Evaluation of the PEG Density in the PEGylated Chitosan Nanoparticles as a Drug Carrier for Curcumin and Mitoxantrone

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
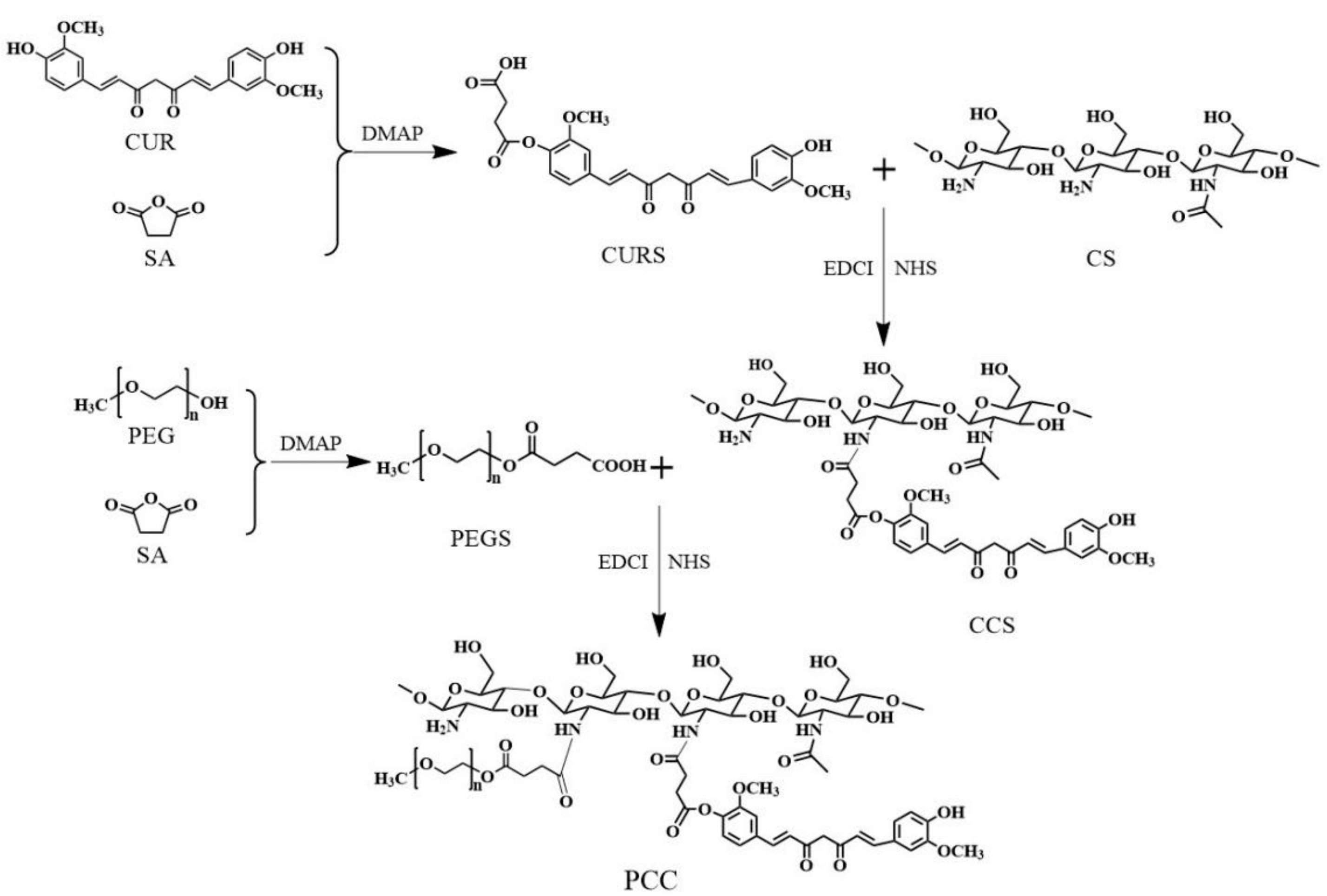
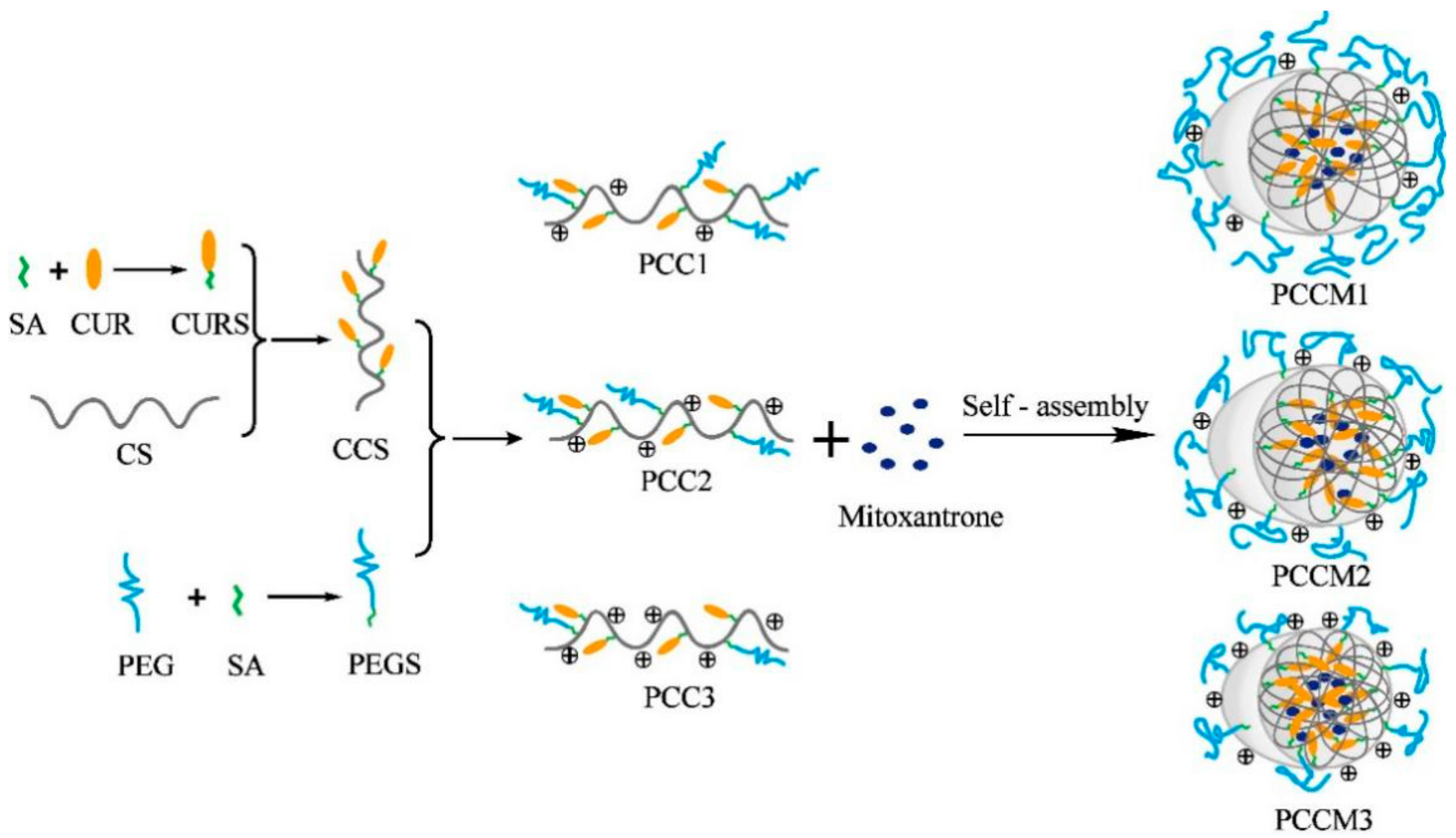
2.2. Synthesis of Polyethylene Glycolated (PEGylated) Curcumin-Grafted-Chitosan (PCC) Conjugates
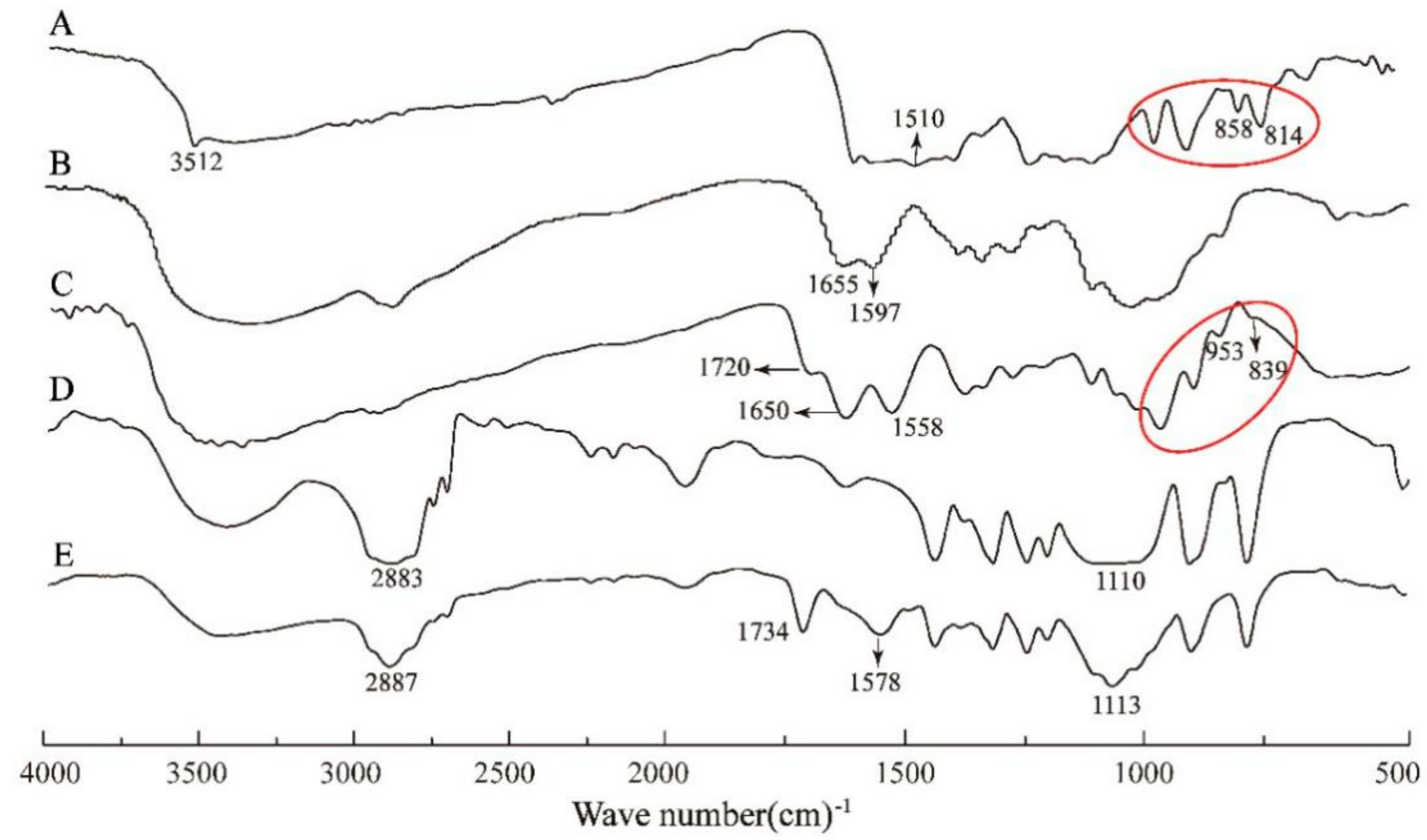
2.3. Fourier Transform Infrared (FTIR), Proton Nuclear Magnetic Resonance (1H NMR) and Ultraviolet (UV-Vis) Spectroscopy
2.4. Preparation PCC Nanoparticles (NPs)
2.5. Dynamic Light Scattering (DLS) and Transmission Electron Microscopy (TEM)
2.6. Preparation and Characterization of Mitoxantrone-loaded PCC NPs (PCCM NPs)
2.7. Determination of Entrapment Efficiency (EE) and Loading Capacity (LC)
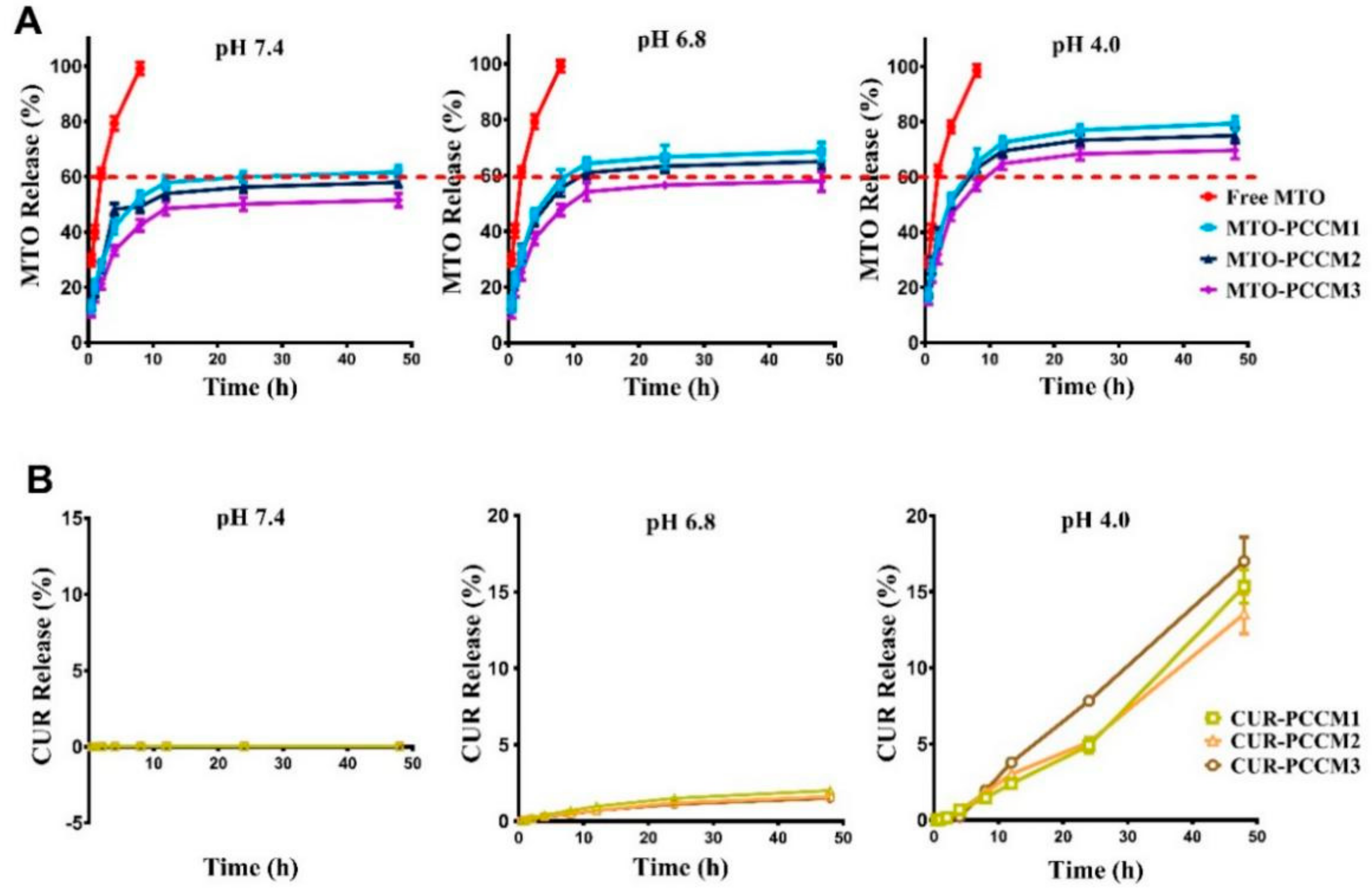
2.8. Determination of Drug Release from PCCM NPs In Vitro
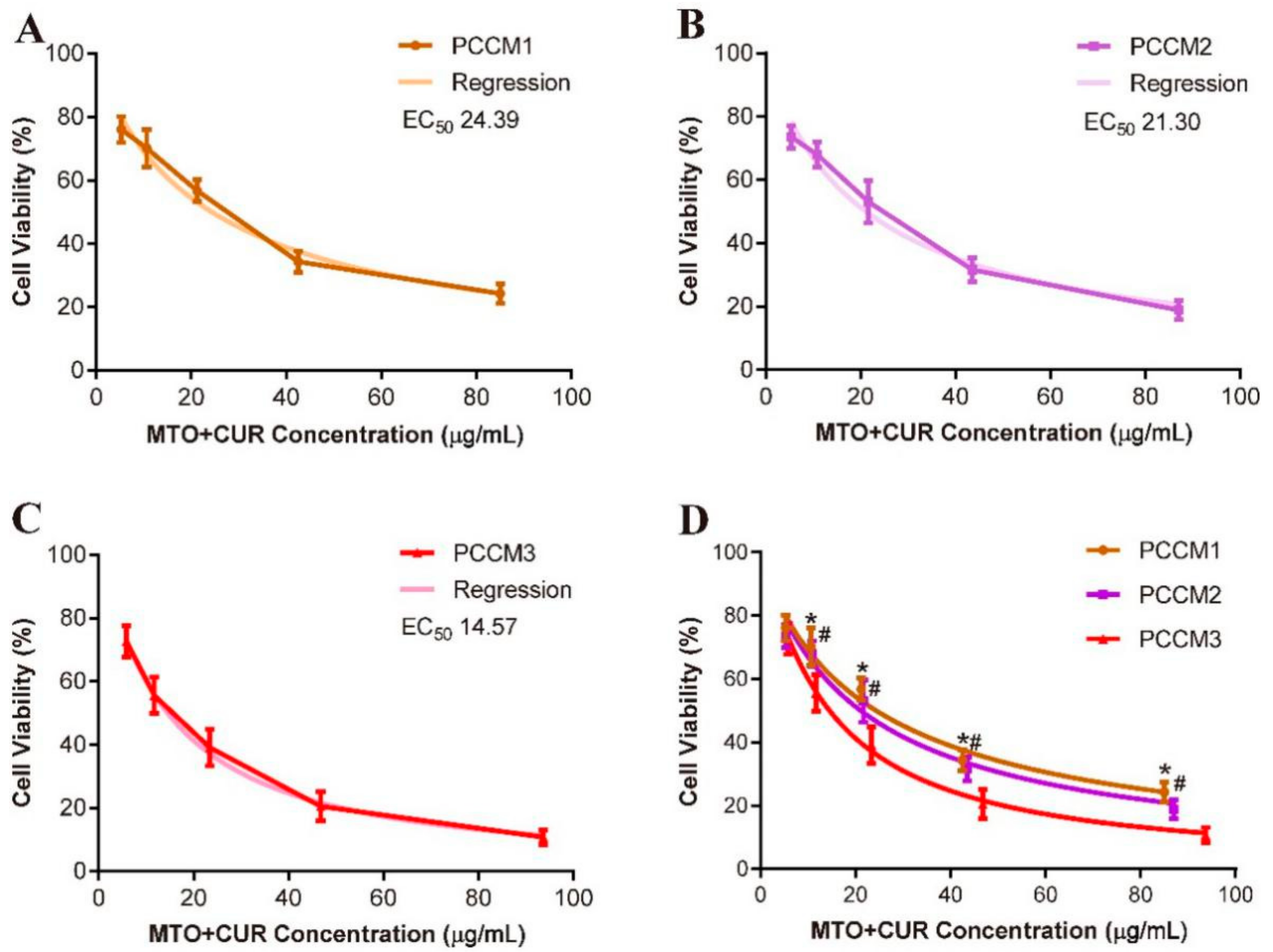
2.9. Determine PCCM NPs’ Cytotoxicity In Vitro
2.10. Calculation of the Synergistic Effect of MTO and CUR
2.11. Statistical Analysis
3. Results
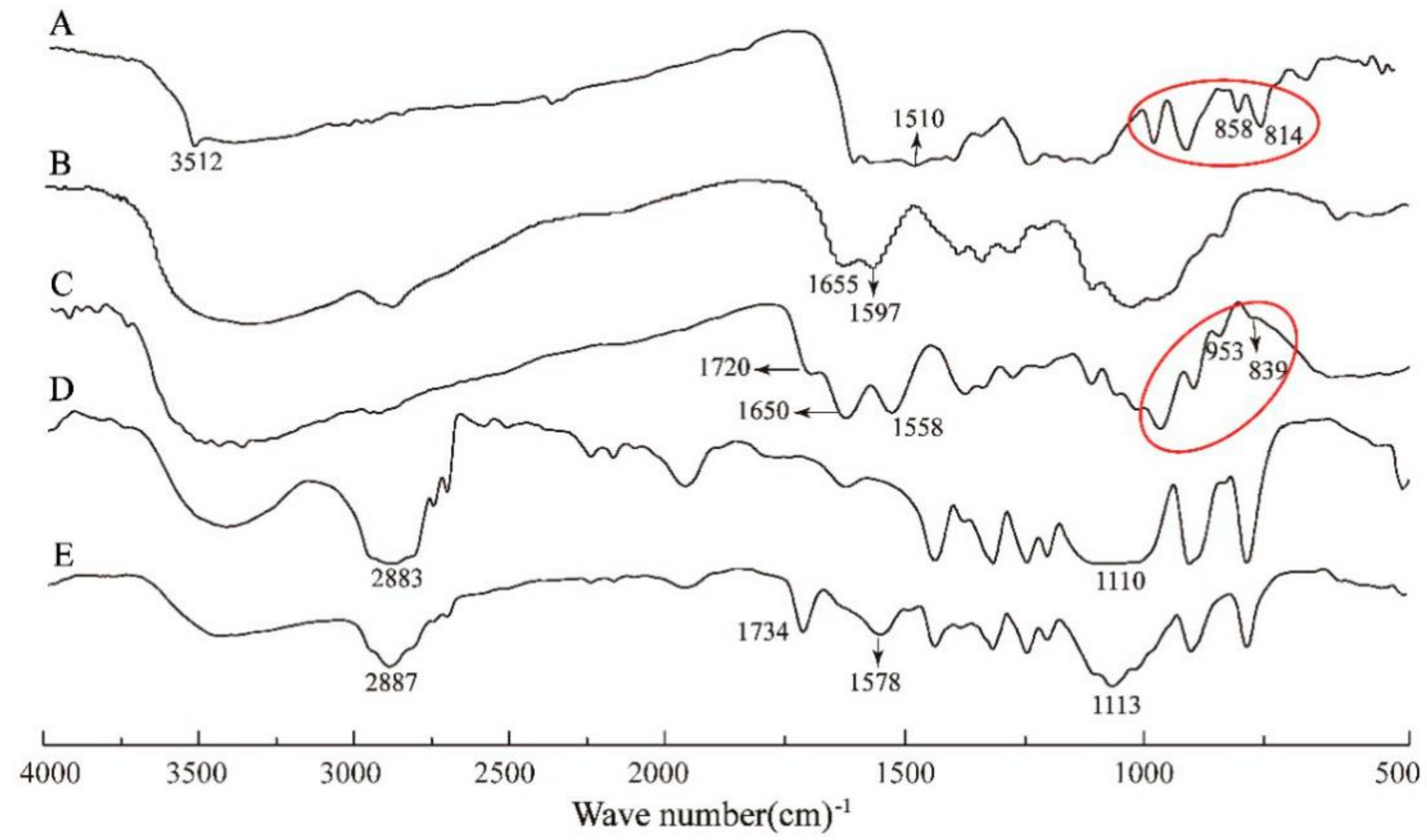
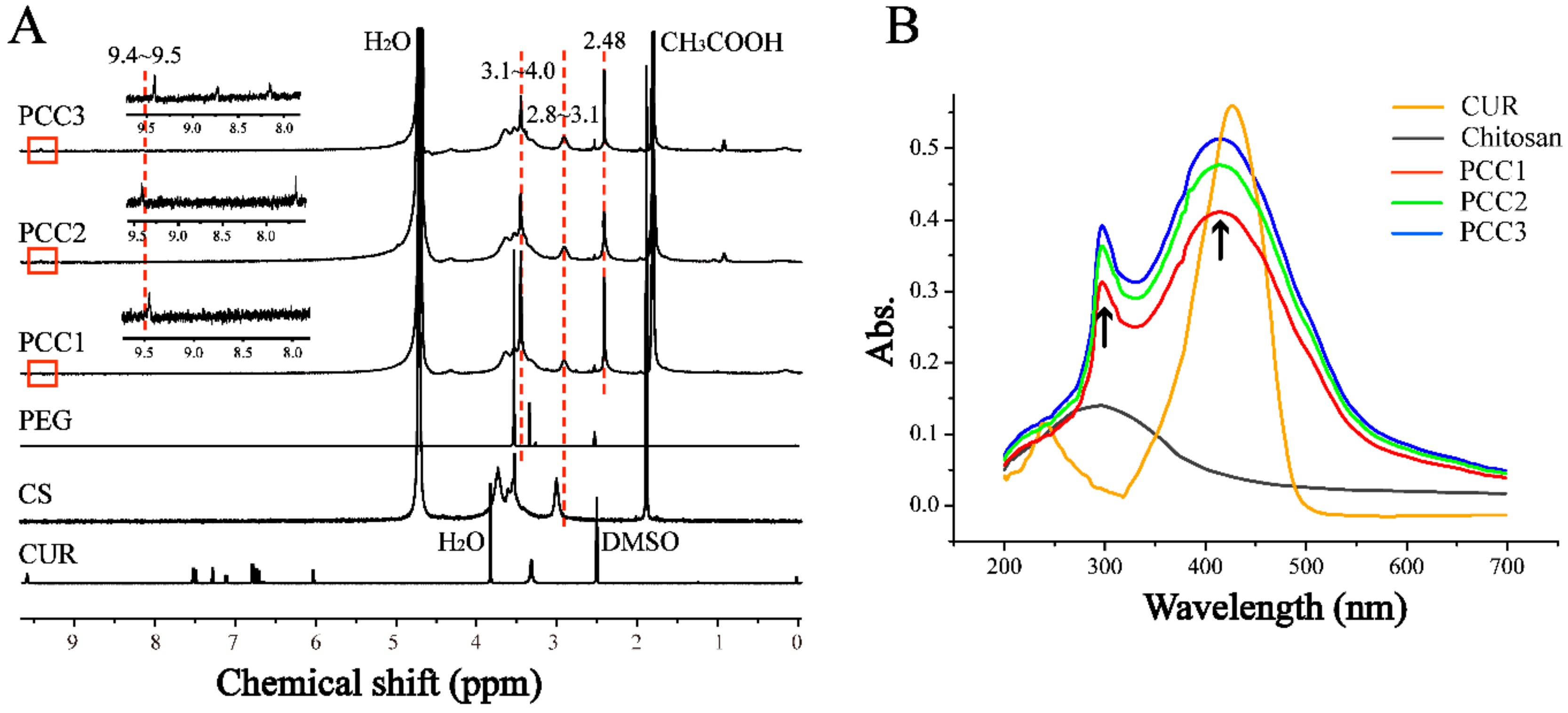
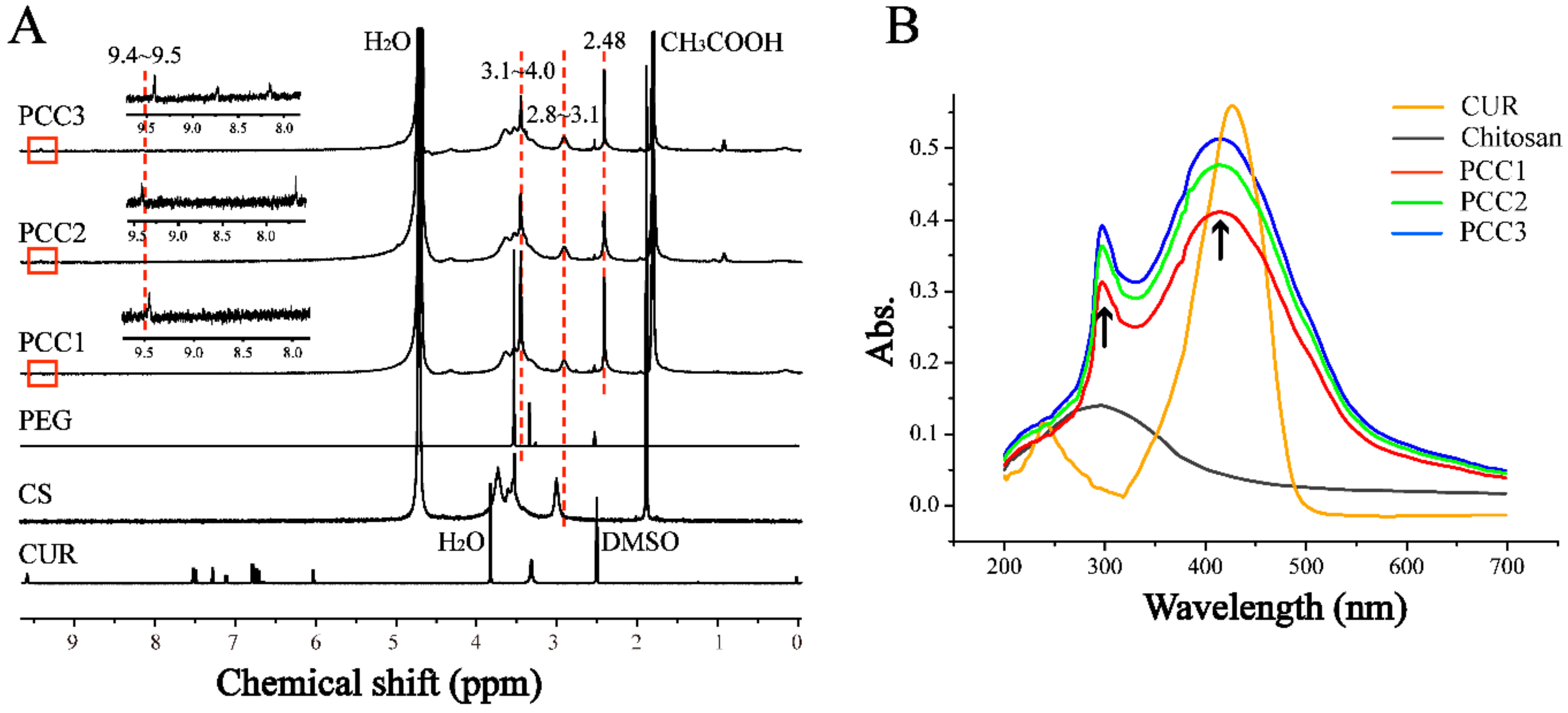
3.1. FTIR, 1H NMR and UV-Vis Spectroscopic Analysis
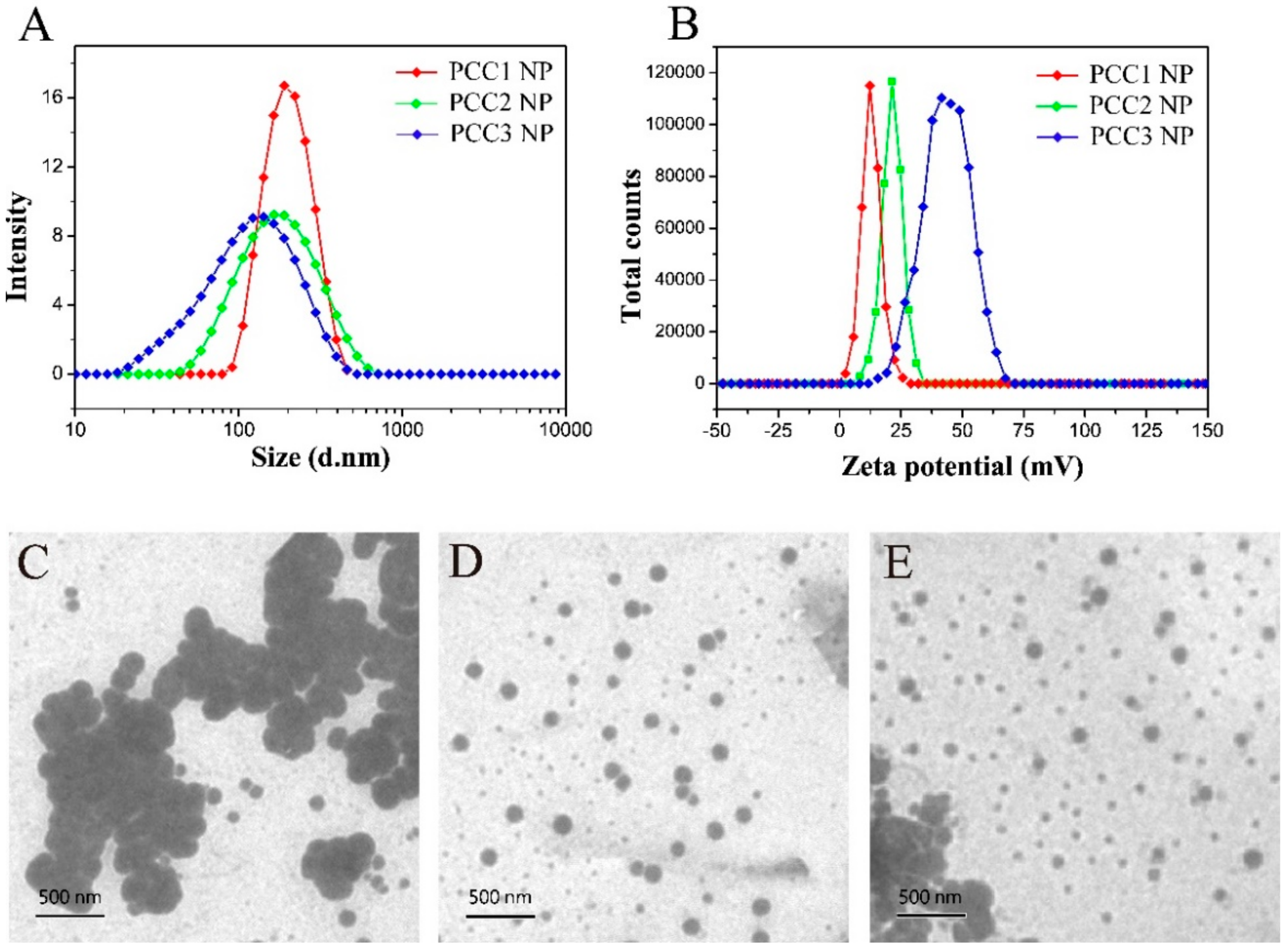
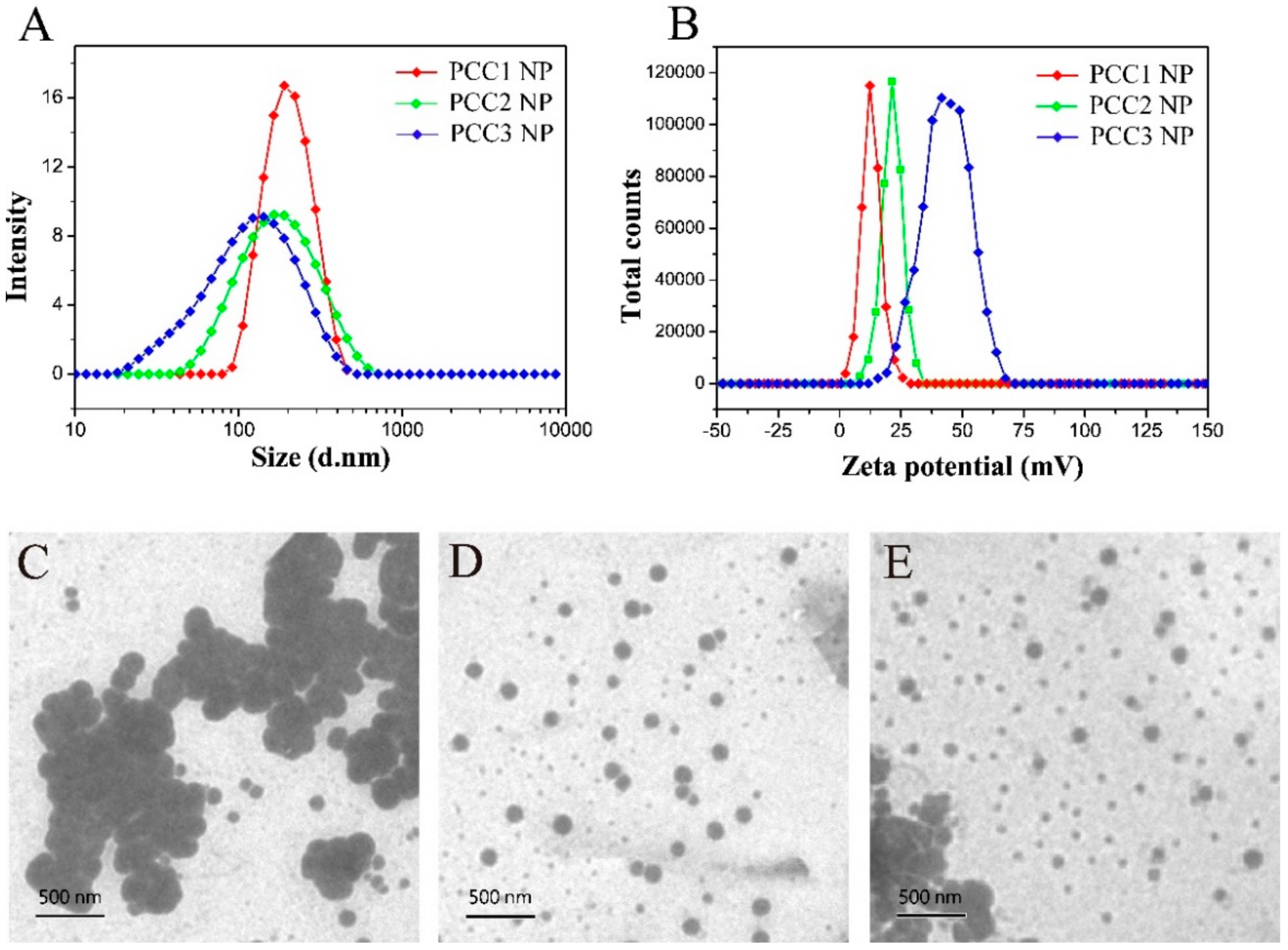
3.2. Size Distribution, Zeta Potential and Morphology of PCC NPs
3.3. Characterizations of PCCM NPs
3.4. Mitoxantrone (MTO) and Curcumin (CUR) Drug Release from PCCM NPs In Vitro
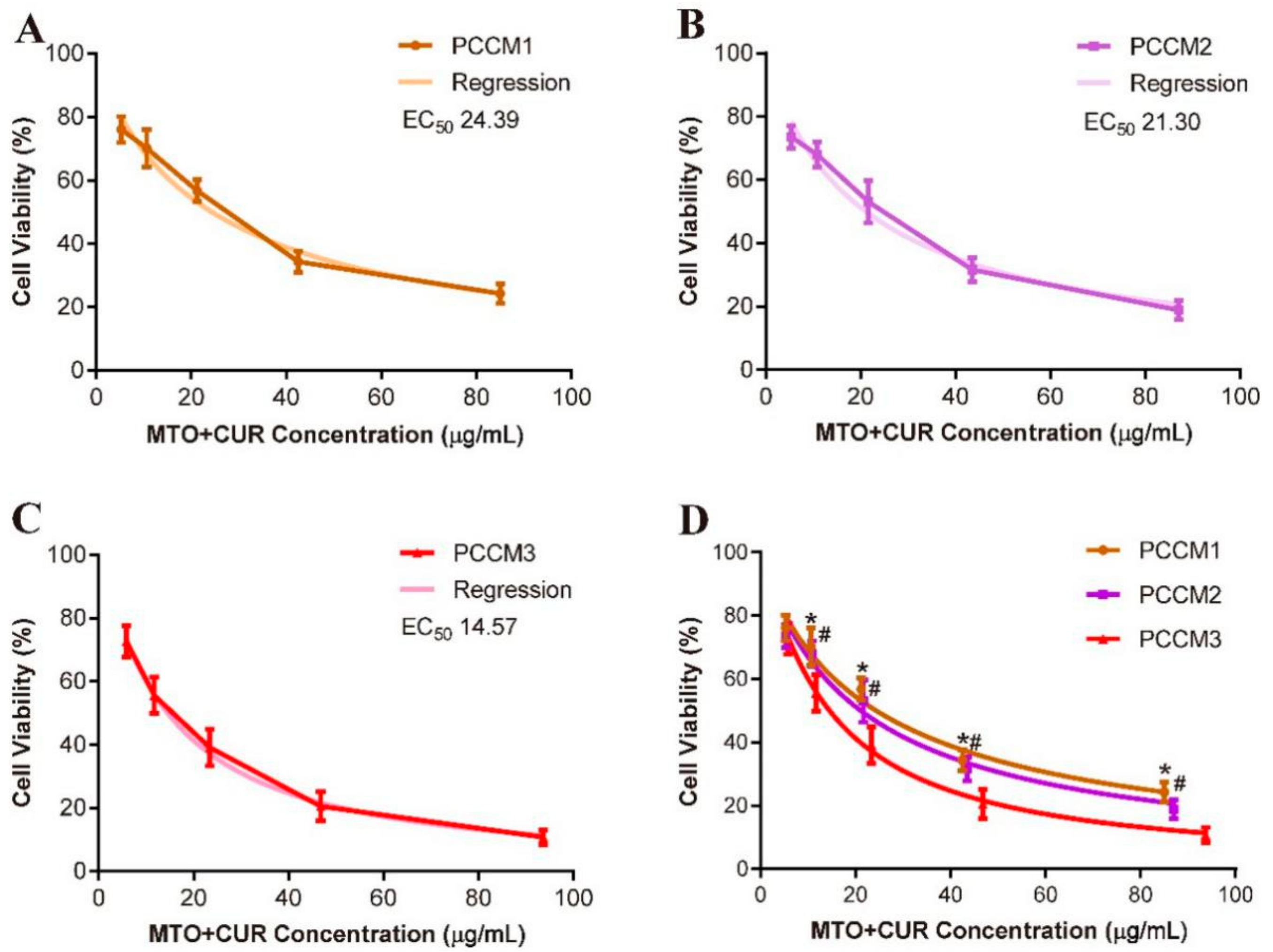
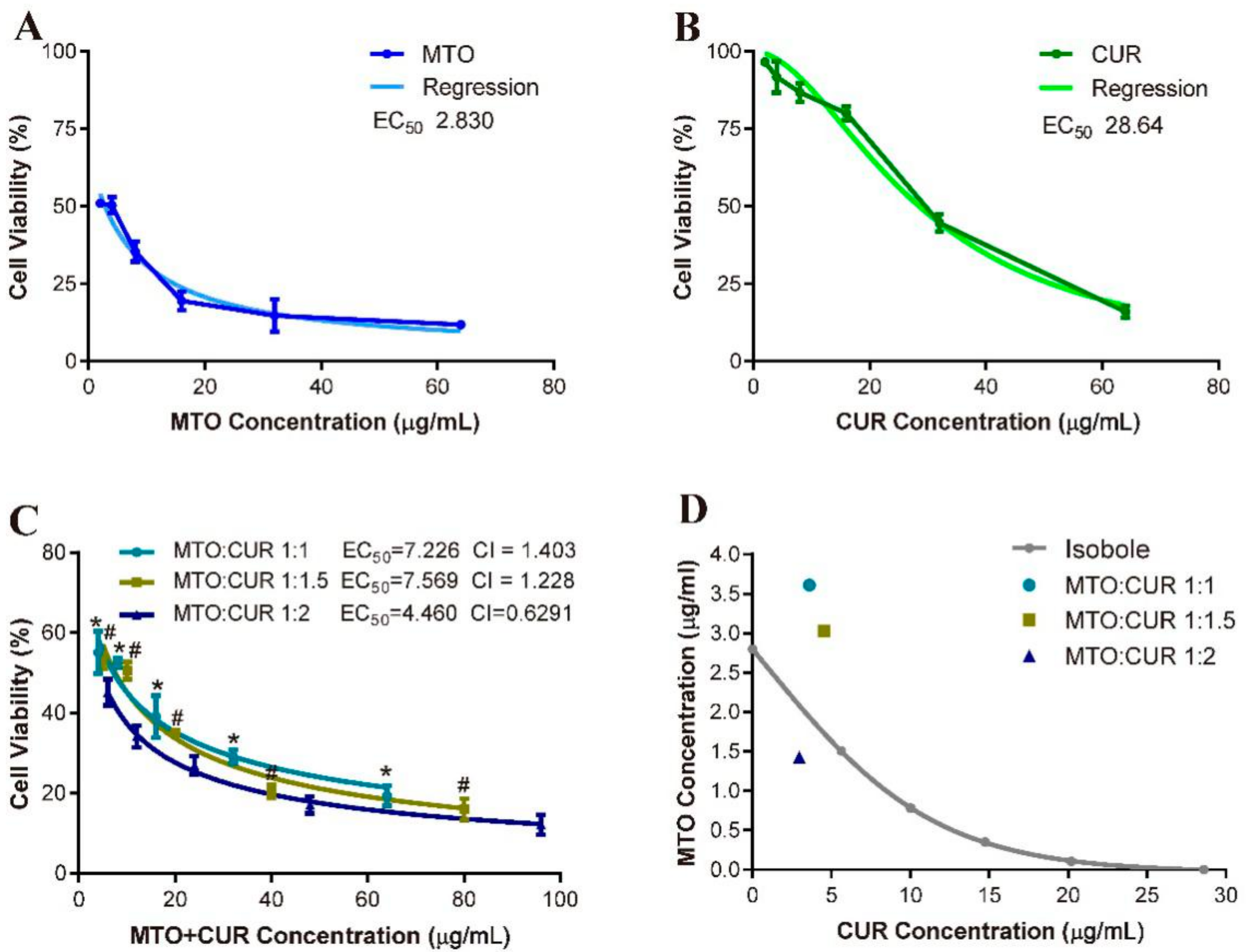
