Computer Simulations of Lipid Nanoparticles



Abstract
:1. Introduction
2. Computational Methods
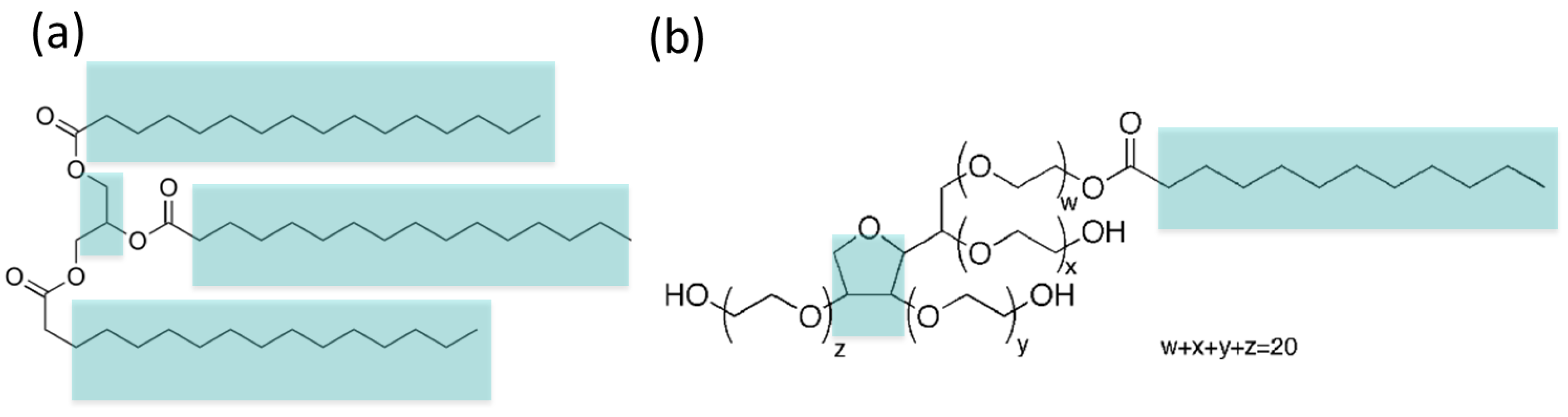
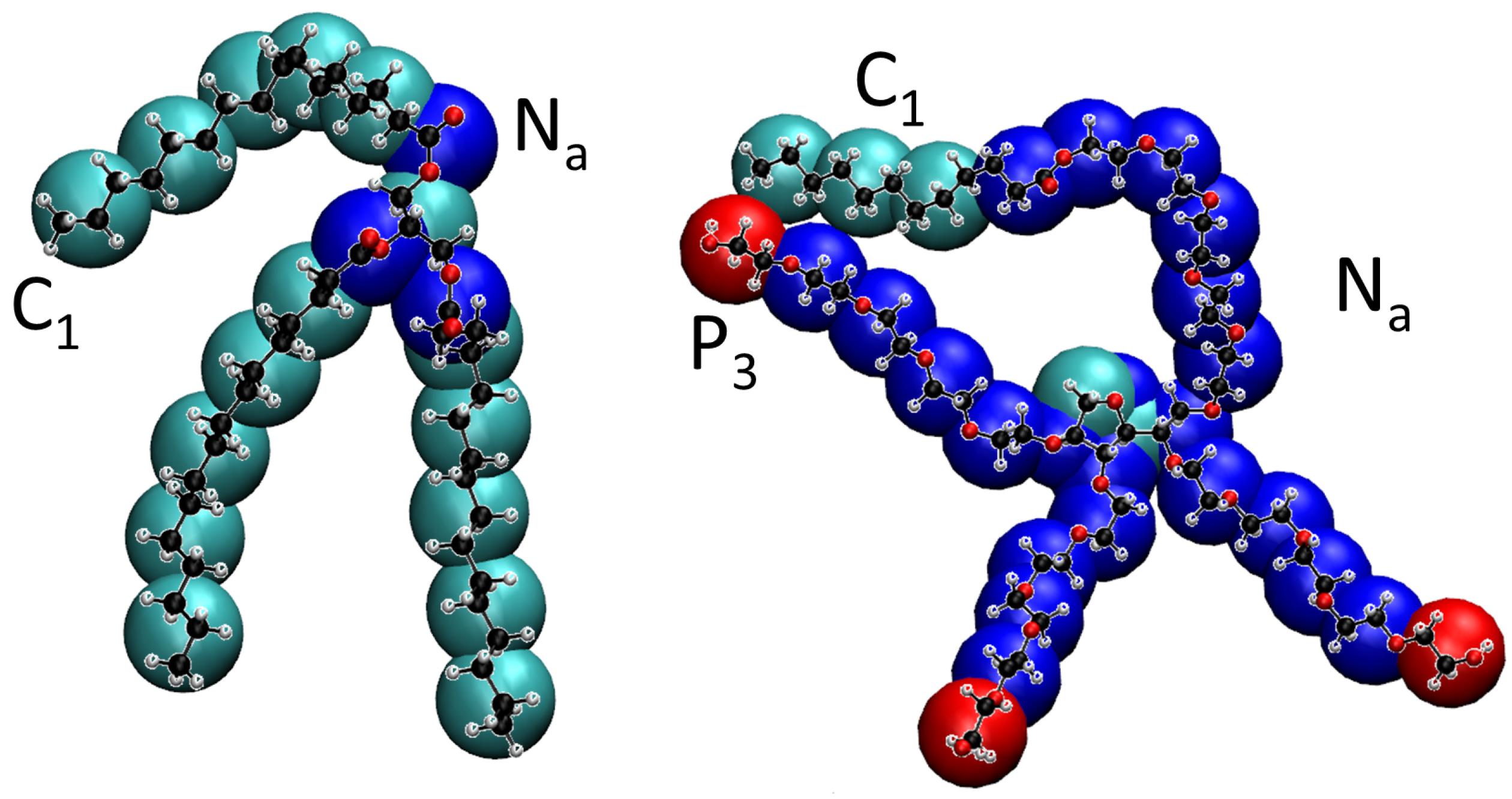
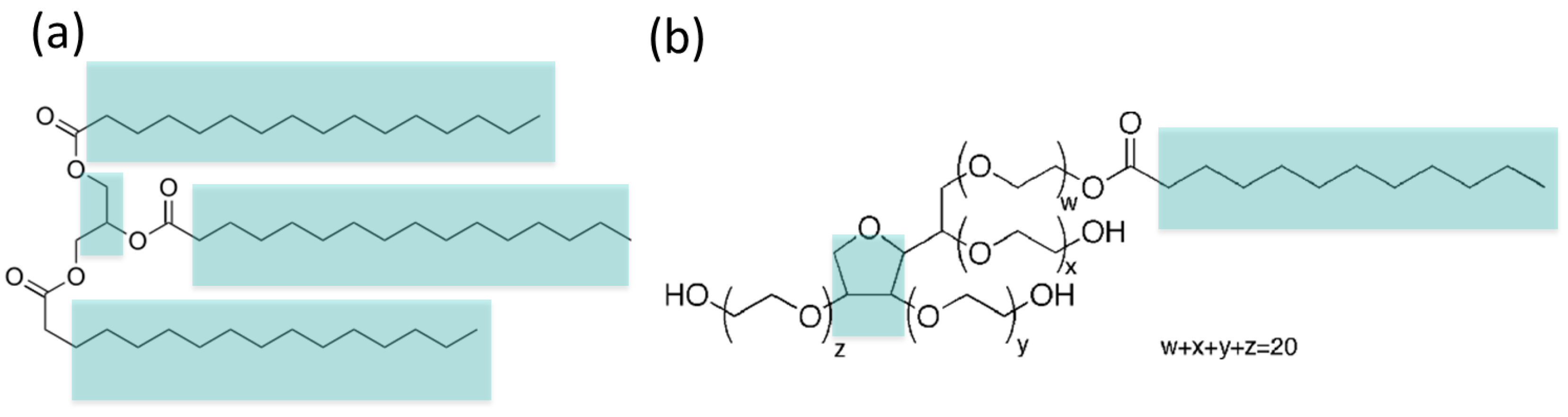
2.1. Coarse-Grain (MARTINI) Model of Tripalmitin and Tween 20
2.2. Methodology for Molecular Dynamics Simulations
2.3. Description of Simulated Systems
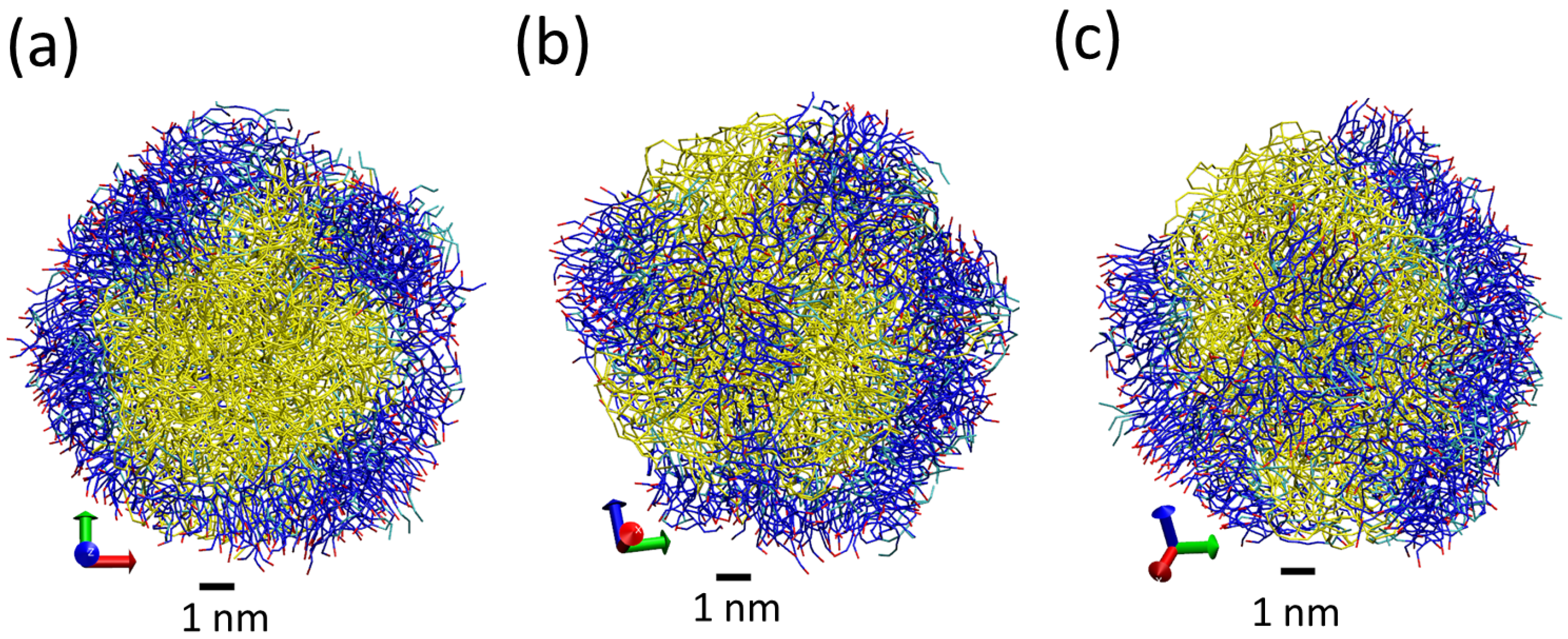
3. Results and Discussion
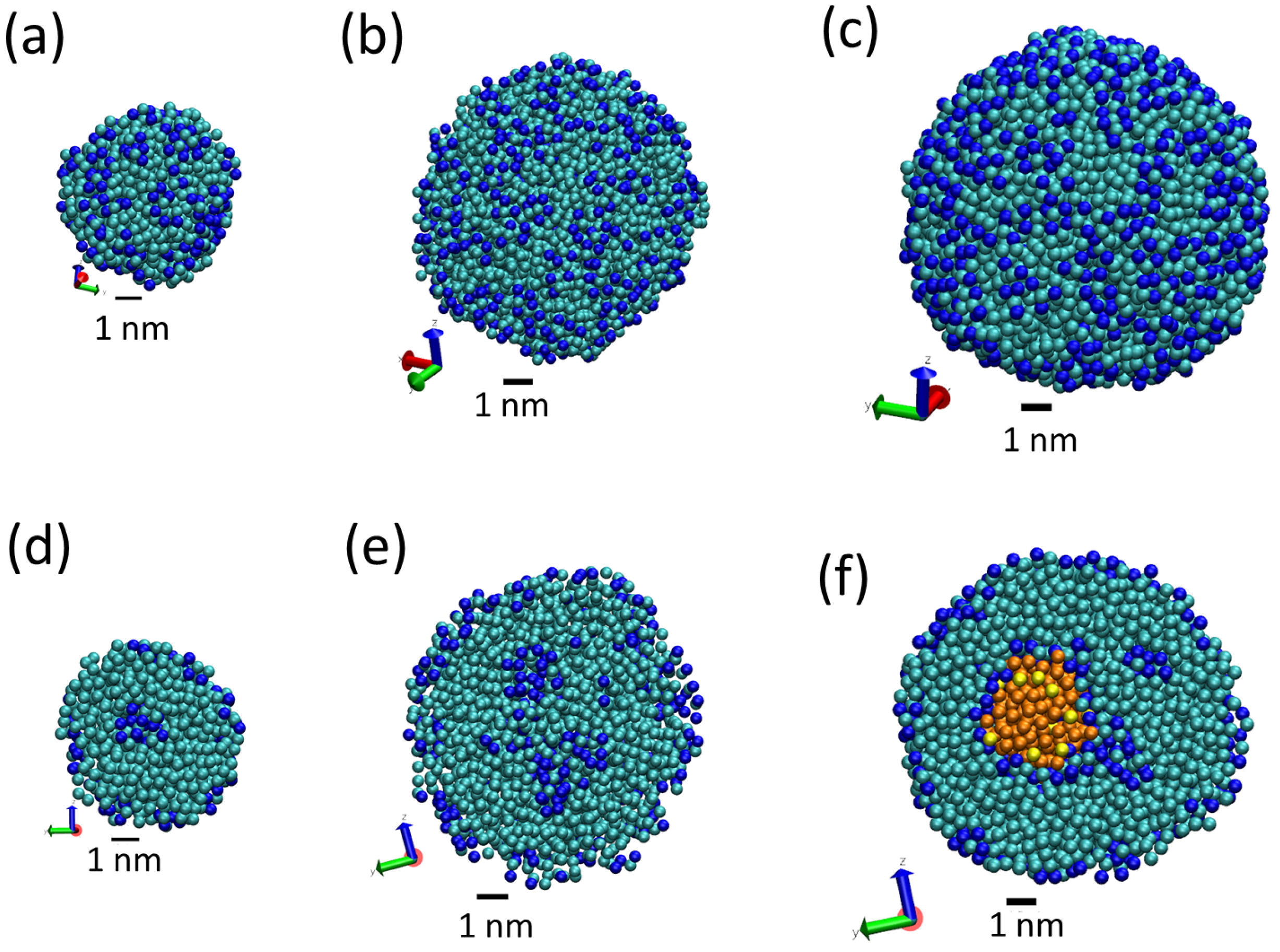
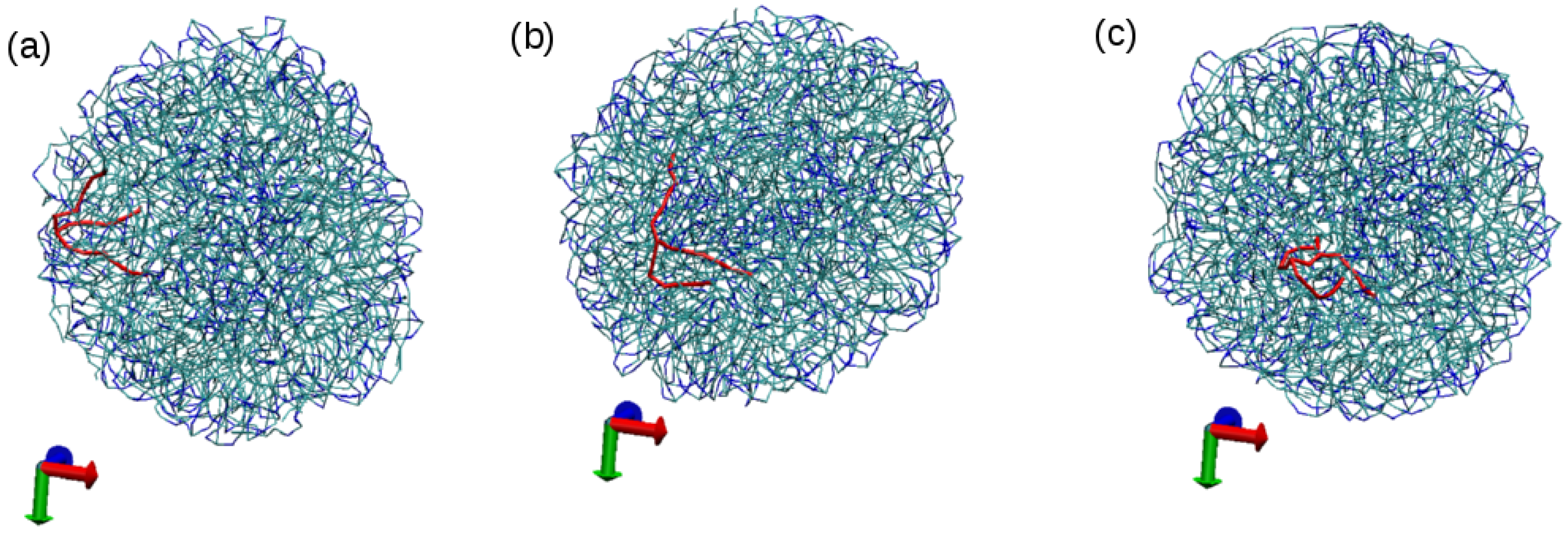
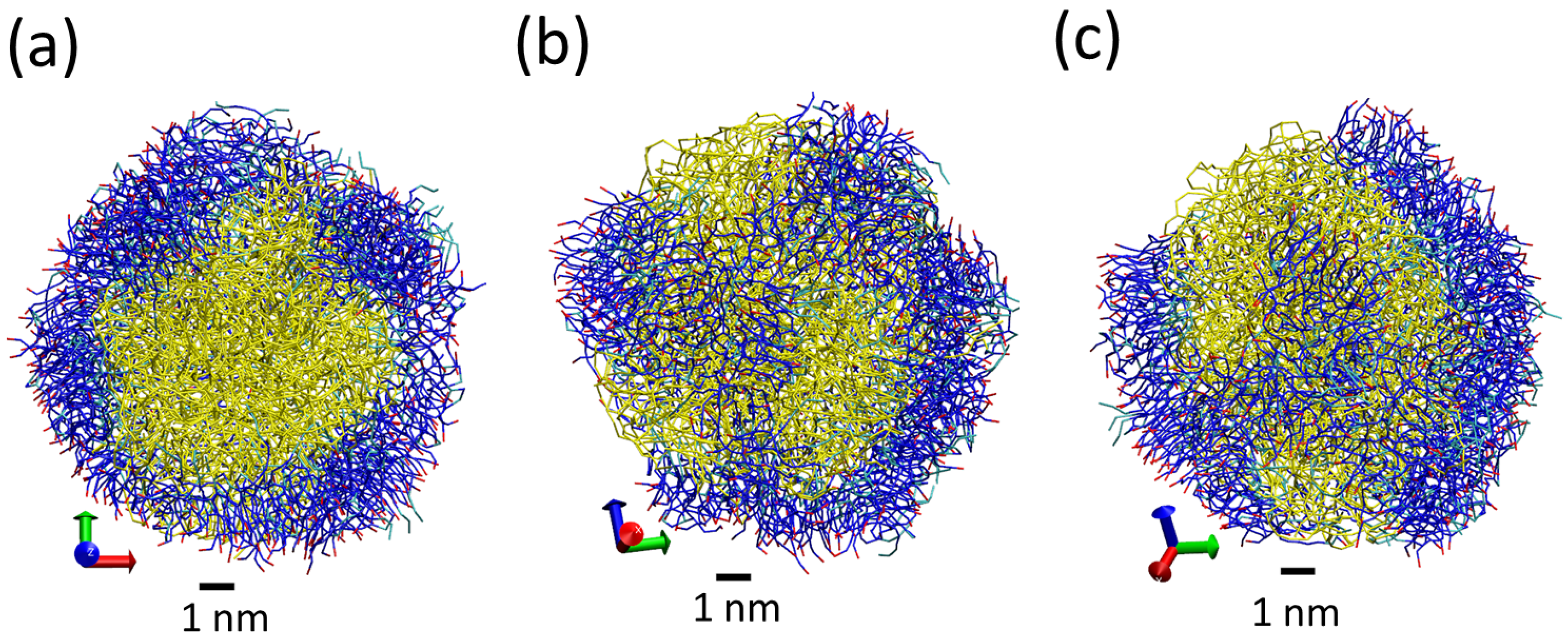
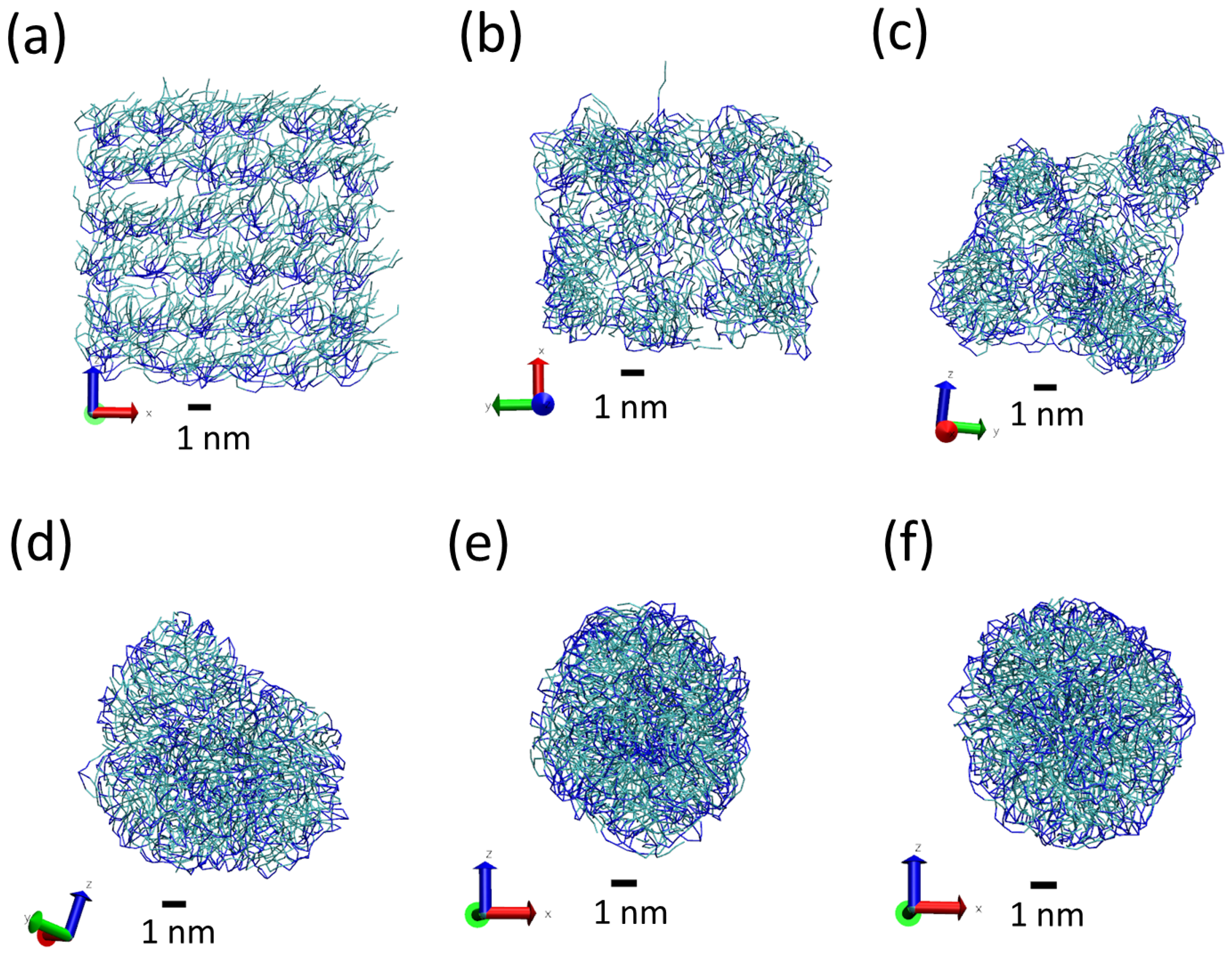
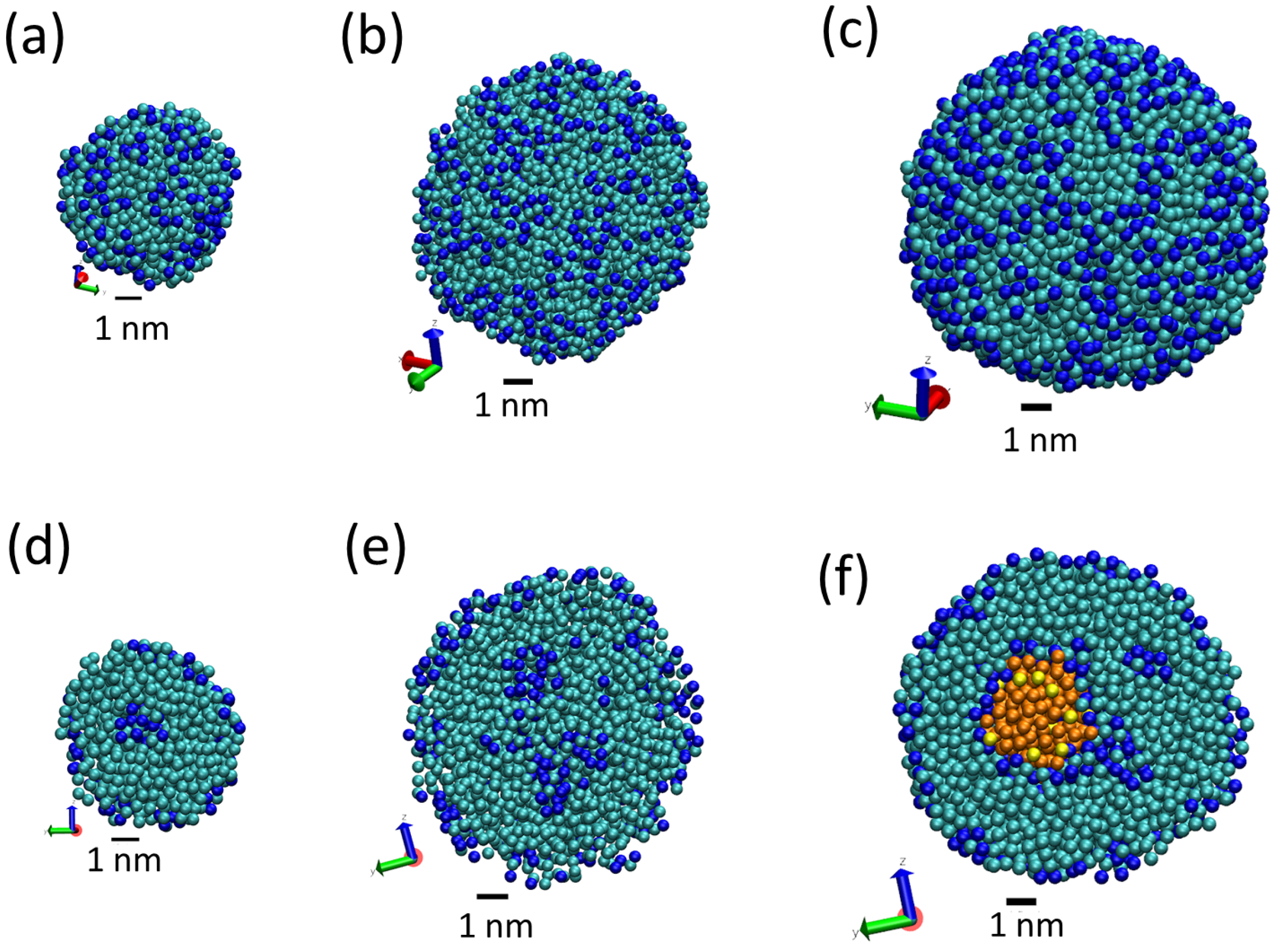
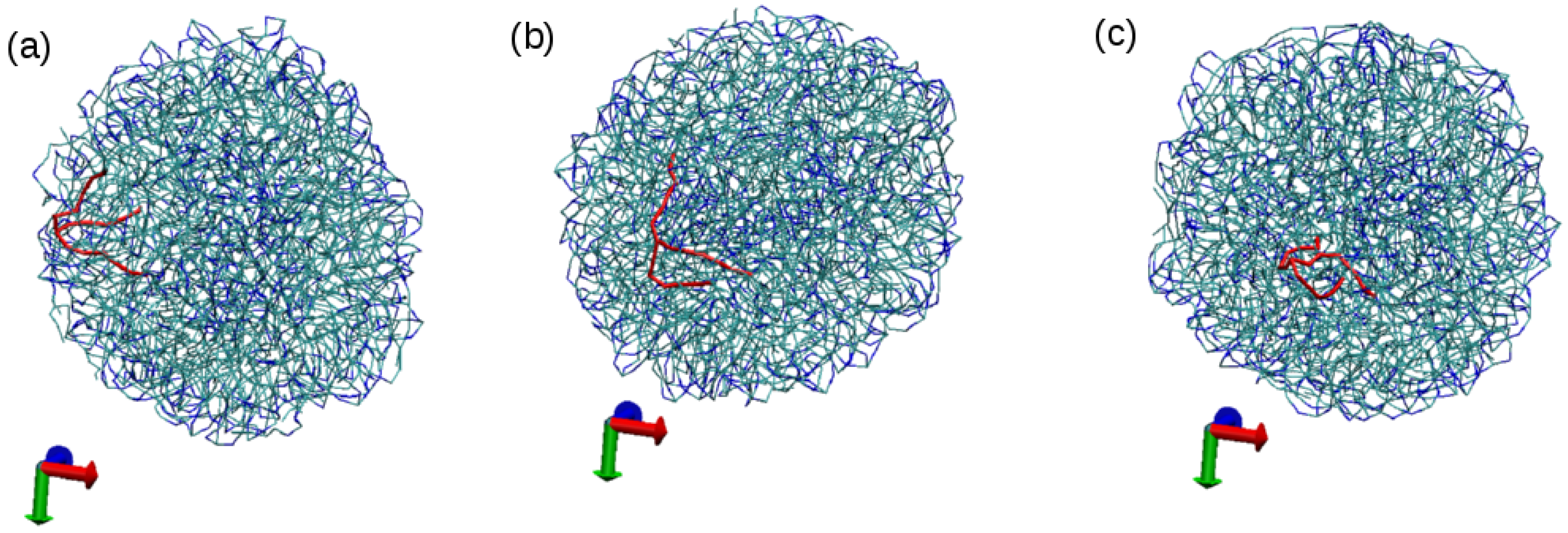
