PEGylation of Superparamagnetic Iron Oxide Nanoparticles with Self-Organizing Polyacrylate-PEG Brushes for Contrast Enhancement in MRI Diagnosis

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
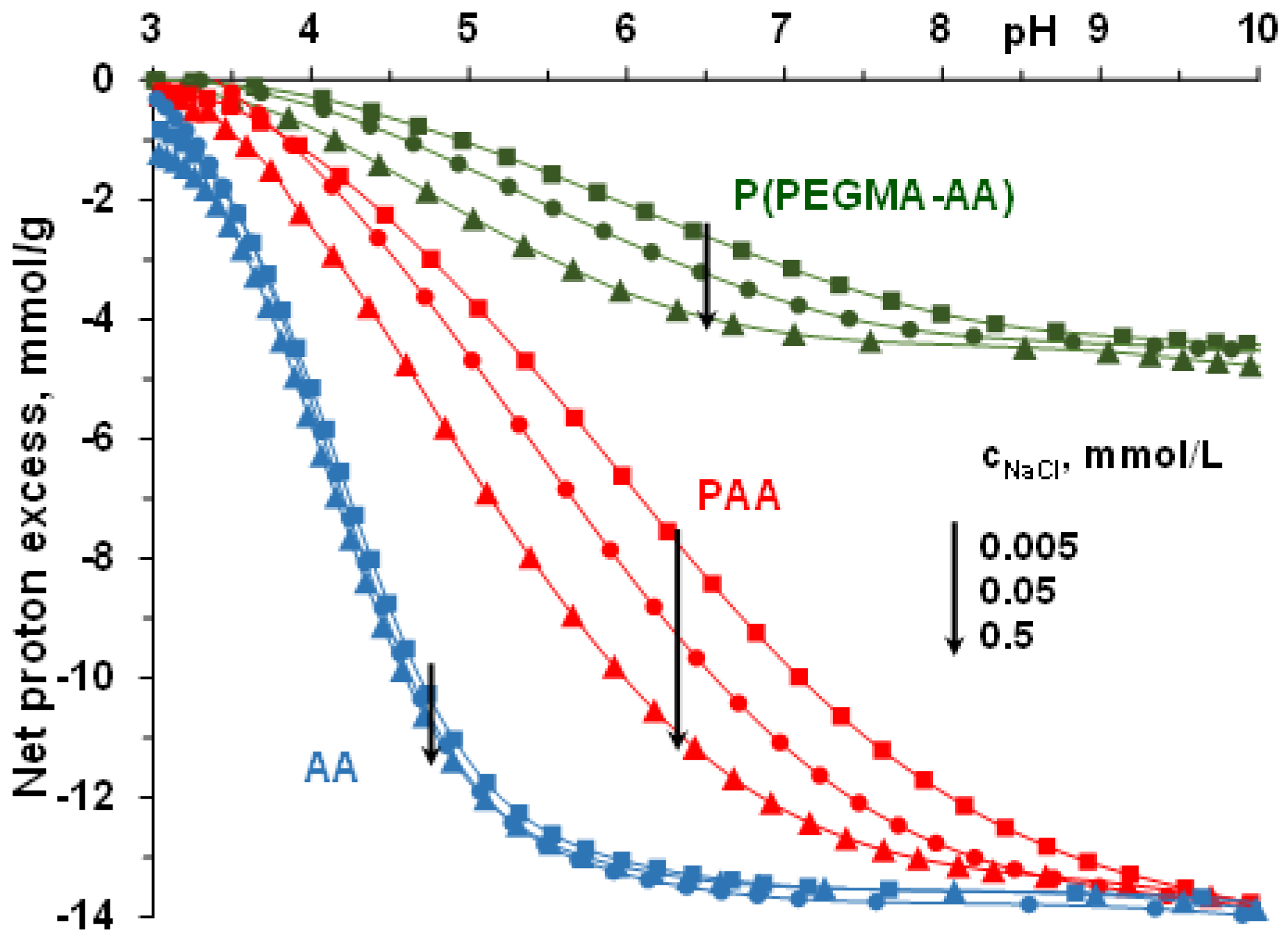
3.1. pH- and Ionic Strength-Dependent Dissociation of P(PEGMA-AA)
3.2. PEGylation of MNPs by Using P(PEGMA-AA)
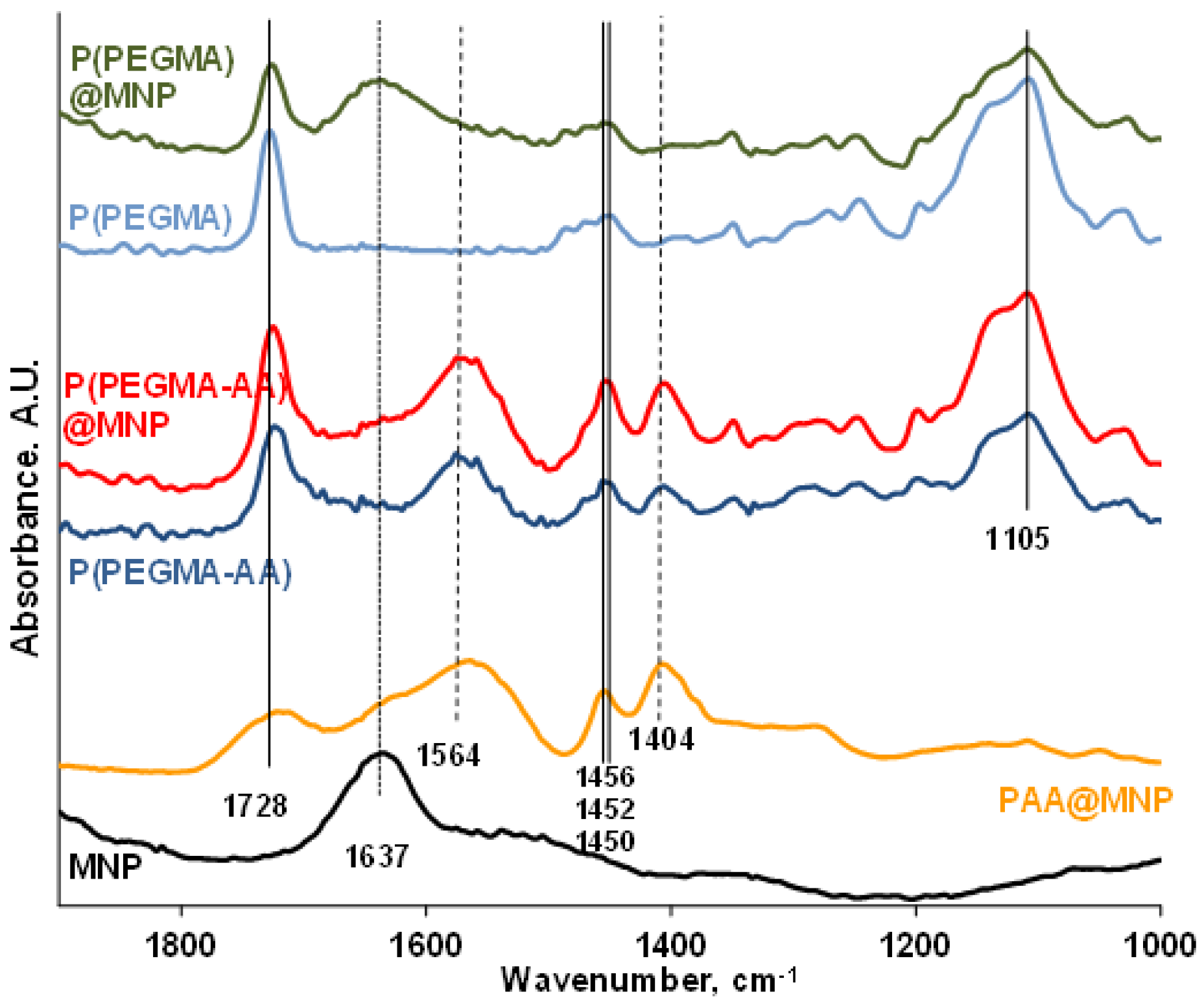
3.3. FT-IR ATR Study of the Binding Chemistry of P(PEGMA-AA) on MNPs
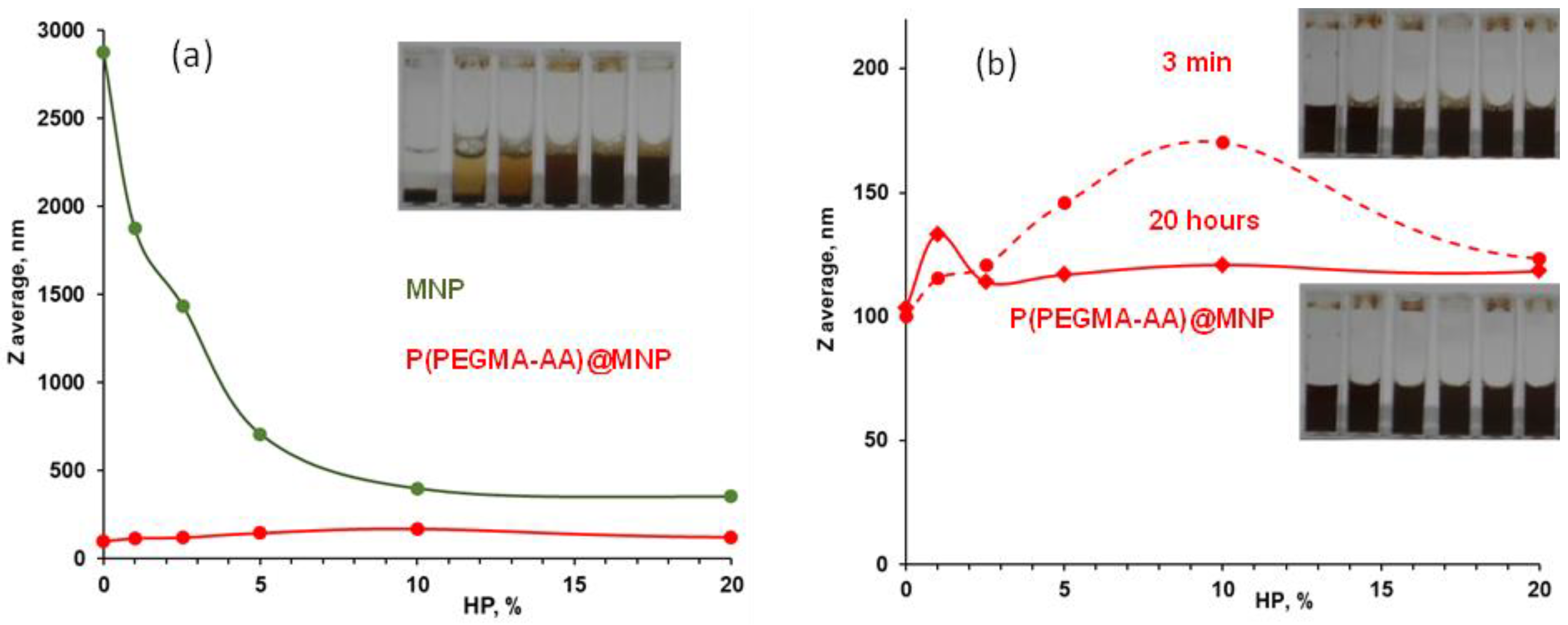
3.4. Evolution of the Colloidal Stability of Core–Shell MNPs
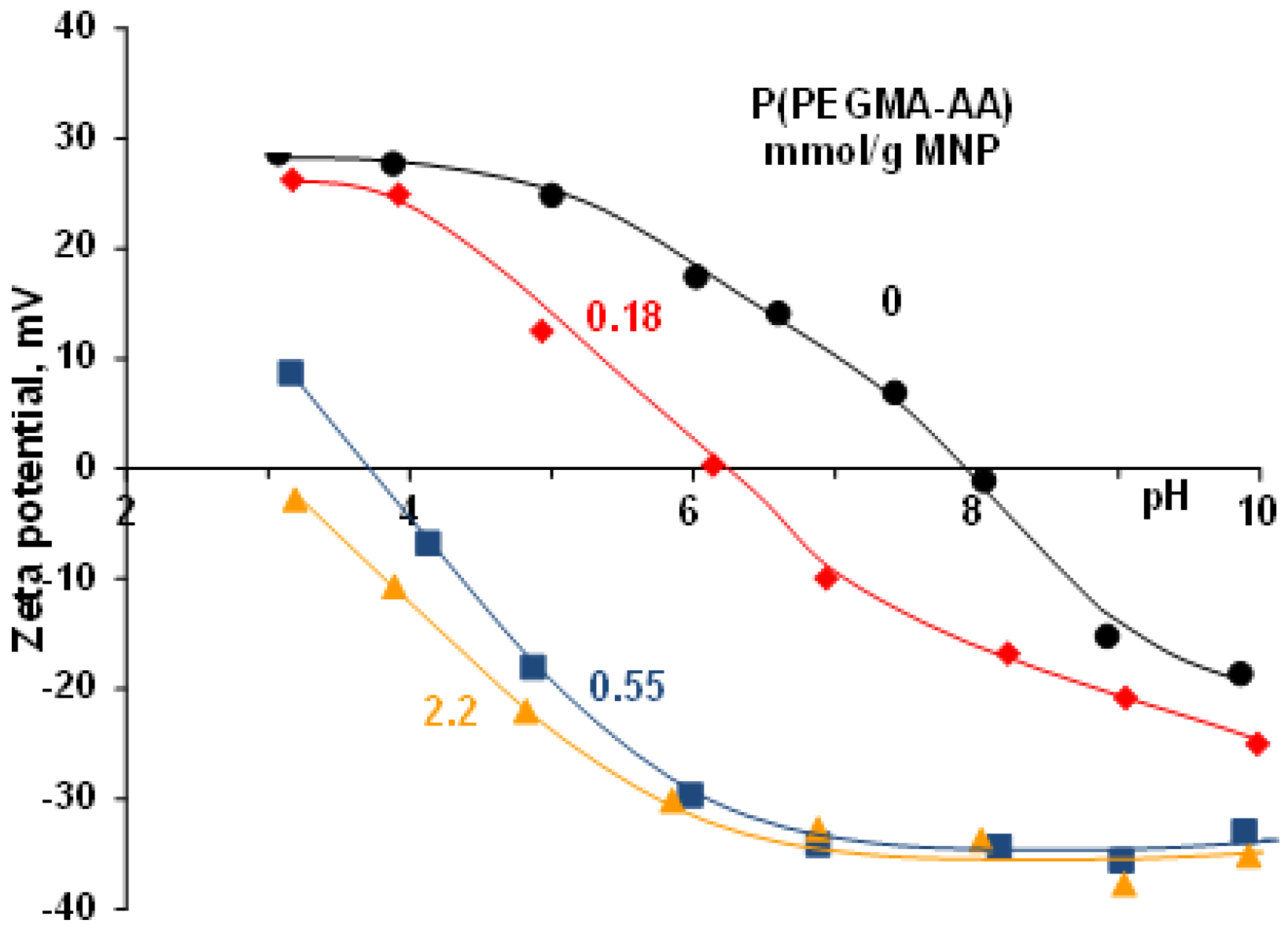
3.5. Salt Tolerance of P(PEGMA-AA)@MNPs
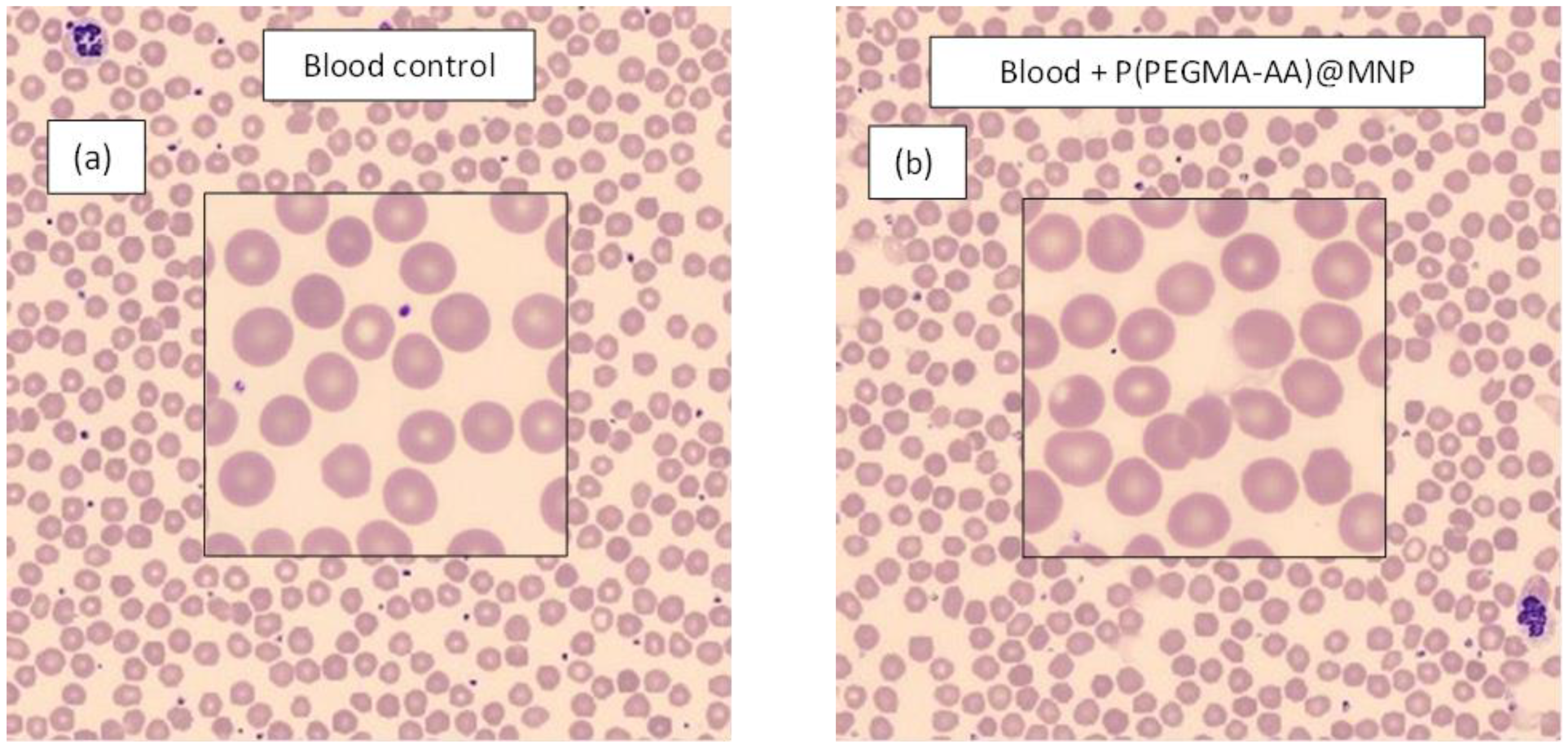
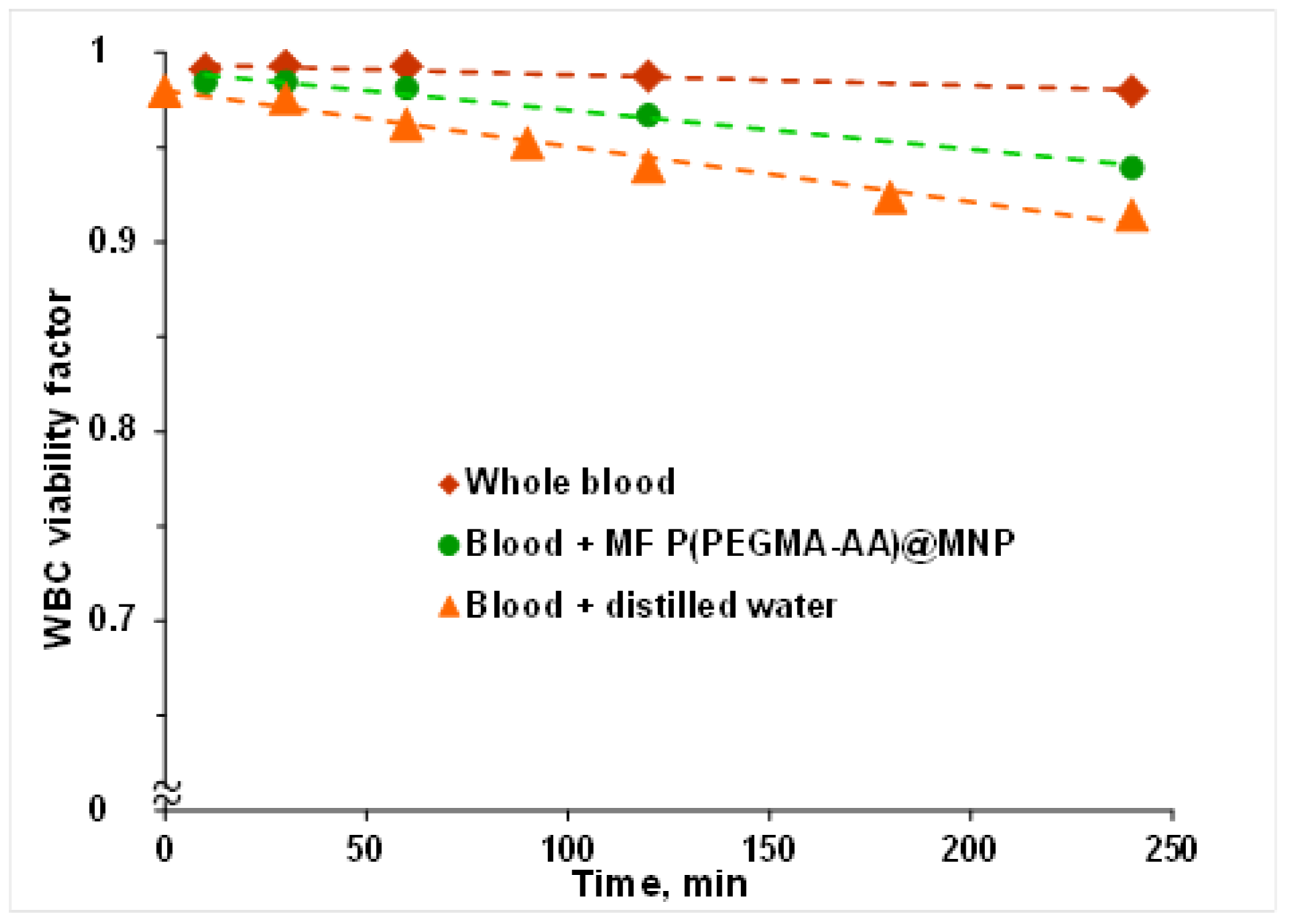
3.6. Hemocompatibility of P(PEGMA-AA)@MNPs
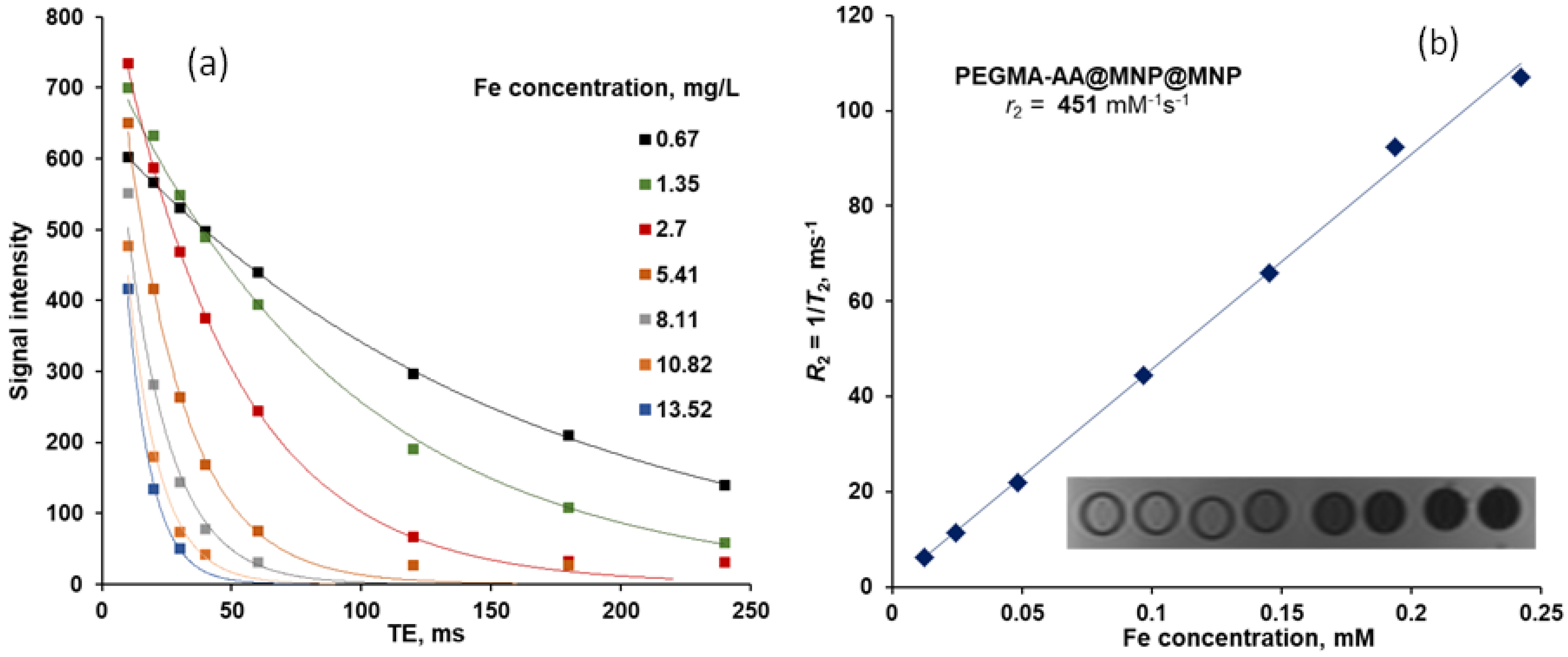
3.7. MRI Contrast Efficiency of P(PEGMA-AA)@MNPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Overchuk, M.; Zheng, G. Overcoming obstacles in the tumor microenvironment: Recent advancements in nanoparticle delivery for cancer theranostics. Biomaterials 2018, 156, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Matusa, M.F.; Vilos, C.; Cisterna, B.A.; Fuentesa, E.; Palomoa, I. Nanotechnology and primary hemostasis: Differential effects of nanoparticles on platelet responses. Vascul. Pharmacol. 