3.5. Cytotoxicity Test In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Duan, J.; Mansour, H.M.; Zhang, Y.; Deng, X.; Chen, Y.; Wang, J.; Pan, Y.; Zhao, J. Reversion of multidrug resistance by co-encapsulation of doxorubicin and curcumin in chitosan/poly(butyl cyanoacrylate) nanoparticles. Int. J. Pharm. 2012, 426, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.D.; Qin, Y.; Yang, C.; Li, L. Effect of curcumin on human colon cancer multidrug resistance in vitro and in vivo. Clinics 2013, 68, 694–701. [Google Scholar] [CrossRef]
- Xiao, B.; Si, X.; Han, M.K.; Viennois, E.; Zhang, M.; Merlin, D. Co-delivery of camptothecin and curcumin by cationic polymeric nanoparticles for synergistic colon cancer combination chemotherapy. J. Mater. Chem. B 2015, 3, 7724–7733. [Google Scholar] [CrossRef] [PubMed]
- Modasiya, M.K.; Patel, V.M. Studies on solubility of curcumin. Int. J. Pharm. Life Sci. 2012, 3, 1490–1497. [Google Scholar]
- Luty, M.; Kwiecień, E.; Firlej, M.; Łabędź-Masłowska, A.; Paw, M.; Madeja, Z.; Czyż, J. Curcumin augments the cytostatic and anti-invasive effects of mitoxantrone on carcinosarcoma cells in vitro. Acta Biochim. Pol. 2016, 63, 397. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yu, L.; Guo, R.; Dong, A.; Lin, C.; Zhang, J. A modular coassembly approach to all-in-one multifunctional nanoplatform for synergistic codelivery of doxorubicin and curcumin. Nanomaterials 2018, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Koeller, J.; Eble, M. Mitoxantrone: A novel anthracycline derivative. Clin. Pharm. 1988, 7, 574–581. [Google Scholar] [PubMed]