3.1. Tripalmitin Nanoparticles of Different Sizes
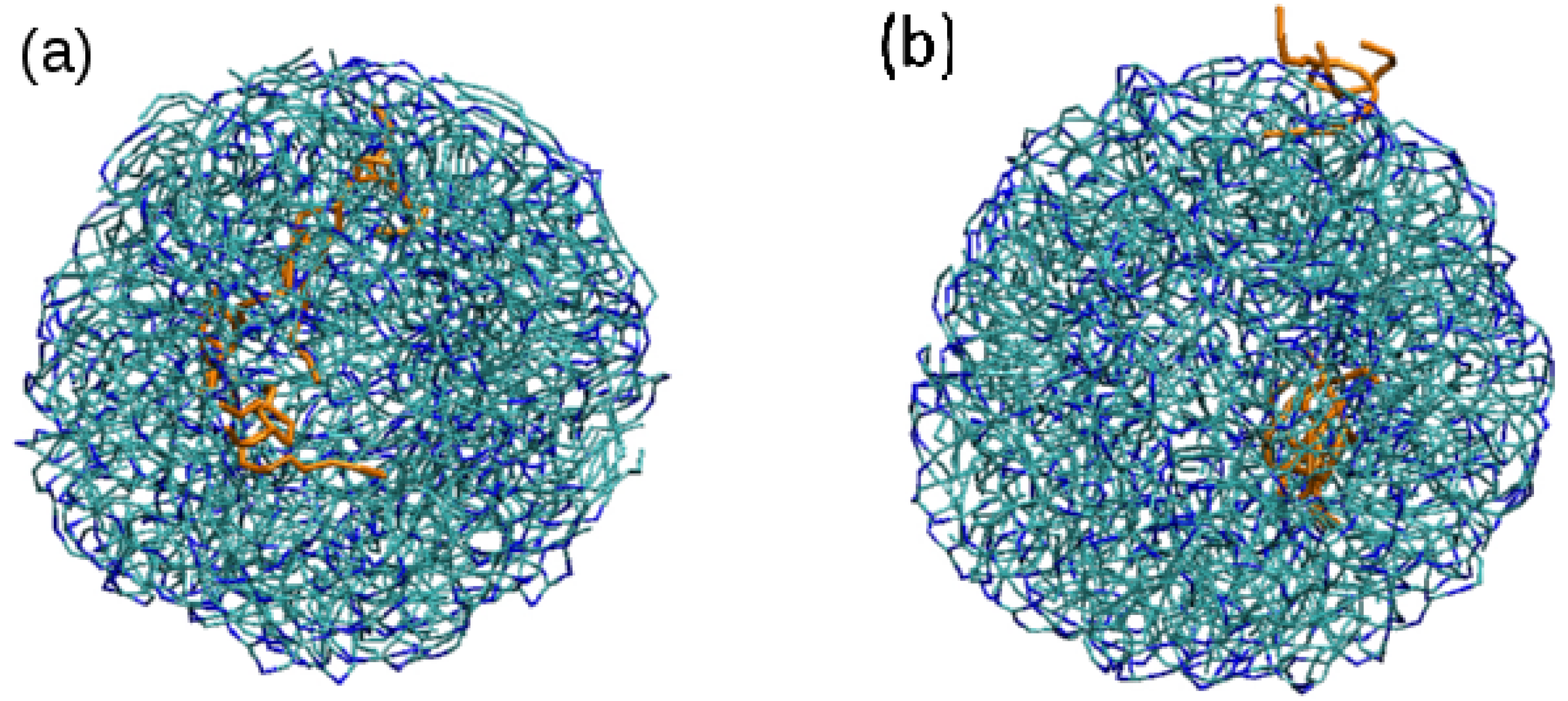
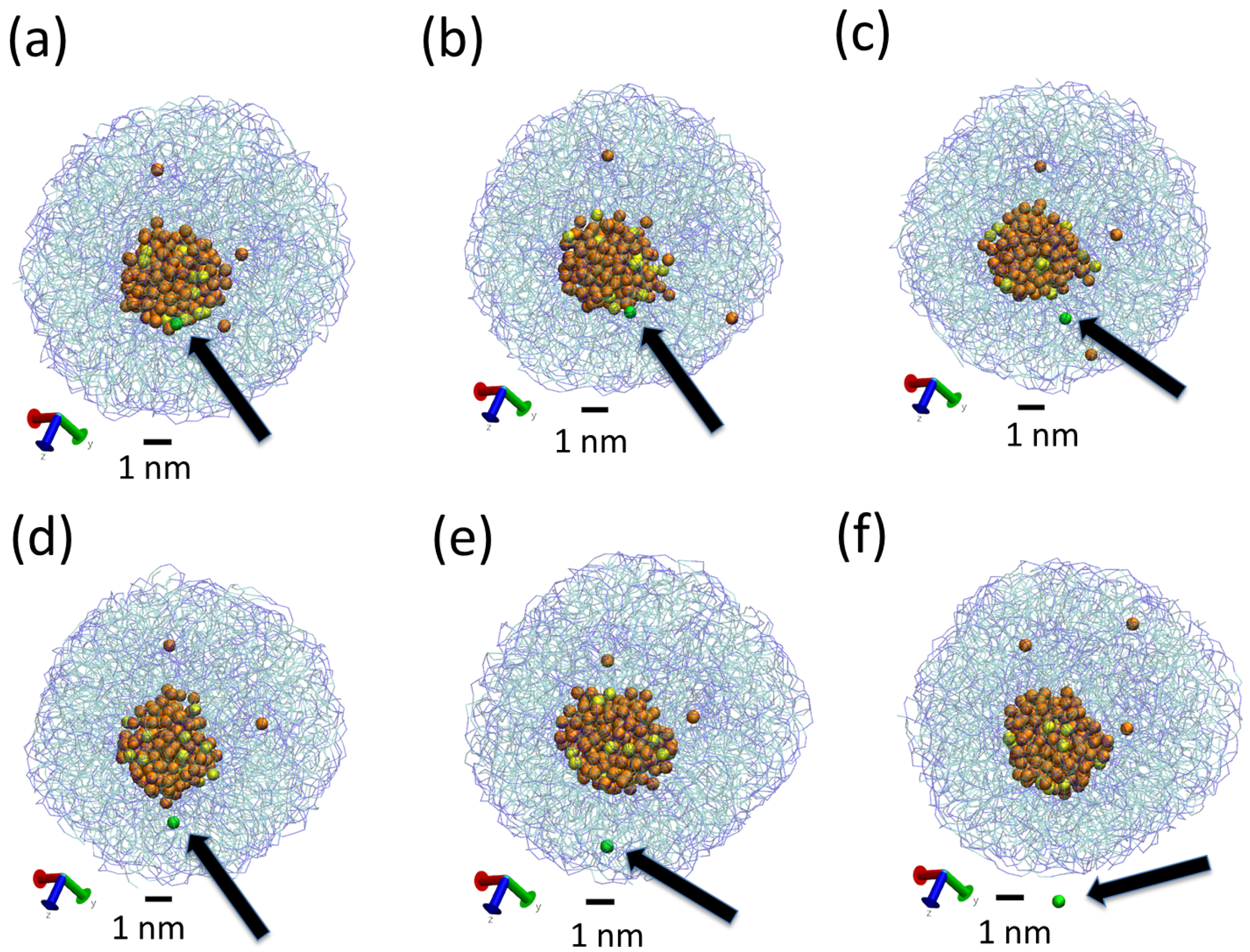
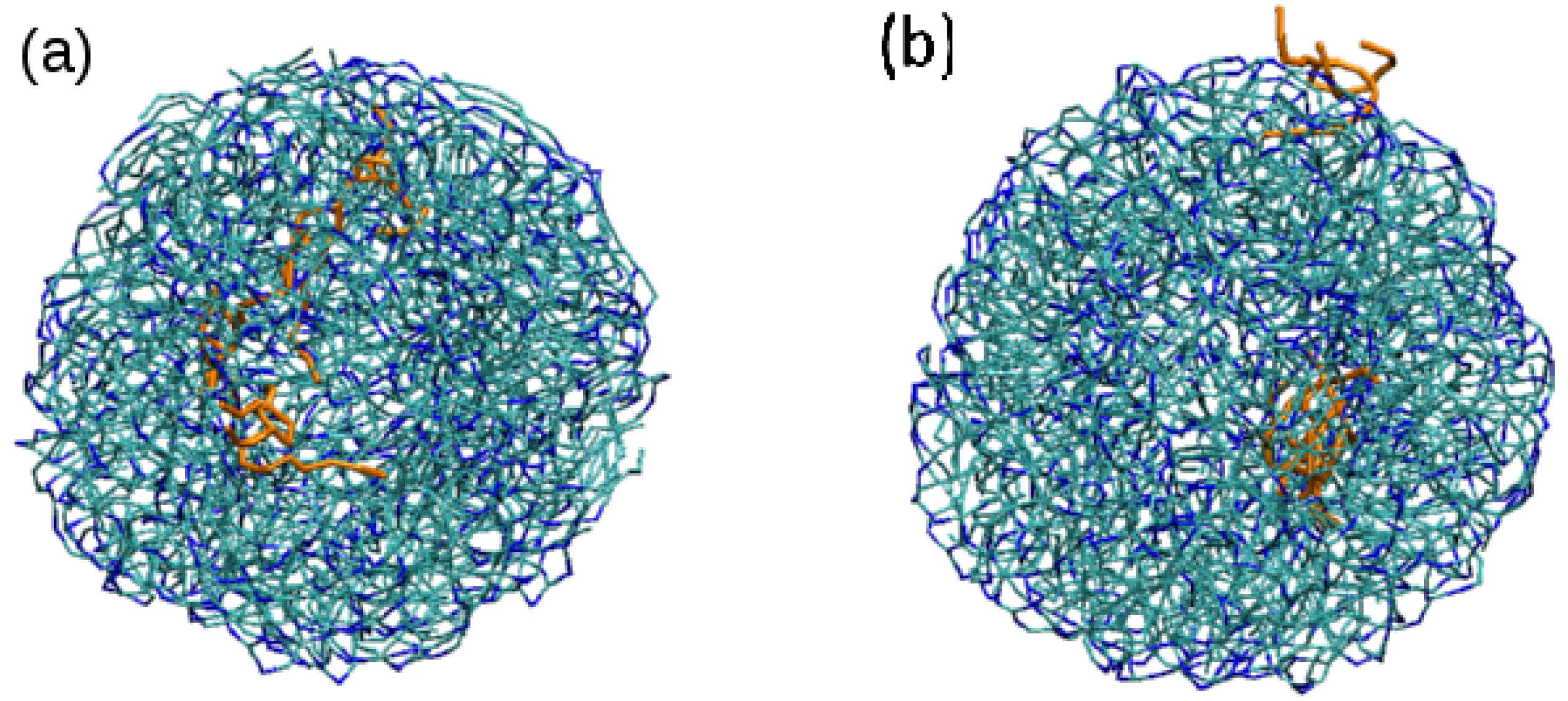
3.2. Effect of Surfactant
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CG | Coarse grained model |
| CPK | Corey, Pauling and Koltun’s convention for the representation of atoms in molecules |
| CTAB | Cetrimonium bromide also known as cetyltrimethylammonium bromide or hexadecyltrimethylammonium bromide |
| LNP | Lipid NanoParticles |
| MD | Molecular Dynamics |
| SDS | Sodium dodecyl sulfate, also known as sodium lauryl sulfate |
References
