DNA-Assisted Assembly of Gold Nanostructures and Their Induced Optical Properties



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structural DNA Self-Assembly Nanotechnology
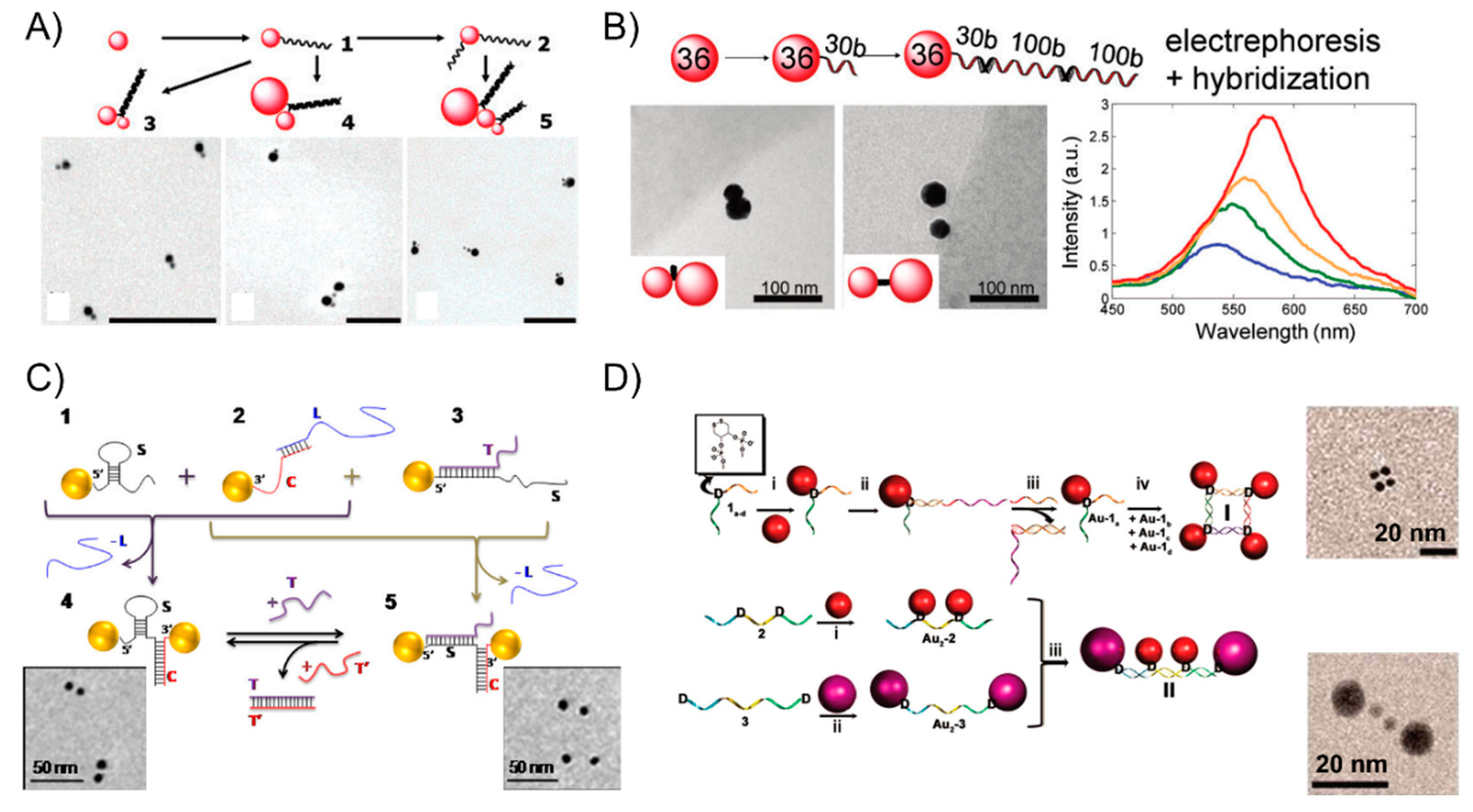
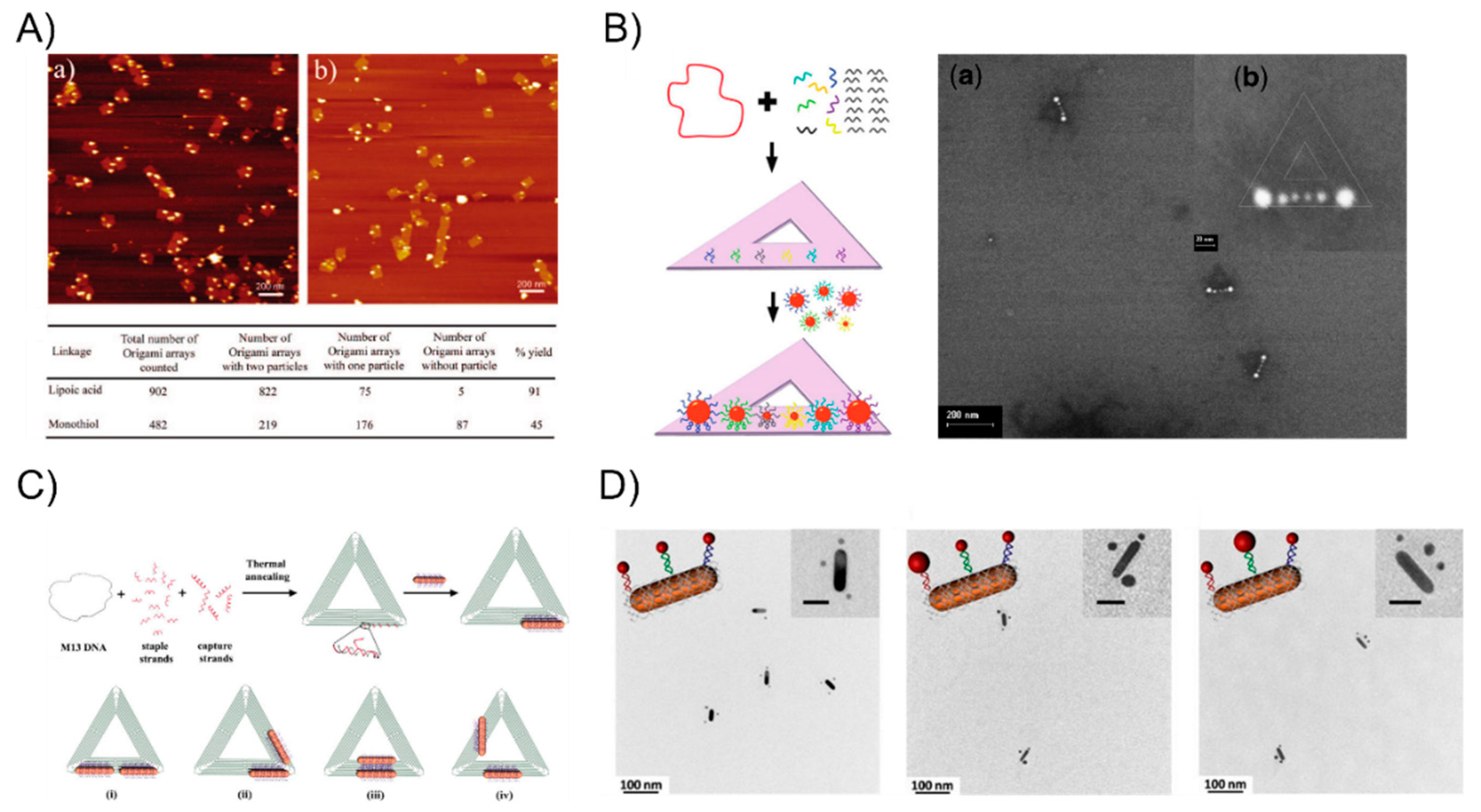
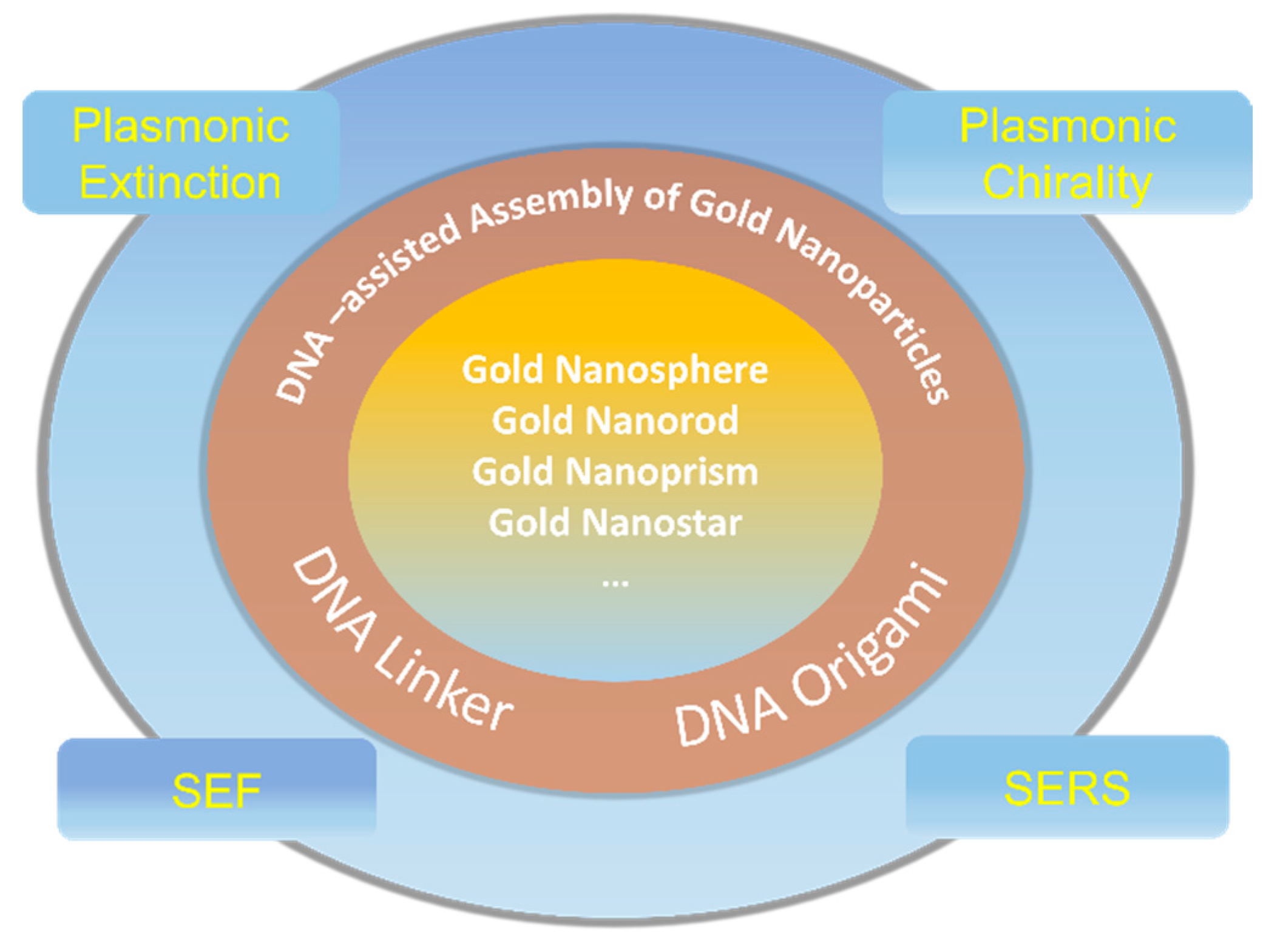
3. The DNA-Assisted Self-Assembly of AuNP Nanoarchitectures
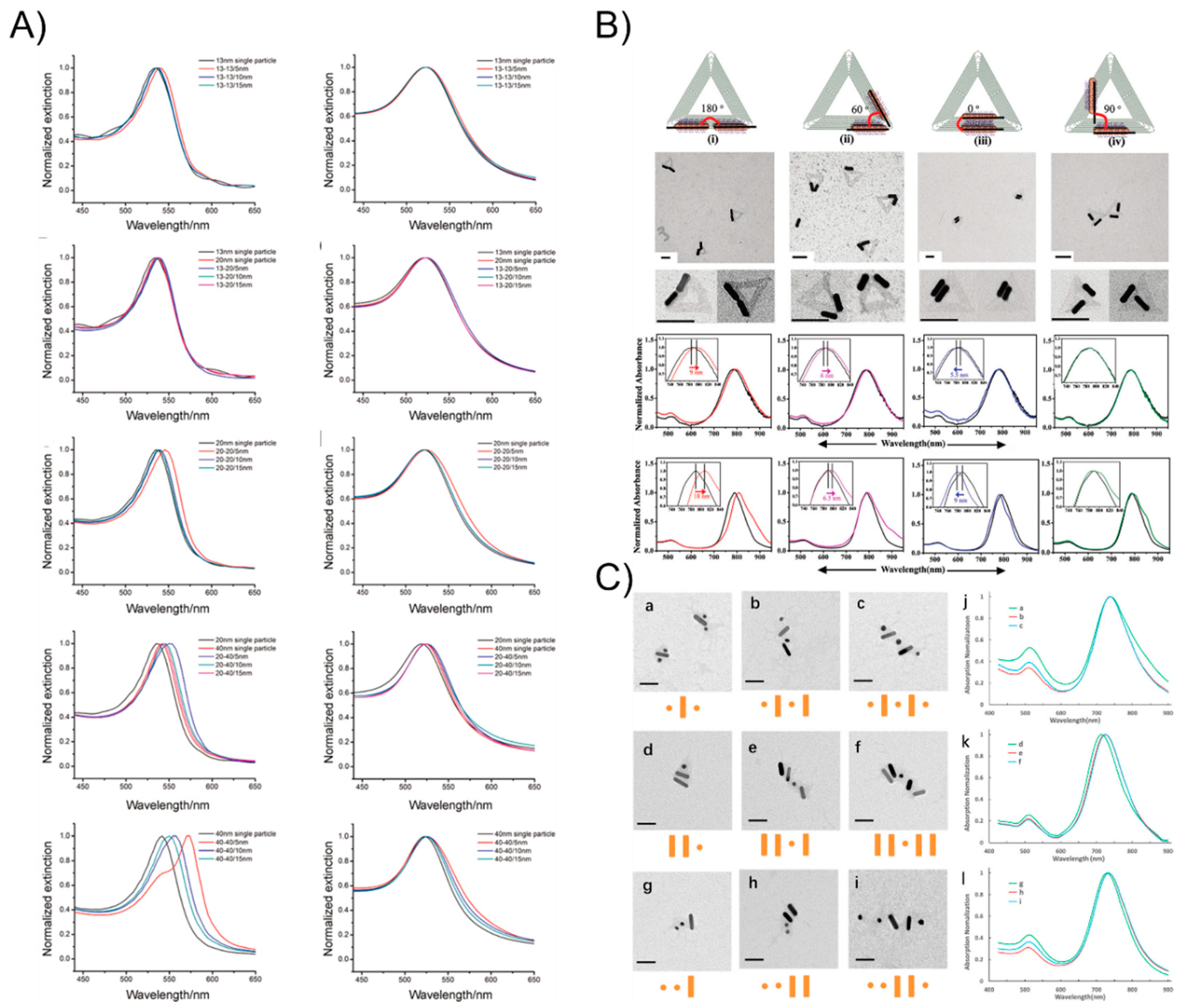
4. The Plasmonic Extinction of DNA-Assisted Self-Assembly of AuNP Nanoarchitectures
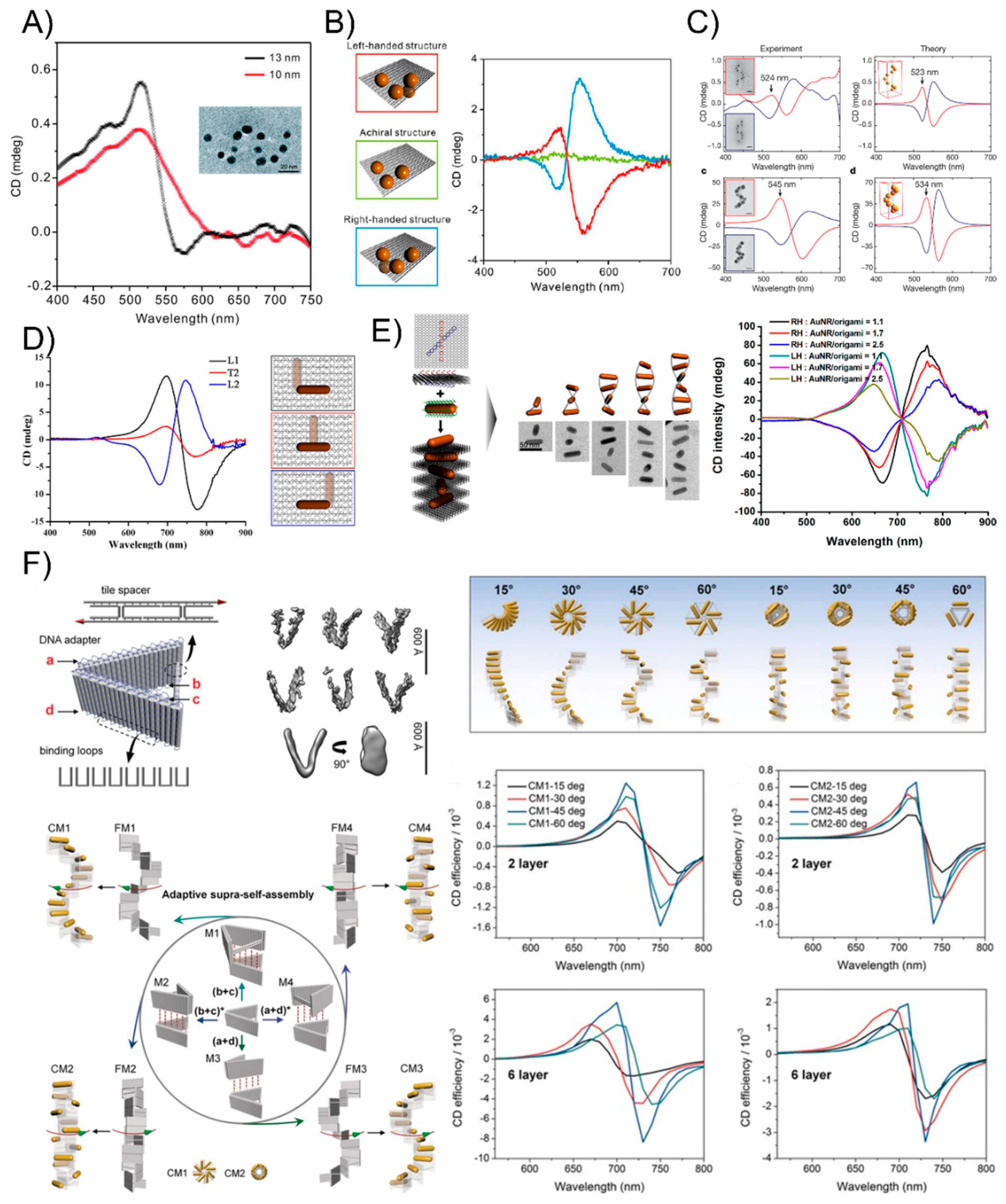
5. The Plasmonic Chirality of DNA-Directed AuNP Nanoarchitectures
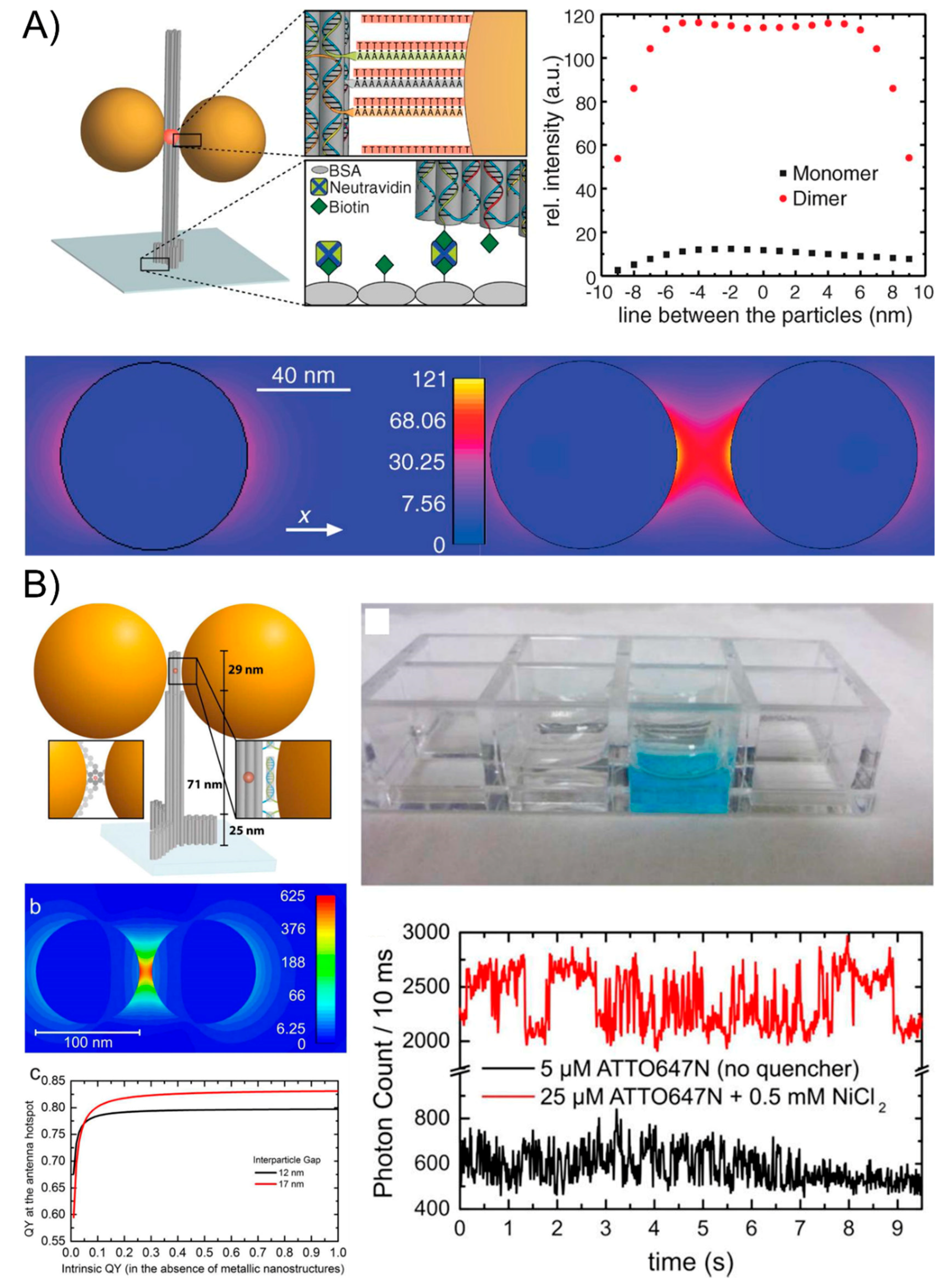
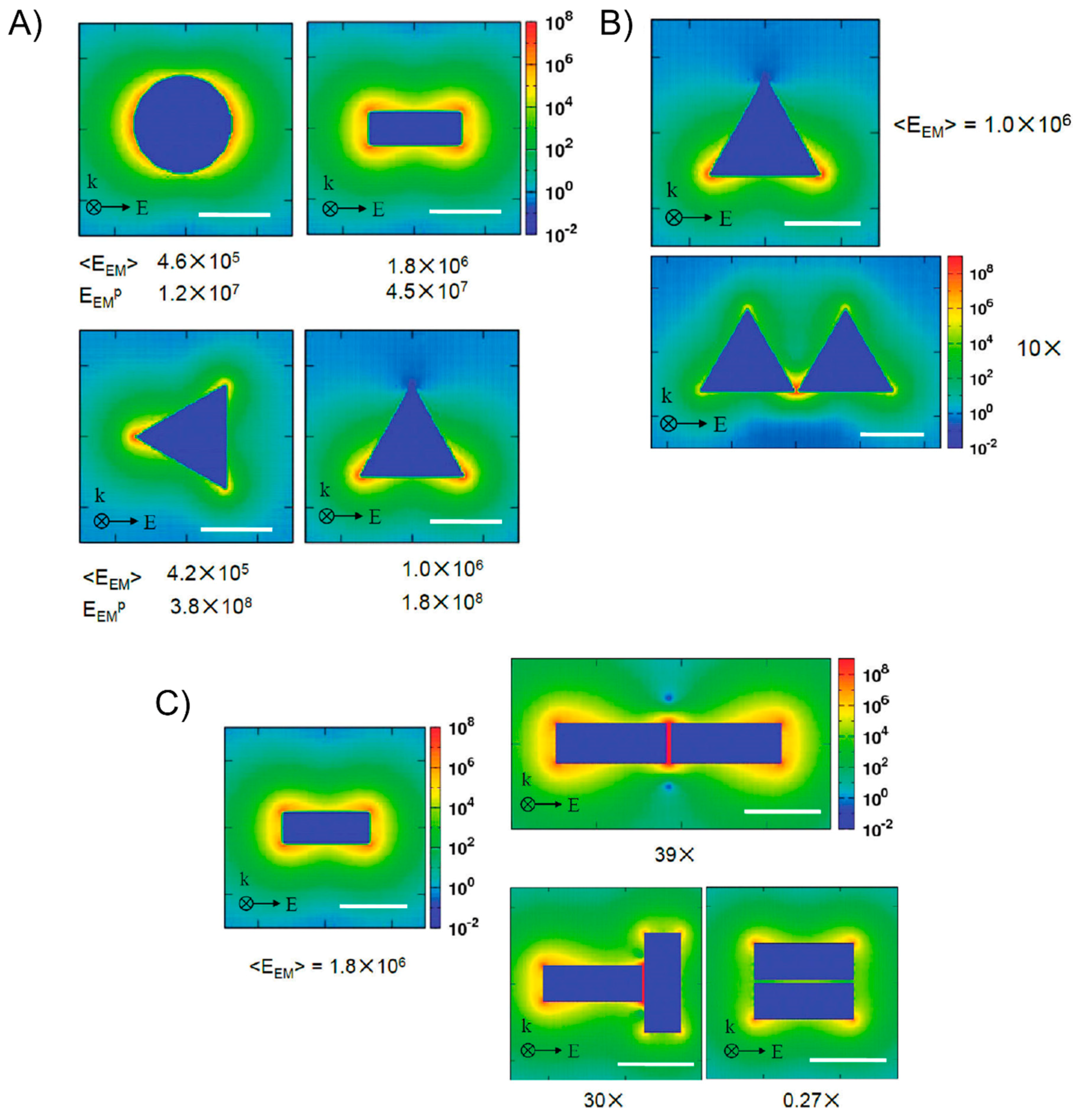
6. The Surface-Enhanced Fluorescence of DNA-Directed AuNP Nanoarchitectures
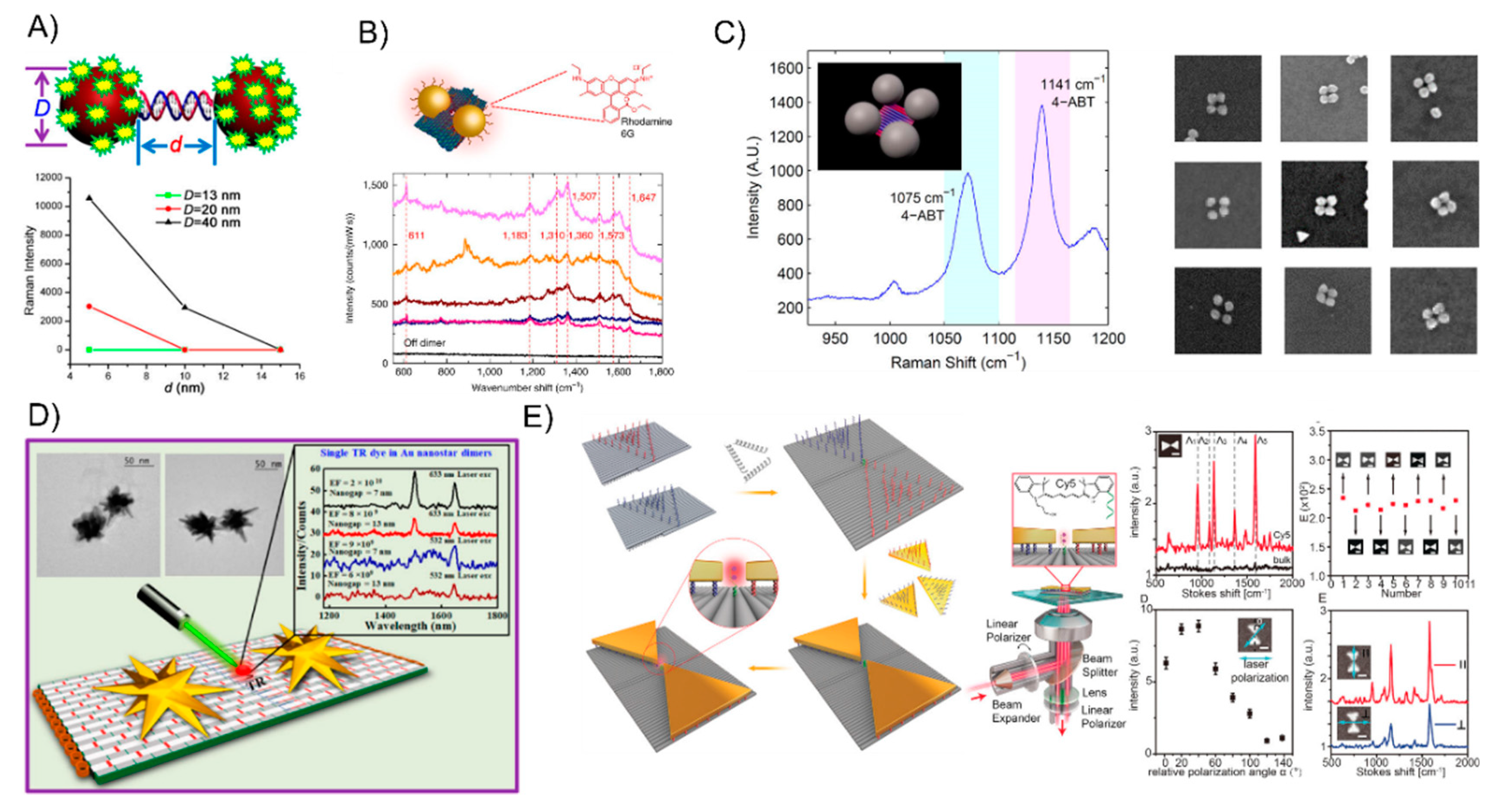
7. The Surface-Enhanced Raman Scattering of DNA-Directed AuNP Nanoarchitectures
8. Challenges and Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nie, Z.; Petukhova, A.; Kumacheva, E. Properties and emerging applications of self-assembled structures made from inorganic nanoparticles. Nat. Nanotechnol. 2010, 5, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Frens, G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nat. Phys. Sci. 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Jana, N.R.; Gearheart, L.; Murphy, C.J. Seeding growth for size control of 5–40 nm diameter gold nanoparticles. Langmuir 2001, 17, 6782–6786. [Google Scholar] [CrossRef]
- Hussain, I.; Graham, S.; Wang, Z.; Tan, B.; Sherrington, D.C.; Rannard, S.P.; Cooper, A.I.; Brust, M. Size-controlled synthesis of near-monodisperse gold nanoparticles in the 1–4 nm range using polymeric stabilizers. J. Am. Chem. Soc. 2005, 127, 16398–16399. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Fernández, J.; Pérez-Juste, J.; García de Abajo, F.J.; Liz-Marzán, L.M. Seeded growth of submicron Au colloids with quadrupole plasmon resonance modes. Langmuir 2006, 22, 7007–7010. [Google Scholar] [CrossRef] [PubMed]
- Perrault, S.D.; Chan, W.C. Synthesis and surface modification of highly monodispersed, spherical gold nanoparticles of 50–200 nm. J. Am. Chem. Soc. 2009, 131, 17042–17043. [Google Scholar] [CrossRef] [PubMed]
- Haiss, W.; Thanh, N.T.; Aveyard, J.; Fernig, D.G. Determination of size and concentration of gold nanoparticles from UV–Vis spectra. Anal. Chem. 2007, 79, 4215–4221. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-Y.; Chang, S.-S.; Lee, C.-L.; Wang, C.C. Gold nanorods: Electrochemical synthesis and optical properties. J. Phys. Chem. B 1997, 101, 6661–6664. [Google Scholar] [CrossRef]
- Jana, N.R.; Gearheart, L.; Murphy, C.J. Wet chemical synthesis of high aspect ratio cylindrical gold nanorods. J. Phys. Chem. B 2001, 105, 4065–4067. [Google Scholar] [CrossRef]
- Jana, N.R.; Gearheart, L.; Murphy, C.J. Seed-mediated growth approach for shape-controlled synthesis of spheroidal and rod-like gold nanoparticles using a surfactant template. Adv. Mater. 2001, 13, 1389–1393. [Google Scholar] [CrossRef]
- Jana, N.R.; Gearheart, L.; Obare, S.O.; Murphy, C.J. Anisotropic chemical reactivity of gold spheroids and nanorods. Langmuir 2002, 18, 922–927. [Google Scholar] [CrossRef]
- Nikoobakht, B.; El-Sayed, M.A. Preparation and growth mechanism of gold nanorods (NRs) using seed-mediated growth method. Chem. Mater. 2003, 15, 1957–1962. [Google Scholar] [CrossRef]