- Wiseman, L.R.; Spencer, C.M. Mitoxantrone: A review of its pharmacology and clinical efficacy in the management of hormone-resistant advanced prostate cancer. Drugs Aging 1997, 10, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Limtrakul, P.; Chearwae, W.; Shukla, S.; Phisalphong, C.; Ambudkar, S.V. Modulation of function of three ABC drug transporters, p-glycoprotein (ABCB1), mitoxantrone resistance protein (ABCG2) and multidrug resistance protein 1 (ABCC1) by tetrahydrocurcumin, a major metabolite of curcumin. Mol. Cell. Biochem. 2007, 296, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Pridgen, E.M.; Langer, R.; Farokhzad, O.C. Biodegradable, polymeric nanoparticle delivery systems for cancer therapy. Nanomedicine 2007, 2, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, E.S.; Oh, K.T.; Gao, Z.G.; Bae, Y.H. Doxorubicin-loaded polymeric micelle overcomes multidrug resistance of cancer by double-targeting folate receptor and early endosomal pH. Small 2008, 4, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Li, A.; Zhang, A.; Sun, Y.; Liu, J. Polymer-based nanocarriers for co-delivery and combination of diverse therapies against cancers. Nanomaterials 2018, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Asamoah-Asare, J. Novel carboxymethyl chitosan-β-cyclodextrin nanoparticles as a drug delivery system; preparation and characterization. Int. J. Eng. Sci. Res. Technol. 2014, 3, 141–146. [Google Scholar]
- Cheung, R.; Ng, T.; Wong, J.; Chan, W. Chitosan: An update on potential biomedical and pharmaceutical applications. Mar. Drugs 2015, 13, 5156–5186. [Google Scholar] [CrossRef] [PubMed]
- Kean, T.; Thanou, M. Biodegradation, biodistribution and toxicity of chitosan. Adv. Drug Deliv. Rev. 2010, 62, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.G.; Wei, W.; Lv, P.P.; Yue, H.; Wang, L.Y.; Su, Z.G.; Ma, G.H. Surface charge affects cellular uptake and intracellular trafficking of chitosan-based nanoparticles. Biomacromolecules 2011, 12, 2440. [Google Scholar] [CrossRef] [PubMed]
