
Exploration of the Electronic and Catalytic Properties of [Co5MS8(PEt3)5]1+ Nanoclusters: A Computational Study



Abstract
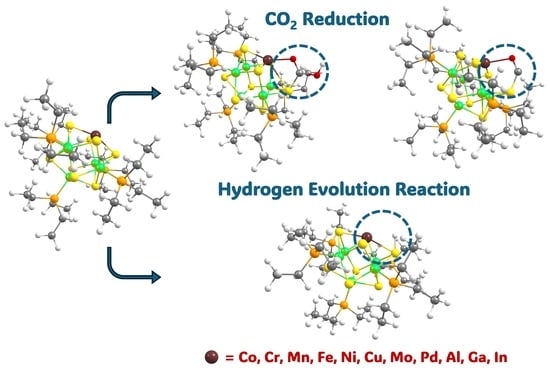
1. Introduction
2. Methods
Computational Details
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qin, L.; Ma, G.; Wang, L.; Tang, Z. Atomically precise metal nanoclusters for (photo)electroreduction of CO2: Recent advances, challenges and opportunities. J. Energy Chem. 2021, 57, 359–370. [Google Scholar] [CrossRef]
- Kagalwala, H.N.; Gottlieb, E.; Li, G.; Li, T.; Jin, R.; Bernhard, S. Photocatalytic Hydrogen Generation System Using a Nickel-Thiolate Hexameric Cluster. Inorg. Chem. 2013, 52, 9094–9101. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.-P.; Xu, Y.; Zhang, X.-G.; Zhang, H.; Yao, L.; Wang, R.; Zang, S.-Q. Copper-Sulfur-Nitrogen Cluster Providing a Local Proton for Efficient Carbon Dioxide Photoreduction. Angew. Chem. Int. Ed. 2023, 62, e202313648. [Google Scholar] [CrossRef]
- Deepika; Gholipour-Ranjbar, H.; Fang, H.; Sertse, L.; Laskin, J.; Jena, P. Atomically Precise Core-Tailored Metal Chalcogenide Nanoclusters: Tuning the Electronic Structure and Magnetic Properties. J. Phys. Chem. C 2022, 126, 6512–6522. [Google Scholar] [CrossRef]
- Kumar, B.; Kawawaki, T.; Shimizu, N.; Imai, Y.; Suzuki, D.; Hossain, S.; Nair, L.V.; Negishi, Y. Gold nanoclusters as electrocatalysts: Size, ligands, heteroatom doping, and charge dependences. Nanoscale 2020, 12, 9969–9979. [Google Scholar] [CrossRef]
- Yubero Valdivielso, D.; Kerpal, C.; Schöllkopf, W.; Meijer, G.; Fielicke, A. IR spectra and structures of saturated ruthenium cluster carbonyl cations Run(CO)m+ (n = 1–6). Dalton Trans. 2023, 52, 9929–9939. [Google Scholar] [CrossRef]
- Vaari, J.; Lahtinen, J.; Hautojärvi, P. A bimetallic Ru Co surface prepared by Ru3(CO)12 adsorption on Co(0001). Surf. Sci. 1996, 346, 11–17. [Google Scholar] [CrossRef]
- Cao, Y.; Fung, V.; Yao, Q.; Chen, T.; Zang, S.; Jiang, D.-E.; Xie, J. Control of single-ligand chemistry on thiolated Au25 nanoclusters. Nat. Commun. 2020, 11, 5498. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Jiang, D.-E.; Mann, A.K.P.; Mullins, D.R.; Qiao, Z.-A.; Allard, L.F.; Zeng, C.; Jin, R.; Overbury, S.H. Thiolate Ligands as a Double-Edged Sword for CO Oxidation on CeO2 Supported Au25(SCH2CH2Ph)18 Nanoclusters. J. Am. Chem. Soc. 2014, 136, 6111–6122. [Google Scholar] [CrossRef]
- Liu, G.; Chauhan, V.; Aydt, A.P.; Ciborowski, S.M.; Pinkard, A.; Zhu, Z.; Roy, X.; Khanna, S.N.; Bowen, K.H. Ligand Effect on the Electronic Structure of Cobalt Sulfide Clusters: A Combined Experimental and Theoretical Study. J. Phys. Chem. C 2019, 123, 25121–25127. [Google Scholar] [CrossRef]
- Li, X.; Havenridge, S.; Gholipour-Ranjbar, H.; Forbes, D.; Crain, W.; Liu, C.; Laskin, J. Structural Changes in Metal Chalcogenide Nanoclusters Associated with Single Heteroatom Incorporation. J. Phys. Chem. A 2025, 129, 1310–1317. [Google Scholar] [CrossRef]
- Gholipour-Ranjbar, H.; Deepika; Jena, P.; Laskin, J. Gas-phase fragmentation of single heteroatom-incorporated Co5MS8(PEt3)6+ (M = Mn, Fe, Co, Ni) nanoclusters. Commun. Chem. 2022, 5, 130. [Google Scholar] [CrossRef]