2018, 101, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, P.; Subramony, J.A. Nanotherapeutics in oral and parenteral drug delivery: Key learnings and future outlooks as we think small. J. Control. Release 2018, 272, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Aftab, S.; Shah, A.; Nadhman, A.; Kurbanoglu, S.; Ozkan, S.A.; Dionysiou, D.D.; Shukla, S.S.; Aminabhavi, T.M. Nanomedicine: An effective tool in cancer therapy. Int. J. Pharm. 2018, 540, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, S.; Shi, X.; Shen, M. Aqueous-phase synthesis of iron oxide nanoparticles and composites for cancer diagnosis and therapy. Adv. Colloid Interface Sci. 2017, 249, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Pugazhendhi, A.; Edison, T.N.J.I.; Karuppusamy, I.; Kathirvel, B. Inorganic nanoparticles: A potential cancer therapy for human welfare. Int. J. Pharm. 2018, 539, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Kunz-Schughart, L.A.; Dubrovska, A.; Peitzsch, C.; Ewe, A.; Aigner, A.; Schellenburg, S.; Muders, M.H.; Hampel, S.; Cirillo, G.; Iemma, F.; et al. Nanoparticles for radiooncology: Mission, vision, challenges. Biomaterials 2017, 120, 155–184. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Nejadnik, H.; Daldrup-Link, H.E. Next-generation superparamagnetic iron oxide nanoparticles for cancer theranostics. Drug Discov. Today 2017, 22, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Pankhurst, Q.A.; Connolly, J.; Jones, S.K.; Dobson, J. Applications of magnetic nanoparticles in biomedicine. J. Phys. D Appl. Phys. 2003, 36, R167–R181. [Google Scholar] [CrossRef]