- Gorzelanny, C.; Pöppelmann, B.; Pappelbaum, K.; Moerschbacher, B.M.; Schneider, S.W. Human macrophage activation triggered by chitotriosidase-mediated chitin and chitosan degradation. Biomaterials 2010, 31, 8556–8563. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.Q.; Wang, T.Y.; Webster, T.J.; Duan, H.J.; Qiu, J.Y.; Zhao, Z.M.; Yin, X.X.; Zheng, C.L. Intracellular disposition of chitosan nanoparticles in macrophages: Intracellular uptake, exocytosis, and intercellular transport. Int. J. Nanomed. 2017, 12, 6383. [Google Scholar] [CrossRef] [PubMed]
- Rudzinski, W.E.; Palacios, A.; Ahmed, A.; Lane, M.A.; Aminabhavi, T.M. Targeted delivery of small interfering rna to colon cancer cells using chitosan and pegylated chitosan nanoparticles. Carbohydr. Polym. 2016, 147, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Gao, S.; Dagnaes-Hansen, F.; Jakobsen, M.; Kjems, J. Impact of peg chain length on the physical properties and bioactivity of pegylated chitosan/sirna nanoparticles in vitro and in vivo. ACS Appl. Mater. Interfaces 2017, 9, 12203–12216. [Google Scholar] [CrossRef] [PubMed]
- Termsarasab, U.; Yoon, I.S.; Park, J.H.; Moon, H.T.; Cho, H.J.; Kim, D.D. Polyethylene glycol-modified arachidyl chitosan-based nanoparticles for prolonged blood circulation of doxorubicin. Int. J. Pharm. 2014, 464, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Boal, A.K.; Ilhan, F.; Derouchey, J.E.; Thurn-Albrecht, T.; Russell, T.P.; Rotello, V.M. Self-assembly of nanoparticles into structured spherical and network aggregates. Nature 2000, 404, 746. [Google Scholar] [CrossRef] [PubMed]