- Sun, Y.; Xia, Y. Shape-controlled synthesis of gold and silver nanoparticles. Science 2002, 298, 2176–2179. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kou, X.; Yang, Z.; Ni, W.; Wang, J. Shape-and size-dependent refractive index sensitivity of gold nanoparticles. Langmuir 2008, 24, 5233–5237. [Google Scholar] [CrossRef] [PubMed]
- Hanarp, P.; Käll, M.; Sutherland, D.S. Optical properties of short range ordered arrays of nanometer gold disks prepared by colloidal lithography. J. Phys. Chem. B 2003, 107, 5768–5772. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, C.; Zhu, Y.; Chen, Z. A novel ultraviolet irradiation technique for shape-controlled synthesis of gold nanoparticles at room temperature. Chem. Mater. 1999, 11, 2310–2312. [Google Scholar] [CrossRef]
- Hao, F.; Nehl, C.L.; Hafner, J.H.; Nordlander, P. Plasmon resonances of a gold nanostar. Nano Lett. 2007, 7, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Maye, M.M.; Luo, J.; Lim, I.-I.S.; Han, L.; Kariuki, N.N.; Rabinovich, D.; Liu, T.; Zhong, C.-J. Size-controlled assembly of gold nanoparticles induced by a tridentate thioether ligand. J. Am. Chem. Soc. 2003, 125, 9906–9907. [Google Scholar] [CrossRef] [PubMed]
- Mendes, P.M.; Jacke, S.; Critchley, K.; Plaza, J.; Chen, Y.; Nikitin, K.; Palmer, R.E.; Preece, J.A.; Evans, S.D.; Fitzmaurice, D. Gold nanoparticle patterning of silicon wafers using chemical e-beam lithography. Langmuir 2004, 20, 3766–3768. [Google Scholar] [CrossRef] [PubMed]
- Werts, M.H.; Lambert, M.; Bourgoin, J.-P.; Brust, M. Nanometer scale patterning of Langmuir–Blodgett films of gold nanoparticles by electron beam lithography. Nano Lett. 2002, 2, 43–47. [Google Scholar] [CrossRef]
- Kim, F.; Kwan, S.; Akana, J.; Yang, P. Langmuir–Blodgett nanorod assembly. J. Am. Chem. Soc. 2001, 123, 4360–4361. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, P.; Baumberg, J.; Birkin, P.R.; Ghanem, M.; Netti, M. Highly ordered macroporous gold and platinum films formed by electrochemical deposition through templates assembled from submicron diameter monodisperse polystyrene spheres. Chem. Mater. 2002, 14, 2199–2208. [Google Scholar] [CrossRef]
- Schmid, G.; Bäumle, M.; Beyer, N. Ordered Two-Dimensional Monolayers of Au55 Clusters. Angew. Chem. Int. Ed. 2000, 39, 181–183. [Google Scholar] [CrossRef]
- Wilkins, M.H.F.; Stokes, A.R.; Wilson, H.R. Molecular structure of nucleic acids: Molecular structure of deoxypentose nucleic acids. Nature 1953, 171, 738–740. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. Nucleic acid junctions and lattices. J. Theor. Biol. 1982, 99, 237–247. [Google Scholar] [CrossRef]
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Alivisatos, A.P.; Johnsson, K.P.; Peng, X.; Wilson, T.E.; Loweth, C.J.; Bruchez, M.P., Jr.; Schultz, P.G. Organization of ‘nanocrystal molecules’ using DNA. Nature 1996, 382, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, P.W. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Dietz, H.; Liedl, T.; Högberg, B.; Graf, F.; Shih, W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 2009, 459, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Seeman, N.C.; Mirkin, C.A. Programmable materials and the nature of the DNA bond. Science 2015, 347, 1260901. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, A.V.; Han, D.; Shih, W.M.; Yan, H. Challenges and opportunities for structural DNA nanotechnology. Nat. Nanotechnol. 2011, 6, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.J.; Campolongo, M.J.; Luo, D.; Cheng, W. Building plasmonic nanostructures with DNA. Nat. Nanotechnol. 