- Wahajuddin; Arora, S. Superparamagnetic iron oxide nanoparticles: Magnetic nanoplatforms as drug carriers. Int. J. Nanomed. 2012, 7, 3445–3471. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Eurell, T.; Gunawan, R.; Leckband, D. Chain-length dependence of the protein and cell resistance of oligo (ethylene glycol)-terminated self-assembled monolayers on gold. J. Biomed. Mater. Res. 2001, 56, 406–416. [Google Scholar] [CrossRef]
- Price, M.E.; Cornelius, R.M.; Brash, J.L. Protein adsorption to polyethylene glycol modified liposomes from fibrinogen solution and from plasma. BBA Biomembr. 2001, 1512, 191–205. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Allen, T.M.; Gabizon, A.; Mayhew, E.; Matthay, K.; Huang, S.K.; Lee, K.D.; Woodle, M.C.; Lasic, D.D.; Redemann, C. Sterically stabilized liposomes: Improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. USA 1991, 88, 11460–11464. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, J.; Yang, Y.; Shen, J.; Moten, A.; Chen, C.; Shen, H.; Ferrari, M.; Zhao, Y. The nano-plasma interface: Implications of the protein corona. Colloids Surf. B 2014, 124, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Saei, A.A.; Yazdani, M.; Lohse, S.E.; Bakhtiary, Z.; Serpooshan, V.; Ghavami, M.; Asadian, M.; Mashaghi, S.; Dreaden, E.C.; Mashaghi, A.; et al. Nanoparticle surface functionality dictates cellular and systemic toxicity. Chem. Mater. 2017, 29, 6578–6595. [Google Scholar] [CrossRef]
- Xiao, K.; Li, Y.; Luo, J.; Lee, J.S.; Xiao, W.; Gonik, A.M.; Agarwal, R.; Lam, K.S. The effect of surface charge on in vivo biodistribution of PEG oligocholic acid based micellar nanoparticles. Biomaterials 2011, 32, 3435–3446. [Google Scholar] [CrossRef] [PubMed]
- Sadat, S.M.A.; Jahan, S.T.; Haddadi, A. Effects of size and surface charge of polymeric nanoparticles on in vitro and in vivo applications. J. Biomater. Nanobiotechnol. 2016, 7, 91–108. [Google Scholar] [CrossRef]
- Shubayev, V.I.; Pisanic, T.R.; Jin, S. Magnetic nanoparticles for theragnostics. Adv. Drug Deliv. Rev. 2009, 61, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Patel, B.B.; Tiwari, S. Colloidal nanocarriers: A review on formulation technology, types and applications toward targeted drug delivery. Nanomedicine 2010, 6, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Jokerst, J.V.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Nayak, B.; Dey, R.K. PEGylation in anti-cancer therapy: An overview. Asian J. Pharm. Sci. 2016, 11, 337–348. [Google Scholar] [CrossRef]