- Grzelczak, M.; Vermant, J.; Furst, E.M.; Liz-Marzán, L.M. Directed self-assembly of nanoparticles. ACS Nano 2010, 4, 3591–3605. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Xie, Y.; Zhang, Q.; Qiu, X.; Yuan, L.; Wen, Y.; Li, M.; Yang, X.; Tao, T.; Xie, M. Cholesterol-modified amino-pullulan nanoparticles as a drug carrier: Comparative study of cholesterol-modified carboxyethyl pullulan and pullulan nanoparticles. Nanomaterials 2016, 6, 165. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Zhang, Q.; Yang, W.; Zhang, Q. The interaction between human serum albumin and cholesterol-modified pullulan nanoparticle. Curr. Nanosci. 2012, 8, 830–837. [Google Scholar] [CrossRef]
- Tan, Y.L.; Liu, C.G. Self-aggregated nanoparticles from linoleic acid modified carboxymethyl chitosan: Synthesis, characterization and application in vitro. Colloids Surf. B Biointerfaces 2009, 69, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhang, S.; Kong, D.; Gao, X.; Zhao, Y.; Wang, Z. Biodegradable polymer-curcumin conjugate micelles enhance the loading and delivery of low-potency curcumin. Pharm. Res. 2012, 29, 3512–3525. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.I.; Kim, D.G.; Jang, M.K.; Nah, J.W. Preparation and spectroscopic characterization of methoxy poly(ethylene glycol)-grafted water-soluble chitosan. Carbohydr. Res. 2008, 343, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym Regul. 1984, 22, 27. [Google Scholar] [CrossRef]
- Loewe, S. The problem of synergism and antagonism of combined drugs. Arzneimittel-Forschung 1953, 3, 285. [Google Scholar] [PubMed]