2011, 6, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Macfarlane, R.J.; Lee, B.; Zhang, J.; Young, K.L.; Senesi, A.J.; Mirkin, C.A. DNA-nanoparticle superlattices formed from anisotropic building blocks. Nat. Mater. 2010, 9, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Wilner, O.I.; Willner, I. Functionalized DNA nanostructures. Chem. Rev. 2012, 112, 2528–2556. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Zuo, X.; Zhu, D.; Huang, Q.; Fan, C. Functional DNA nanostructures for theranostic applications. Acc. Chem. Res. 2013, 47, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Kallenbach, N.R.; Ma, R.-I.; Seeman, N.C. An immobile nucleic acid junction constructed from oligonucleotides. Nature 1983, 305, 829–831. [Google Scholar] [CrossRef]
- Chen, J.; Seeman, N.C. Synthesis from DNA of a molecule with the connectivity of a cube. Nature 1991, 350, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.J.; Seeman, N.C. DNA double-crossover molecules. Biochemistry 1993, 32, 3211–3220. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, X.; Qi, J.; Seeman, N.C. Antiparallel DNA double crossover molecules as components for nanoconstruction. J. Am. Chem. Soc. 1996, 118, 6131–6140. [Google Scholar] [CrossRef]
- Mao, C.; LaBean, T.H.; Reif, J.H.; Seeman, N.C. Logical computation using algorithmic self-assembly of DNA triple-crossover molecules. Nature 2000, 407, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Winfree, E.; Liu, F.; Wenzler, L.A.; Seeman, N.C. Design and self-assembly of two-dimensional DNA crystals. Nature 1998, 394, 539–544. [Google Scholar] [CrossRef] [PubMed]
- LaBean, T.H.; Yan, H.; Kopatsch, J.; Liu, F.; Winfree, E.; Reif, J.H.; Seeman, N.C. Construction, analysis, ligation, and self-assembly of DNA triple crossover complexes. J. Am. Chem. Soc. 2000, 122, 1848–1860. [Google Scholar] [CrossRef]
- He, Y.; Chen, Y.; Liu, H.; Ribbe, A.E.; Mao, C. Self-assembly of hexagonal DNA two-dimensional (2D) arrays. J. Am. Chem. Soc. 2005, 127, 12202–12203. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Tian, Y.; Ribbe, A.E.; Mao, C. Highly connected two-dimensional crystals of DNA six-point-stars. J. Am. Chem. Soc. 2006, 128, 15978–15979. [Google Scholar] [CrossRef] [PubMed]
- Bidault, S.; García de Abajo, F.J.; Polman, A. Plasmon-based nanolenses assembled on a well-defined DNA template. J. Am. Chem. Soc. 2008, 130, 2750–2751. [Google Scholar] [CrossRef] [PubMed]
- Busson, M.P.; Rolly, B.; Stout, B.; Bonod, N.; Larquet, E.; Polman, A.; Bidault, S. Optical and topological characterization of gold nanoparticle dimers linked by a single DNA double strand. Nano Lett. 2011, 11, 5060–5065. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Dong, Y.; Sun, Y.; Chen, P.; Yang, Y.; Zhou, C.; Xu, L.; Yang, Z.; Liu, D. DNA bimodified gold nanoparticles. Langmuir 2011, 28, 1966–1970. [Google Scholar] [CrossRef] [PubMed]
- Lermusiaux, L.; Sereda, A.; Portier, B.; Larquet, E.; Bidault, S. Reversible Switching of the Interparticle Distance in DNA-Templated Gold Nanoparticle Dimers. ACS Nano 2012, 6, 10992–10998. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; McLaughlin, C.K.; Lo, P.K.; Yang, H.; Sleiman, H.F. Stable gold nanoparticle conjugation to internal DNA positions: Facile generation of discrete gold nanoparticle–DNA assemblies. Bioconjug. Chem. 2010, 21, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Chhabra, R.; Andersen, C.S.; Gothelf, K.V.; Yan, H.; Liu, Y. Toward Reliable Gold Nanoparticle Patterning On Self-Assembled DNA Nanoscaffold. J. Am. Chem. Soc. 2008, 130, 7820–7821. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Deng, Z.; Yan, H.; Cabrini, S.; Zuckermann, R.N.; Bokor, J. Gold Nanoparticle Self-Similar Chain Structure Organized by DNA Origami. J. Am. Chem. Soc. 2010, 132, 3248–3249. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Deng, Z.; Wang, H.; Zou, S.; Liu, Y.; Yan, H. DNA directed self-assembly of anisotropic plasmonic nanostructures. J. Am. Chem. Soc. 2011, 133, 17606–17609. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Lan, X.; Lu, X.; Meyer, T.A.; Ni, W.; Ke, Y.; Wang, Q. Site-Specific Surface Functionalization of Gold Nanorods Using DNA Origami Clamps. J. Am. Chem. Soc. 2016, 138, 1764–1767. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, M.; Dong, J.; Zhou, C.; Wang, Q. Modular Assembly of Plasmonic Nanoparticles Assisted by DNA Origami. Langmuir 2018. [Google Scholar] [CrossRef] [PubMed]
- Halas, N.J.; Lal, S.; Chang, W.-S.; Link, S.; Nordlander, P. Plasmons in strongly coupled metallic nanostructures. Chem. Rev. 2011, 111, 3913–3961. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, S.; Haldar, K.K.; Sen, T. DNA Origami Directed Au Nanostar Dimers for Single-Molecule Surface-Enhanced Raman Scattering. J. Am. Chem. Soc. 2017, 139, 17639–17648. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Wen, T.; Wang, Z.G.; He, Y.; Shi, J.; Wang, T.; Liu, X.; Lu, G.; Ding, B. DNA Origami Directed Assembly of Gold Bowtie Nanoantennas for Single-Molecule Surface-Enhanced Raman Scattering. Angew. Chem. Int. Ed. 2018, 57, 2846–2850. [Google Scholar] [CrossRef] [PubMed]
- Krajcar, R.; Siegel, J.; Lyutakov, O.; Slepička, P.; Švorčík, V. Optical response of anisotropic silver nanostructures on polarized light. Mater Lett. 2014, 137, 72–74. [Google Scholar] [CrossRef]
- Krajcar, R.; Siegel, J.; Slepička, P.; Fitl, P.; Švorčík, V. Silver nanowires prepared on PET structured by laser irradiation. Mater Lett. 2014, 117, 184–187. [Google Scholar] [CrossRef]
- Barb, R.-A.; Hrelescu, C.; Dong, L.; Heitz, J.; Siegel, J.; Slepicka, P.; Vosmanska, V.; Svorcik, V.; Magnus, B.; Marksteiner, R. Laser-induced periodic surface structures on polymers for formation of gold nanowires and activation of human cells. Appl. Phys. A 2014, 117, 295–300. [Google Scholar] [CrossRef]
- Wei, Q.-H.; Su, K.-H.; Durant, S.; Zhang, X. Plasmon resonance of finite one-dimensional Au nanoparticle chains. Nano Lett. 2004, 4, 1067–1071. [Google Scholar] [CrossRef]
- Su, K.-H.; Wei, Q.-H.; Zhang, X.; Mock, J.; Smith, D.R.; Schultz, S. Interparticle coupling effects on plasmon resonances of nanogold particles. Nano Lett. 2003, 3, 1087–1090. [Google Scholar] [CrossRef]
- Reinhard, B.M.; Siu, M.; Agarwal, H.; Alivisatos, A.P.; Liphardt, J. Calibration of dynamic molecular rulers based on plasmon coupling between gold nanoparticles. Nano Lett. 2005, 5, 2246–2252. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Chen, Z.; Liu, B.J.; Ren, B.; Henzie, J.; Wang, Q. DNA-Directed Gold Nanodimers with Tunable Sizes and Interparticle Distances and Their Surface Plasmonic Properties. Small 2013, 9, 2308–2315. [Google Scholar] [CrossRef] [PubMed]
- Plum, E.; Zhou, J.; Dong, J.; Fedotov, V.; Koschny, T.; Soukoulis, C.; Zheludev, N. Metamaterial with negative index due to chirality. Phys. Rev. B 2009, 79, 035407. [Google Scholar] [CrossRef]