- Emilsson, G.; Schoch, R.L.; Feuz, L.; Höök, F.; Lim, R.Y.H.; Dahlin, A.B. Strongly stretched protein resistant poly(ethylene glycol) brushes prepared by grafting-to. ACS Appl. Mater. Interfaces 2015, 7, 7505–7515. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.D.; Allen, C.; Doppen, A.-M.; Anantha, M.; Cox, K.A.K.; Gallagher, R.C.; Karlsson, G.; Edwards, K.; Kenner, G.; Samuels, L.; et al. Influence of poly(ethylene glycol) grafting density and polymer length on liposomes: Relating plasma circulation lifetimes to protein binding. Biochim. Biophys. Acta 2007, 1768, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Masoudi, A.; Hosseini, H.R.M.; Shokrgozar, M.A.; Ahmadi, R.; Oghabian, M.A. The effect of poly (ethylene glycol) coating on colloidal stability of superparamagnetic iron oxide nanoparticles as potential MRI contrast agent. Int. J. Pharm. 2012, 433, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Huang, L. Stealth nanoparticles: High density but sheddable PEG is a key for tumor targeting. J. Control. Release 2010, 145, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Sant, S.; Poulin, S.; Hildgen, P. Effect of polymer architecture on surface properties, plasma protein adsorption, and cellular interactions of pegylated nanoparticles. J. Biomed. Mater. Res. A 2008, 78, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Grenier, P.; Mahmoudi, M.; Lima, E.M.; Appel, E.A.; Dormont, F.; Lim, J.