- Tallarida, R.J. Quantitative methods for assessing drug synergism. Genes Cancer 2011, 2, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Chaoran, X.; Wei, H.; Yaqi, L.; Chao, Q.; Lingjia, S.; Lifang, Y. Self-assembled nanoparticles from hyaluronic acid-paclitaxel prodrugs for direct cytosolic delivery and enhanced antitumor activity. Int. J. Pharm. 2015, 493, 172–181. [Google Scholar]
- Prego, C.; Torres, D.; Fernandezmegia, E.; Novoacarballal, R.; Quiñoá, E.; Alonso, M.J. Chitosan-peg nanocapsules as new carriers for oral peptide delivery. Effect of chitosan pegylation degree. J. Control. Release 2006, 111, 299. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Zheng, F.; Zhong, S.; Tao, X.; Zhang, Y.; Gao, F.; Yao, F.; Chen, J.; Chen, Y.; Shi, G. Self-assembled nanoparticles of glycyrrhetic acid-modified pullulan as a novel carrier of curcumin. Molecules 2014, 19, 13305–13318. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, A.; Katzir, I.; Dekel, E.; Mayo, A.E.; Alon, U. Prediction of multidimensional drug dose responses based on measurements of drug pairs. Proc. Natl. Acad. Sci. USA 2016, 113, 10442–10447. [Google Scholar] [CrossRef] [PubMed]
- Bareford, L.M.; Swaan, P.W. Endocytic mechanisms for targeted drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Cheah, Y.H.; Nordin, F.J.; Sarip, R.; Tee, T.T.; Azimahtol, H.L.P.; Sirat, H.M.; Rashid, B.A.A.; Abdullah, N.R.; Ismail, Z. Combined xanthorrhizol-curcumin exhibits synergistic growth inhibitory activity via apoptosis induction in human breast cancer cells mda-mb-231. Cancer Cell Int. 2009, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Wang, T.; Cochrane, C.; Mccarron, P. Modulation of surface charge, particle size and morphological properties of chitosan-TPP nanoparticles intended for gene delivery. Colloids Surf. B Biointerfaces 2005, 44, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Qiang, G.; Ying, L.; Wang, X.; Wang, J.; Tu, P. Co-assembly of doxorubicin and curcumin targeted micelles for synergistic delivery and improving anti-tumor efficacy. Eur. J. Pharm. Biopharm. 