- Gansel, J.K.; Thiel, M.; Rill, M.S.; Decker, M.; Bade, K.; Saile, V.; von Freymann, G.; Linden, S.; Wegener, M. Gold helix photonic metamaterial as broadband circular polarizer. Science 2009, 325, 1513–1515. [Google Scholar] [CrossRef]
- Hendry, E.; Carpy, T.; Johnston, J.; Popland, M.; Mikhaylovskiy, R.; Lapthorn, A.; Kelly, S.; Barron, L.; Gadegaard, N.; Kadodwala, M. Ultrasensitive detection and characterization of biomolecules using superchiral fields. Nat. Nanotechnol. 2010, 5, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xu, L.; Liu, L.; Ma, W.; Yin, H.; Kuang, H.; Wang, L.; Xu, C.; Kotov, N.A. Unexpected chirality of nanoparticle dimers and ultrasensitive chiroplasmonic bioanalysis. J. Am. Chem. Soc. 2013, 135, 18629–18636. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Wang, Q. Self-Assembly of Chiral Plasmonic Nanostructures. Adv. Mater. 2016, 28, 10499–10507. [Google Scholar] [CrossRef] [PubMed]
- Mastroianni, A.J.; Claridge, S.A.; Alivisatos, A.P. Pyramidal and chiral groupings of gold nanocrystals assembled using DNA scaffolds. J. Am. Chem. Soc. 2009, 131, 8455–8459. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Song, C.; Wang, J.; Shi, D.; Wang, Z.; Liu, N.; Ding, B. Rolling Up Gold Nanoparticle-Dressed DNA Origami into Three-Dimensional Plasmonic Chiral Nanostructures. J. Am. Chem. Soc. 2012, 134, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Asenjo-Garcia, A.; Liu, Q.; Jiang, Q.; García de Abajo, F.J.; Liu, N.; Ding, B. Three-Dimensional Plasmonic Chiral Tetramers Assembled by DNA Origami. Nano Lett. 2013, 13, 2128–2133. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Lu, X.; Chen, Z.; Meng, C.; Ni, W.; Wang, Q. DNA Origami-Directed, Discrete Three-Dimensional Plasmonic Tetrahedron Nanoarchitectures with Tailored Optical Chirality. ACS Appl. Mater. Interfaces 2014, 6, 5388–5392. [Google Scholar] [CrossRef] [PubMed]
- Kuzyk, A.; Schreiber, R.; Fan, Z.; Pardatscher, G.; Roller, E.-M.; Högele, A.; Simmel, F.C.; Govorov, A.O.; Liedl, T. DNA-based self-assembly of chiral plasmonic nanostructures with tailored optical response. Nature 2012, 483, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Chen, Z.; Dai, G.; Lu, X.; Ni, W.; Wang, Q. Bifacial DNA origami-directed discrete, three-dimensional, anisotropic plasmonic nanoarchitectures with tailored optical chirality. J. Am. Chem. Soc. 2013, 135, 11441–11444. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Lu, X.; Shen, C.; Ke, Y.; Ni, W.; Wang, Q. Au nanorod helical superstructures with designed chirality. J. Am. Chem. Soc. 2014, 137, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Su, Z.; Zhou, Y.; Meyer, T.; Ke, Y.; Wang, Q.; Chiu, W.; Liu, N.; Zou, S.; Yan, H. Programmable Supra-Assembly of a DNA Surface Adapter for Tunable Chiral Directional Self-Assembly of Gold Nanorods. Angew. Chem. Int. Ed. 2017, 129, 14824–14828. [Google Scholar] [CrossRef]
- Chen, Z.; Lan, X.; Wang, Q. DNA Origami Directed Large-Scale Fabrication of Nanostructures Resembling Room Temperature Single-Electron Transistors. Small 2013, 9, 3567–3571. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lan, X.; Chiu, Y.-C.; Lu, X.; Ni, W.; Gao, H.; Wang, Q. Strong chiroptical activities in gold nanorod dimers assembled using DNA origami templates. ACS Photonics 2015, 2, 392–397. [Google Scholar] [CrossRef]