-M.; Karnik, R.; Langer, R.; Farokhzad, O.C. Mechanistic understanding of in vivo protein corona formation on polymeric nanoparticles and impact on pharmacokinetics. Nat. Commun. 2017, 8, 777. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, E.; Kim, D.K.; Jang, N.K.; Jeong, Y.Y.; Jon, S. Antibiofouling polymer-coated superparamagnetic iron oxide nanoparticles as potential magnetic resonance contrast agents for in vivo cancer imaging. J. Am. Chem. Soc. 2006, 128, 7383–7389. [Google Scholar] [CrossRef] [PubMed]
- Giamblanco, N.; Marletta, G.; Graillot, A.; Bia, N.; Loubat, C.; Berret, J.-F. Serum protein resistant behavior of multisite-bound poly(ethylene glycol) chains onto iron oxide surfaces. ACS Omega 2017, 2, 1309–1320. [Google Scholar] [CrossRef]
- Illés, E.; Szekeres, M.; Tóth, I.Y.; Szabó, Á.; Iván, B.; Turcu, R.; Vékás, L.; Zupkó, I.; Jaics, G.; Tombácz, E. Multifunctional PEG-carboxylate copolymer coated superparamagnetic iron oxide nanoparticles for biomedical application. J. Magn. Magn. Mater. 2018, 451, 710–720. [Google Scholar] [CrossRef]
- Illés, E.; Tombácz, E.; Szekeres, M.; Tóth, I.Y.; Szabó, Á.; Iván, B. Novel carboxylated PEG-coating on magnetite nanoparticles designed for biomedical applications. J. Magn. Magn. Mater. 2015, 380, 132–139. [Google Scholar] [CrossRef]