2016, 112, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, Q.; Liu, W.; Li, Y.; Tang, H.; Liu, X.; Yang, X. Codelivery of doxorubicin and curcumin with lipid nanoparticles results in improved efficacy of chemotherapy in liver cancer. Int. J. Nanomed. 2015, 10, 257–270. [Google Scholar]
- Esfandiarpour-Boroujeni, S.; Bagheri-Khoulenjani, S.; Mirzadeh, H.; Amanpour, S. Fabrication and study of curcumin loaded nanoparticles based on folate-chitosan for breast cancer therapy application. Carbohydr. Polym. 2017, 168, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Zou, L.; Liu, W.; Li, Z.; Liu, W.; Hu, X.; Chen, X.; Liu, C. Hybrid liposomes composed of amphiphilic chitosan and phospholipid: Preparation, stability and bioavailability as a carrier for curcumin. Carbohydr. Polym. 2016, 156, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, B.; Del, P.P.; Maffre, P.; Hartmann, R.; Gallego, M.; Rivera-Fernández, S.; Jm, D.L.F.; Nienhaus, G.U.; Parak, W.J. Surface functionalization of nanoparticles with polyethylene glycol: Effects on protein adsorption and cellular uptake. ACS Nano 2015, 9, 6996. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R.H. ’Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (peg): Influences of the corona (peg chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Feed PEG/CS Molar Ratio | PEG Molar DS | EE% | LCM% | LCC% | Zeta Potential (mV) | Size (nm) | PDI |
|---|---|---|---|---|---|---|---|---|