- Shen, C.; Lan, X.; Lu, X.; Ni, W.; Wang, Q. Tuning the structural asymmetries of three-dimensional gold nanorod assemblies. Chem. Commun. 2015, 51, 13627–13629. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Liu, T.; Wang, Z.; Govorov, A.O.; Yan, H.; Liu, Y. DNA-Guided Plasmonic Helix with Switchable Chirality. J. Am. Chem. Soc. 2018, 140, 11763–11770. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Liu, Q.; Shi, Y.; Wang, Z.-G.; Zhan, P.; Liu, J.; Liu, C.; Wang, H.; Shi, X.; Zhang, L. Stimulus-Responsive Plasmonic Chiral Signals of Gold Nanorods Organized on DNA Origami. Nano Lett. 2017, 17, 7125–7130. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Choi, C.K.K.; Wang, Q. Origin of the Plasmonic Chirality of Gold Nanorod Trimers Templated by DNA Origami. ACS Appl. Mater. Interfaces 2018, 10, 26835–26840. [Google Scholar] [CrossRef] [PubMed]
- Dulkeith, E.; Ringler, M.; Klar, T.; Feldmann, J.; Munoz Javier, A.; Parak, W. Gold nanoparticles quench fluorescence by phase induced radiative rate suppression. Nano Lett. 2005, 5, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Jennings, T.; Singh, M.; Strouse, G. Fluorescent lifetime quenching near d = 1.5 nm gold nanoparticles: Probing NSET validity. J. Am. Chem. Soc. 2006, 128, 5462–5467. [Google Scholar] [CrossRef] [PubMed]
- Acuna, G.P.; Bucher, M.; Stein, I.H.; Steinhauer, C.; Kuzyk, A.; Holzmeister, P.; Schreiber, R.; Moroz, A.; Stefani, F.D.; Liedl, T. Distance dependence of single-fluorophore quenching by gold nanoparticles studied on DNA origami. ACS Nano 2012, 6, 3189–3195. [Google Scholar] [CrossRef] [PubMed]
- Anger, P.; Bharadwaj, P.; Novotny, L. Enhancement and quenching of single-molecule fluorescence. Phys. Rev. Lett. 2006, 96, 113002. [Google Scholar] [CrossRef] [PubMed]
- Kühn, S.; Håkanson, U.; Rogobete, L.; Sandoghdar, V. Enhancement of single-molecule fluorescence using a gold nanoparticle as an optical nanoantenna. Phys. Rev. Lett. 2006, 97, 017402. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Khatua, S.; Zijlstra, P.; Yorulmaz, M.; Orrit, M. Thousand-fold enhancement of single-molecule fluorescence near a single gold nanorod. Angew. Chem. Int. Ed. 2013, 52, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Taminiau, T.; Stefani, F.; Segerink, F.B.; Van Hulst, N. Optical antennas direct single-molecule emission. Nat. Photonics 2008, 2, 234–237. [Google Scholar] [CrossRef]
- Curto, A.G.; Volpe, G.; Taminiau, T.H.; Kreuzer, M.P.; Quidant, R.; van Hulst, N.F. Unidirectional emission of a quantum dot coupled to a nanoantenna. Science 2010, 329, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Pellegrotti, J.V.; Acuna, G.P.; Puchkova, A.; Holzmeister, P.; Gietl, A.; Lalkens, B.; Stefani, F.D.; Tinnefeld, P. Controlled reduction of photobleaching in DNA origami–gold nanoparticle hybrids. Nano Lett. 2014, 14, 2831–2836. [Google Scholar] [CrossRef] [PubMed]
- Vasilev, K.; Stefani, F.D.; Jacobsen, V.; Knoll, W.; Kreiter, M. Reduced photobleaching of chromophores close to a metal surface. J. Chem. Phys. 2004, 120, 6701–6704. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Zhong, Y.; Ming, T.; Wang, J.; Wong, K.S. Extraordinary surface plasmon coupled emission using core/shell gold nanorods. J. Phys. Chem. C 2012, 116, 9259–9264. [Google Scholar] [CrossRef]
- Le Ru, E.; Etchegoin, P.; Grand, J.; Felidj, N.; Aubard, J.; Levi, G. Mechanisms of spectral profile modification in surface-enhanced fluorescence. J. Phys. Chem. C 2007, 111, 16076–16079. [Google Scholar] [CrossRef]
- Schuller, J.A.; Barnard, E.S.; Cai, W.; Jun, Y.C.; White, J.S.; Brongersma, M.L. Plasmonics for extreme light concentration and manipulation. Nat. Mater. 2010, 9, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Novotny, L.; Van Hulst, N. Antennas for light. Nat. Photonics 2011, 5, 83–90. [Google Scholar] [CrossRef]
