, L.J.; Tung, V.C.; Tan, A.T.L.; Goins, P.E.; Wu, J.; Huang, J. Graphene oxide nanocolloids. J. Am. Chem. Soc. 2010, 132, 17667–17669. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, J.; Sang, X.; Kang, X.; Zhang, B.; Luo, T.; Tan, X.; Han, B.; Zheng, L.; Zhang, J. CO2/water emulsions stabilized by partially reduced graphene oxide. ACS Appl. Mater. Interfaces 2017, 9, 17613–17619. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, X.; Jia, W.; Li, Z.; Zhao, Y.; Ren, S. Demulsification of crude oil-in-water emulsions driven by graphene oxide nanosheets. Energy Fuels 2015, 29, 4644–4653. [Google Scholar] [CrossRef]
- Bai, H.; Li, C.; Wang, X.; Shi, G. On the gelation of graphene oxide. J. Phys. Chem. C 2011, 115, 5545–5551. [Google Scholar] [CrossRef]
- Park, S.; Lee, K.-S.; Bozoklu, G.; Cai, W.; Nguyen, S.T.; Ruoff, R.S. Graphene oxide papers modified by divalent ions—Enhancing mechanical properties via chemical cross-linking. ACS Nano 2008, 2, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Potluri, V.K.; McLeod, M.C.; Wang, Y.; Liu, J.; Enick, R.M.; Hamilton, A.D.; Roberts, C.B.; Johnson, J.K.; Beckman, E.J. Oxygenated hydrocarbon ionic surfactants exhibit CO2 solubility. J. Am. Chem. Soc. 2005, 127, 11754–11762. [Google Scholar] [CrossRef] [PubMed]
- Kilic, S.; Michalik, S.; Wang, Y.; Johnson, J.K.; Enick, R.M.; Beckman, E.J. Effect of grafted lewis base groups on the phase behavior of model poly (dimethyl siloxanes) in CO2. Ind. Eng. Chem. Res. 2003, 42, 6415–6424. [Google Scholar] [CrossRef]
- Chabert, M.; Morvan, M.; Nabzar, L. Advanced screening technologies for the selection of dense CO2 foaming surfactants. In Proceedings of the SPE Improved Oil Recovery Symposium, Society of Petroleum Engineers, Tulsa, OK, USA, 2012. [Google Scholar]
- Aarra, M.G.; Skauge, A.; Solbakken, J.; Ormehaug, P.A. Properties of N2- and CO2-foams as a function of pressure. J. Pet. Sci. Eng. 2014, 116, 72–80. [Google Scholar] [CrossRef]
- Chen, Y.; Elhag, A.S.; Poon, B.M.; Cui, L.; Ma, K.; Liao, S.Y.; Omar, A.; Worthen, A.; Hirasaki, G.; Nguyen, P.; et al. Ethoxylated cationic surfactants for CO2 eor in high temperature, high salinity reservoirs. In Proceedings of the SPE Improved Oil Recovery Symposium, Tulsa, OK, USA, 14–18 April 2012. [Google Scholar]
- Alvarez, J.M.; Rivas, H.J.; Rossen, W.R. Unified model for steady-state foam behavior at high and low foam qualities. In Proceedings of the SPE Annual Technical Conference and Exhibition, The Hague, The Netherlands, 21–22 May 2001. [Google Scholar]
- Batôt, G.; Fleury, M.; Nabzar, L. Reducing CO2 flow using foams. Energy Procedia 2017, 114, 4129–4139. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO (%) | rGO (%) | |
|---|---|---|
| Carbon | 49–56 | 77–87 |
| Hydrogen | 0–1 | 0–1 |
| Nitrogen | 0–1 | 0–1 |
| Sulphur | 2–4 | 0 |
| Oxygen | 41–50 | 13–22 |
| Salt | Concentration (g/L) |
|---|---|
| NaCl | 23.612 |
| CaCl2·2H2O | 1.911 |
| MgCl2·6H2O | 9.149 |
| KCl | 0.746 |
| Na2SO4 | 3.407 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrabino, A.; Holt, T.; Lindeberg, E. An Evaluation of Graphene Oxides as Possible Foam Stabilizing Agents for CO2 Based Enhanced Oil Recovery. Nanomaterials 2018, 8, 603. https://doi.org/10.3390/nano8080603
Barrabino A, Holt T, Lindeberg E. An Evaluation of Graphene Oxides as Possible Foam Stabilizing Agents for CO2 Based Enhanced Oil Recovery. Nanomaterials. 2018; 8(8):603. https://doi.org/10.3390/nano8080603
Chicago/Turabian StyleBarrabino, Albert, Torleif Holt, and Erik Lindeberg. 2018. "An Evaluation of Graphene Oxides as Possible Foam Stabilizing Agents for CO2 Based Enhanced Oil Recovery" Nanomaterials 8, no. 8: 603. https://doi.org/10.3390/nano8080603
APA StyleBarrabino, A., Holt, T., & Lindeberg, E. (2018). An Evaluation of Graphene Oxides as Possible Foam Stabilizing Agents for CO2 Based Enhanced Oil Recovery. Nanomaterials, 8(8), 603. https://doi.org/10.3390/nano8080603