- Mitchell, B.S.; Chirila, A.; Kephart, J.A.; Boggiano, A.C.; Krajewski, S.M.; Rogers, D.; Kaminsky, W.; Velian, A. Metal–Support Interactions in Molecular Single-Site Cluster Catalysts. J. Am. Chem. Soc. 2022, 144, 18459–18469. [Google Scholar] [CrossRef]
- Gholipour-Ranjbar, H.; Samayoa-Oviedo, H.Y.; Laskin, J. Controlled Formation of Fused Metal Chalcogenide Nanoclusters Using Soft Landing of Gaseous Fragment Ions. ACS Nano 2023, 17, 17427–17435. [Google Scholar] [CrossRef]
- Rybacki, M.; Nagarajan, A.V.; Mpourmpakis, G. Ligand removal energetics control CO2 electroreduction selectivity on atomically precise, ligated alloy nanoclusters. Environ. Sci. Nano 2022, 9, 2032–2040. [Google Scholar] [CrossRef]
- Havenridge, S.; Li, X.; Laskin, J.; Liu, C. Effect of heteroatom incorporation on electronic communication in metal chalcogenide nanoclusters. Phys. Chem. Chem. Phys. 2025, 27, 12577–12583. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F. Hartree–Fock exchange fitting basis sets for H to Rn. J. Comput. Chem. 2008, 29, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Helmich-Paris, B.; de Souza, B.; Neese, F.; Izsák, R. An improved chain of spheres for exchange algorithm. J. Chem. Phys. 2021, 155, 104109. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Stevens, W.J.; Krauss, M.; Basch, H.; Jasien, P.G. Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms. Can. J. Chem. 1992, 70, 612–630. [Google Scholar] [CrossRef]
- Stevens, W.J.; Basch, H.; Krauss, M. Compact effective potentials and efficient shared-exponent basis sets for the first- and second-row atoms. J. Chem. Phys. 1984, 81, 6026–6033. [Google Scholar] [CrossRef]
- Webster, A.J.; Mueller, C.M.; Foegen, N.P.; Sit, P.H.L.; Speetzen, E.D.; Cunningham, D.W.; D’Acchioli, J.S. Oxidation states “naturally”: A Natural Bond Orbital method for determining transition metal oxidation states. Polyhedron 2016, 114, 128–132. [Google Scholar] [CrossRef]
- Sit, P.H.L.; Car, R.; Cohen, M.H.; Selloni, A. Simple, Unambiguous Theoretical Approach to Oxidation State Determination via First-Principles Calculations. Inorg. Chem. 2011, 50, 10259–10267. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Lu, Z.; Jafari, M.G.; Hernández-Prieto, C.; Zatsepin, P.; Mindiola, D.J.; Kaphan, D.M.; Delferro, M.; Kropf, A.J.; Liu, C. Integrated Experimental and Computational K-Edge X-ray Absorption Near-Edge Structure Analysis of Vanadium Catalysts. J. Phys. Chem. C 2022, 126, 11949–11962. [Google Scholar] [CrossRef]
- Andre Clayborne, P.; Lopez-Acevedo, O.; Whetten, R.L.; Grönbeck, H.; Häkkinen, H. Evidence of superatom electronic shells in ligand-stabilized aluminum clusters. J. Chem. Phys. 2011, 135, 094701. [Google Scholar] [CrossRef] [PubMed]
- Roy, X.; Lee, C.-H.; Crowther, A.C.; Schenck, C.L.; Besara, T.; Lalancette, R.A.; Siegrist, T.; Stephens, P.W.; Brus, L.E.; Kim, P.; et al. Nanoscale Atoms in Solid-State Chemistry. Science 2013, 341, 157–160. [Google Scholar] [CrossRef]