- Acuna, G.; Möller, F.; Holzmeister, P.; Beater, S.; Lalkens, B.; Tinnefeld, P. Fluorescence enhancement at docking sites of DNA-directed self-assembled nanoantennas. Science 2012, 338, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Puchkova, A.; Vietz, C.; Pibiri, E.; Wünsch, B.; Sanz Paz, M.A.; Acuna, G.P.; Tinnefeld, P. DNA origami nanoantennas with over 5000-fold fluorescence enhancement and single-molecule detection at 25 μM. Nano Lett. 2015, 15, 8354–8359. [Google Scholar] [CrossRef] [PubMed]
- Jeanmaire, D.L.; Van Duyne, R.P. Surface Raman spectroelectrochemistry: Part I. Heterocyclic, aromatic, and aliphatic amines adsorbed on the anodized silver electrode. J. Electroanal. Chem. Interfacial Electrochem. 1977, 84, 1–20. [Google Scholar] [CrossRef]
- Camden, J.P.; Dieringer, J.A.; Wang, Y.; Masiello, D.J.; Marks, L.D.; Schatz, G.C.; Van Duyne, R.P. Probing the structure of single-molecule surface-enhanced Raman scattering hot spots. J. Am. Chem. Soc. 2008, 130, 12616–12617. [Google Scholar] [CrossRef] [PubMed]
- Michaels, A.M.; Jiang, J.; Brus, L. Ag nanocrystal junctions as the site for surface-enhanced Raman scattering of single rhodamine 6G molecules. J. Phys. Chem. B 2000, 104, 11965–11971. [Google Scholar] [CrossRef]
- Wustholz, K.L.; Henry, A.-I.; McMahon, J.M.; Freeman, R.G.; Valley, N.; Piotti, M.E.; Natan, M.J.; Schatz, G.C.; Van Duyne, R.P. Structure−Activity Relationships in Gold Nanoparticle Dimers and Trimers for Surface-Enhanced Raman Spectroscopy. J. Am. Chem. Soc. 2010, 132, 10903–10910. [Google Scholar] [CrossRef] [PubMed]
- McMahon, J.M.; Li, S.; Ausman, L.K.; Schatz, G.C. Modeling the effect of small gaps in surface-enhanced Raman spectroscopy. J. Phys. Chem. C 2011, 116, 1627–1637. [Google Scholar] [CrossRef]
- Klar, T.; Perner, M.; Grosse, S.; Von Plessen, G.; Spirkl, W.; Feldmann, J. Surface-plasmon resonances in single metallic nanoparticles. Phys. Rev. Lett. 1998, 80, 4249. [Google Scholar] [CrossRef]
- Jain, P.K.; El-Sayed, M.A. Plasmonic coupling in noble metal nanostructures. Chem. Phys. Lett. 2010, 487, 153–164. [Google Scholar] [CrossRef]
- Lan, X.; Chen, Z.; Lu, X.; Dai, G.; Ni, W.; Wang, Q. DNA-Directed Gold Nanodimers with Tailored Ensemble Surface-Enhanced Raman Scattering Properties. ACS Appl. Mater. Interfaces 2013, 5, 10423–10427. [Google Scholar] [CrossRef] [PubMed]
- Kühler, P.; Roller, E.-M.; Schreiber, R.; Liedl, T.; Lohmüller, T.; Feldmann, J. Plasmonic DNA-origami nanoantennas for surface-enhanced Raman spectroscopy. Nano Lett. 2014, 14, 2914–2919. [Google Scholar] [CrossRef] [PubMed]
- Thacker, V.V.; Herrmann, L.O.; Sigle, D.O.; Zhang, T.; Liedl, T.; Baumberg, J.J.; Keyser, U.F. DNA origami based assembly of gold nanoparticle dimers for surface-enhanced Raman scattering. Nat. Commun. 2014, 5, 3448. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-Z.; Wang, X.; Xing, Y.-K.; Ren, S.-K.; Teng, N.; Wang, J.; Chao, J.; Wang, L.-H. DNA origami-templated assembly of plasmonic nanostructures with enhanced Raman scattering. Nucl. Sci. Tech. 2018, 29, 6. [Google Scholar] [CrossRef]
- Pilo-Pais, M.; Watson, A.; Demers, S.; LaBean, T.; Finkelstein, G. Surface-enhanced Raman scattering plasmonic enhancement using DNA origami-based complex metallic nanostructures. Nano Lett. 2014, 14, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Khoury, C.G.; Vo-Dinh, T. Gold nanostars for surface-enhanced Raman scattering: Synthesis, characterization and optimization. J. Phys. Chem. C 2008, 112, 18849–18859. [Google Scholar] [CrossRef]