- Lu, D.; Aksimentiev, A.; Shih, A.Y.; Cruz-Chu, E.; Freddolino, P.L.; Arkhipov, A.; Schulten, K. The role of molecular modeling in bionanotechnology. Phys. Biol. 2006, 3, S40–S53. [Google Scholar] [CrossRef] [PubMed]
- Soussan, E.; Cassel, S.; Blanzat, M.; Rico-Lattes, I. Drug delivery by soft matter: Matrix and vesicular carriers. Angew. Chem. Int. Ed. 2009, 48, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, I.; Abasolo, I.; Corchero, J.L.; Elizondo, E.; Gil, P.R.; Moreno, E.; Faraudo, J.; Sala, S.; Bueno, D.; González-Mira, E.; et al. α-Galactosidase-A Loaded-Nanoliposomes with Enhanced Enzymatic Activity and Intracellular Penetration. Adv. Healthc. Mater. 2016, 5, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Loxley, A. Solid lipid nanoparticles for the delivery of pharmaceutical actives. Drug Deliv. Technol. 2009, 9, 32–37. [Google Scholar]
- Yadav, N.; Khatak, S.; Vir, U.; Sara, S. Solid lipid nanoparticles—A review. Int. J. Appl. Pharm. 2013, 5, 8–18. [Google Scholar]
- Freitas, C.; Müller, R.H. Correlation between long-term stability of solid lipid nanoparticles (SLN™) and crystallinity of the lipid phase. Eur. J. Pharm. Biopharm. 1999, 47, 125–132. [Google Scholar] [CrossRef]
- Helgason, T.; Awad, T.S.; Kristbergsson, K.; McClements, D.J.; Weiss, J. Influence of Polymorphic Transformations on Gelation of Tripalmitin Solid Lipid Nanoparticle Suspensions. J. Am. Oil Chem. Soc. 2008, 85, 501–511. [Google Scholar] [CrossRef]