| PCCM1 | 1/5 | 0.75% | 90.5 ± 2.89 | 7.42 ± 0.16 *,# | 12.3 ± 0.52 *,# | 12.8 ± 4.02 *,# | 250.2 ± 21.5 * | 0.153 |
| PCCM2 | 1/7.5 | 0.45% | 88.6 ± 1.61 | 8.14 ± 0.14 | 14.0 ± 0.87 * | 21.2 ± 4.27 * | 233.1 ± 19.2 * | 0.216 |
| PCCM3 | 1/10 | 0.33% | 87.3 ± 1.74 | 8.30 ± 0.24 | 16.1 ± 0.21 | 34.0 ± 4.52 | 183.1 ± 15.6 | 0.225 |
| Drugs | Concentration (μg/mL) | ||||
|---|---|---|---|---|---|
| PCCM NPs-MTO | 2.00 | 4.00 | 8.00 | 16.00 | 32.00 |
| PCCM1-CUR | 3.32 | 6.63 | 13.26 | 26.52 | 53.05 |
| PCCM2-CUR | 3.44 | 6.88 | 13.70 | 27.50 | 55.00 |
| PCCM3-CUR | 3.85 | 7.71 | 15.40 | 30.80 | 61.70 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Wu, D.; Zhong, W.; Kuang, S.; Luo, Q.; Song, L.; He, L.; Feng, X.; Tao, X. Evaluation of the PEG Density in the PEGylated Chitosan Nanoparticles as a Drug Carrier for Curcumin and Mitoxantrone. Nanomaterials 2018, 8, 486. https://doi.org/10.3390/nano8070486
Chen Y, Wu D, Zhong W, Kuang S, Luo Q, Song L, He L, Feng X, Tao X. Evaluation of the PEG Density in the PEGylated Chitosan Nanoparticles as a Drug Carrier for Curcumin and Mitoxantrone. Nanomaterials. 2018; 8(7):486. https://doi.org/10.3390/nano8070486
Chicago/Turabian StyleChen, Yao, Di Wu, Wu Zhong, Shuwen Kuang, Qian Luo, Liujiang Song, Lihua He, Xing Feng, and Xiaojun Tao. 2018. "Evaluation of the PEG Density in the PEGylated Chitosan Nanoparticles as a Drug Carrier for Curcumin and Mitoxantrone" Nanomaterials 8, no. 7: 486. https://doi.org/10.3390/nano8070486
APA StyleChen, Y., Wu, D., Zhong, W., Kuang, S., Luo, Q., Song, L., He, L., Feng, X., & Tao, X. (2018). Evaluation of the PEG Density in the PEGylated Chitosan Nanoparticles as a Drug Carrier for Curcumin and Mitoxantrone. Nanomaterials, 8(7), 486. https://doi.org/10.3390/nano8070486