- Hrelescu, C.; Sau, T.K.; Rogach, A.L.; Jäckel, F.; Feldmann, J. Single gold nanostars enhance Raman scattering. Appl. Phys. Lett. 2009, 94, 153113. [Google Scholar] [CrossRef]
- Nalbant Esenturk, E.; Hight Walker, A. Surface-enhanced Raman scattering spectroscopy via gold nanostars. J. Raman Spectrosc. 2009, 40, 86–91. [Google Scholar] [CrossRef]
- Scarabelli, L.; Coronado-Puchau, M.; Giner-Casares, J.J.; Langer, J.; Liz-Marzán, L.M. Monodisperse gold nanotriangles: Size control, large-scale self-assembly, and performance in surface-enhanced Raman scattering. ACS Nano 2014, 8, 5833–5842. [Google Scholar] [CrossRef] [PubMed]
- Sönnichsen, C.; Reinhard, B.M.; Liphardt, J.; Alivisatos, A.P. A molecular ruler based on plasmon coupling of single gold and silver nanoparticles. Nat. Biotechnol. 2005, 23, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Do, J.; Roller, E.-M.; Zhang, T.; Schüller, V.J.; Nickels, P.C.; Feldmann, J.; Liedl, T. Hierarchical assembly of metal nanoparticles, quantum dots and organic dyes using DNA origami scaffolds. Nat. Nanotechnol. 2014, 9, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Mangalum, A.; Rahman, M.; Norton, M.L. Site-specific immobilization of single-walled carbon nanotubes onto single and one-dimensional DNA origami. J. Am. Chem. Soc. 2013, 135, 2451–2454. [Google Scholar] [CrossRef] [PubMed]
- Maune, H.T.; Han, S.-P.; Barish, R.D.; Bockrath, M.; Goddard, W.A., III; Rothemund, P.W.; Winfree, E. Self-assembly of carbon nanotubes into two-dimensional geometries using DNA origami templates. Nat. Nanotechnol. 2010, 5, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Barati Farimani, A.; Dibaeinia, P.; Aluru, N.R. DNA Origami–Graphene Hybrid Nanopore for DNA Detection. ACS Appl. Mater. Interfaces 2016, 9, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Gür, F.N.; Schwarz, F.W.; Ye, J.; Diez, S.; Schmidt, T.L. Toward self-assembled plasmonic devices: High-yield arrangement of gold nanoparticles on DNA origami templates. ACS Nano 2016, 10, 5374–5382. [Google Scholar] [CrossRef] [PubMed]
- Bayrak, T.R.; Helmi, S.; Ye, J.; Kauert, D.; Kelling, J.; Schönherr, T.; Weichelt, R.; Erbe, A.; Seidel, R. DNA-mold templated assembly of conductive gold nanowires. Nano Lett. 2018, 18, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mai, B.; Tan, H.; Chen, X. Nucleic acid based nanocomposites and their applications in biomedicine. Compos. Commun. 2018, 10, 194–204. [Google Scholar] [CrossRef]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ou, J.; Tan, H.; Chen, X.; Chen, Z. DNA-Assisted Assembly of Gold Nanostructures and Their Induced Optical Properties. Nanomaterials 2018, 8, 994. https://doi.org/10.3390/nano8120994
Ou J, Tan H, Chen X, Chen Z. DNA-Assisted Assembly of Gold Nanostructures and Their Induced Optical Properties. Nanomaterials. 2018; 8(12):994. https://doi.org/10.3390/nano8120994
Chicago/Turabian StyleOu, Jiemei, Huijun Tan, Xudong Chen, and Zhong Chen. 2018. "DNA-Assisted Assembly of Gold Nanostructures and Their Induced Optical Properties" Nanomaterials 8, no. 12: 994. https://doi.org/10.3390/nano8120994
APA StyleOu, J., Tan, H., Chen, X., & Chen, Z. (2018). DNA-Assisted Assembly of Gold Nanostructures and Their Induced Optical Properties. Nanomaterials, 8(12), 994. https://doi.org/10.3390/nano8120994