- Illés, E.; Tombácz, E. The effect of humic acid adsorption on pH-dependent surface charging and aggregation of magnetite nanoparticles. J. Colloid Interface Sci. 2006, 295, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Tombácz, E.; Illés, E.; Majzik, A.; Hajdú, A.; Rideg, N.; Szekeres, M. Ageing in the inorganic nanoworld: Example of magnetite nanoparticles in aqueous medium. Croat. Chem. Acta 2007, 80, 503–515. [Google Scholar]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003; p. 175. ISBN 3-527-30274-3. [Google Scholar]
- Szekeres, M.; Tombácz, E. Surface charge characterization of metal oxides by potentiometric acid–base titration, revisited theory and experiment. Colloids Surf. A 2012, 414, 302–313. [Google Scholar] [CrossRef]
- Hajdú, A.; Szekeres, M.; Tóth, I.Y.; Bauer, R.A.; Mihály, J.; Zupkó, I.; Tombácz, E. Enhanced stability of polyacrylate-coated magnetite nanoparticles in biorelevant media. Colloids Surf. B 2012, 94, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Tóth, I.Y.; Illés, E.; Bauer, R.A.; Nesztor, D.; Szekeres, M.; Zupkó, I.; Tombácz, E. Designed polyelectrolyte shell on magnetite nanocore for dilution-resistant biocompatible magnetic fluids. Langmuir 2012, 28, 16638–16646. [Google Scholar] [CrossRef] [PubMed]
- Westgard, Q.C. Desirable Biological Variation Database Specifications. Available online: https://www.westgard.com/biodatabase1.htm (accessed on 29 September 2018).