- Helgason, T.; Awad, T.S.; Kristbergsson, K.; McClements, D.J.; Weiss, J. Effect of surfactant surface coverage on formation of solid lipid nanoparticles (SLN). J. Colloid Interface Sci. 2009, 334, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Hathout, R.M.; Metwally, A.A. Towards better modelling of drug-loading in solid lipid nanoparticles: Molecular dynamics, docking experiments and Gaussian Processes machine learning. Eur. J. Pharm. Biopharm. 2016, 108, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Noid, W.G. Perspective: Coarse-grained models for biomolecular systems. J. Chem. Phys. 2013, 139, 090901. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Tieleman, D.P. Perspective on the Martini model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; de Vries, A.H.; Mark, A.E. Coarse Grained Model for Semiquantitative Lipid Simulations. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, J.; Mark, A.E. Simulation of gel phase formation and melting in lipid bilayers using a coarse grained model. Chem. Phys. Lipids 2005, 135, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; Vries, A.H.D. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Rawat, N.; Biswas, P. Size, shape, and flexibility of proteins and DNA. J. Chem. Phys. 2009, 131, 165104. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | N Lipid | N Surfactant | N Water | AF Beads | L | Sim Time | |
|---|---|---|---|---|---|---|---|
| S1 | pure lipid NP | 64 | - | 4176 | 462 | 9.2 nm | 1100 ns |
| S2 | pure lipid NP | 216 | - | 28,003 | 3143 | 16.9 nm | 100 ns |
| S3 | pure lipid NP | 392 | - | 60,210 | 6627 | 21.5 nm | 450 ns |
| S4 | surfacted NP | 216 | 3 | 22,268 | 2420 | 15.6 nm | 327.6 ns |
| S5 | surfacted NP | 216 | 221 | 31,826 | 3518 | 17.9 nm | 201.8 ns |
| Simulation | N | |||
|---|---|---|---|---|
| S1 | 64 | 2.5 nm | 0.98 nm | |
| S2 | 216 | 3.7 nm | 1.02 nm | |
| S3 | 392 | 4.6 nm | 0.96 nm |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Luengo, X.F.; Camacho, J.; Faraudo, J. Computer Simulations of Lipid Nanoparticles. Nanomaterials 2017, 7, 461. https://doi.org/10.3390/nano7120461
Fernandez-Luengo XF, Camacho J, Faraudo J. Computer Simulations of Lipid Nanoparticles. Nanomaterials. 2017; 7(12):461. https://doi.org/10.3390/nano7120461
Chicago/Turabian StyleFernandez-Luengo, Xavier F., Juan Camacho, and Jordi Faraudo. 2017. "Computer Simulations of Lipid Nanoparticles" Nanomaterials 7, no. 12: 461. https://doi.org/10.3390/nano7120461
APA StyleFernandez-Luengo, X. F., Camacho, J., & Faraudo, J. (2017). Computer Simulations of Lipid Nanoparticles. Nanomaterials, 7(12), 461. https://doi.org/10.3390/nano7120461