- Borkovec, M.; Jönsson, B.; Koper, G.J.M. Ionization processes and proton binding in polyprotic systems: Small molecules, proteins, interfaces and polyelectrolytes. Surf. Colloid Sci. 2001, 16, 99–339. [Google Scholar] [CrossRef]
- Tanford, C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes; Krieger Publ. Co.: Malabar, FL, USA, 1991; pp. 5–79. ISBN 10: 0471048933. [Google Scholar]
- Qiao, J.-L.; Yoshimoto, N.; Ishikawa, M.; Morita, M. Acetic acid-doped poly(ethylene oxide)-modified poly (methacrylate): A new proton conducting polymeric gel electrolyte. Electrochim. Acta 2002, 47, 3441–3446. [Google Scholar] [CrossRef]
- Szekeres, M.; Tóth, I.Y.; Illés, E.; Hajdú, A.; Zupkó, I.; Farkas, K.; Oszlánczi, G.; Tiszlavicz, L.; Tombácz, E. Chemical and colloidal stability of carboxylated core-shell magnetite nanoparticles designed for biomedical applications. Int. J. Mol. Sci. 2013, 14, 14550–14574. [Google Scholar] [CrossRef] [PubMed]
- Tombácz, E.; Farkas, K.; Földesi, I.; Szekeres, M.; Illés, E.; Tóth, I.Y.; Nesztor, D.; Szabó, T. Polyelectrolyte coating on superparamagnetic iron oxide nanoparticles as interface between magnetic core and biorelevant media. Interface Focus 2016, 6, 20160068. [Google Scholar] [CrossRef] [PubMed]
- Decuzzi, P.; Ferrari, M. The role of specific and non-specific interactions in receptor-mediated endocytosis of nanoparticles. Biomaterials 2007, 28, 2915–2922. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, M.; Tóth, I.Y.; Turcu, R.; Tombácz, E. The effect of polycarboxylate shell of magnetite nanoparticles on protein corona formation in blood plasma. J. Magn. Magn. Mater. 2017, 427, 95–99. [Google Scholar] [CrossRef]
- Chen, S.; Li, L.; Zhao, C.; Zheng, J. Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer 2010, 51, 5283–5293. [Google Scholar] [CrossRef]
- Stephen, Z.R.; Kievit, F.M.; Zhang, M. Magnetite nanoparticles for medical MR imaging. Mater Today 2011, 14, 330–338. [Google Scholar] [CrossRef]
- Na, H.B.; Song, I.C.; Hyeon, T. Inorganic Nanoparticles for MRI Contrast Agents. Adv. Mater. 2009, 21, 2133–2148. [Google Scholar] [CrossRef]
- Illés, E.; Szekeres, M.; Kupcsik, E.; Tóth, I.Y.; Farkas, K.; Jedlovszky-Hajdú, A.; Tombácz, E. PEGylation of surfacted magnetite core–shell nanoparticles for biomedical application. Colloids Surf. A 2014, 460, 429–440. [Google Scholar] [CrossRef]
- Khalkhali, M.; Rostamizadeh, K.; Sadighian, S.; Khoeini, F.; Naghibi, M.; Hamidi, M. The impact of polymer coatings on magnetite nanoparticles performance as MRI contrast agents: A comparative study. DARU J. Pharm. Sci. 2015, 23, 45. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coating Material | Mn (g/mol) | COOH Functional Groups * (mmol/g) | Molecular Structure |
|---|---|---|---|
| Acrylic acid (AA) small molecule | 72 | 14 |  |
| Polyacrylic acid (PAA) homopolymer | 1800 | 14 |  |
| P(PEGMA) comb-like homopolymer | 12,400 | 0 |  |
| P(PEGMA-AA) comb-like copolymer | 6200 | 5 |  |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Illés, E.; Szekeres, M.; Tóth, I.Y.; Farkas, K.; Földesi, I.; Szabó, Á.; Iván, B.; Tombácz, E. PEGylation of Superparamagnetic Iron Oxide Nanoparticles with Self-Organizing Polyacrylate-PEG Brushes for Contrast Enhancement in MRI Diagnosis. Nanomaterials 2018, 8, 776. https://doi.org/10.3390/nano8100776
Illés E, Szekeres M, Tóth IY, Farkas K, Földesi I, Szabó Á, Iván B, Tombácz E. PEGylation of Superparamagnetic Iron Oxide Nanoparticles with Self-Organizing Polyacrylate-PEG Brushes for Contrast Enhancement in MRI Diagnosis. Nanomaterials. 2018; 8(10):776. https://doi.org/10.3390/nano8100776
Chicago/Turabian StyleIllés, Erzsébet, Márta Szekeres, Ildikó Y. Tóth, Katalin Farkas, Imre Földesi, Ákos Szabó, Béla Iván, and Etelka Tombácz. 2018. "PEGylation of Superparamagnetic Iron Oxide Nanoparticles with Self-Organizing Polyacrylate-PEG Brushes for Contrast Enhancement in MRI Diagnosis" Nanomaterials 8, no. 10: 776. https://doi.org/10.3390/nano8100776
APA StyleIllés, E., Szekeres, M., Tóth, I. Y., Farkas, K., Földesi, I., Szabó, Á., Iván, B., & Tombácz, E. (2018). PEGylation of Superparamagnetic Iron Oxide Nanoparticles with Self-Organizing Polyacrylate-PEG Brushes for Contrast Enhancement in MRI Diagnosis. Nanomaterials, 8(10), 776. https://doi.org/10.3390/nano8100776