- O’Brien, E.S.; Trinh, M.T.; Kann, R.L.; Chen, J.; Elbaz, G.A.; Masurkar, A.; Atallah, T.L.; Paley, M.V.; Patel, N.; Paley, D.W.; et al. Single-crystal-to-single-crystal intercalation of a low-bandgap superatomic crystal. Nat. Chem. 2017, 9, 1170–1174. [Google Scholar] [CrossRef]
- Havenridge, S.; Liu, C. A Theoretical Benchmark of the Geometric and Optical Properties for 3d Transition Metal Nanoclusters via Density Functional Theory. J. Phys. Chem. A 2024, 128, 3947–3956. [Google Scholar] [CrossRef]
- Reber, A.C.; Khanna, S.N. Co6Se8(PEt3)6 superatoms as tunable chemical dopants for two-dimensional semiconductors. npj Comput. Mater. 2018, 4, 33. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Liu, G.; Pinkard, A.; Ciborowski, S.M.; Chauhan, V.; Zhu, Z.; Aydt, A.P.; Khanna, S.N.; Roy, X.; Bowen, K.H. Tuning the electronic properties of hexanuclear cobalt sulfide superatoms via ligand substitution. Chem. Sci. 2019, 10, 1760–1766. [Google Scholar] [CrossRef]
- Ou, L. Competition between Initial CO2 Electroreduction and Hydrogen Evolution Reaction on Cu Catalysts in Acidic Media: Role of Specifically Adsorbed Halide Anions. Langmuir 2024, 40, 13060–13069. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.-Y.; Li, Y.-B.; Xu, J.; Geng, Y.-F.; Zhang, J.-Y.; Xie, J.-L.; Zeng, Q.-D.; Wang, C. Competitive Influence of Hydrogen Bonding and van der Waals Interactions on Self-Assembled Monolayers of Stilbene-Based Carboxylic Acid Derivatives. J. Phys. Chem. C 2014, 118, 28625–28630. [Google Scholar] [CrossRef]
- Østergaard, F.C.; Bagger, A.; Rossmeisl, J. Predicting catalytic activity in hydrogen evolution reaction. Curr. Opin. Electrochem. 2022, 35, 101037. [Google Scholar] [CrossRef]
- Laursen, A.B.; Varela, A.S.; Dionigi, F.; Fanchiu, H.; Miller, C.; Trinhammer, O.L.; Rossmeisl, J.; Dahl, S. Electrochemical Hydrogen Evolution: Sabatier’s Principle and the Volcano Plot. J. Chem. Educ. 2012, 89, 1595–1599. [Google Scholar] [CrossRef]
- Niklas, J.; Kohler, L.; Potocny, A.M.; Mardis, K.L.; Mulfort, K.L.; Poluektov, O.G. Electronic Structure of Molecular Cobalt Catalysts for H2 Production Revealed by Multifrequency EPR. J. Phys. Chem. C 2022, 126, 11889–11899. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C/M-L6 | C/M-L5 | Isomer | ΔE (eV) | ΔEZPE (eV) | ΔG298.15 (eV) | |
|---|---|---|---|---|---|---|
| Co | 1/2 | 1/4 | Co | 2.24 | 2.16 | 1.53 |
| Cr | 1/5, 2/6 | 1/7, 2/6 | Co-Cis, Co-Cis | 2.06, 2.39 | 1.97, 2.25 | 1.23, 1.64 |
| Mn | 1/4 | 1/6 | Mn | 1.96 | 1.88 | 1.20 |
| Fe | 1/3 | 1/5 | Fe | 2.00 | 1.93 | 1.29 |
| Ni | 1/1 | 1/3 | Co-Trans | 2.17 | 2.06 | 1.37 |
| Cu | 1/2 | 1/2 | Cu | 1.49 | 1.44 | 0.71 |
| Mo | 1/3 | 1/3 | Co-Cis | 2.16 | 2.08 | 1.40 |
| Pd | 1/1 | 1/3 | Co-Trans | 2.18 | 2.08 | 1.37 |
| Al | 1/2, 2/3 | 1/2, 2/3 | Al, Co-Cis | 1.86, 2.30 | 1.82, 2.20 | 1.14, 1.53 |
| Ga | 1/2, 2/3 | 1/2, 2/5 * | Ga, Co-Cis | 1.62, 2.22 | 1.60, 2.08 | 1.01, 1.46 |
| In | 1/2, 2/3 | 1/2, 2/3 | In, Co-Cis | 1.49, 2.13 | 1.49, 2.02 | 0.82, 1.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Havenridge, S.; Miller, A.G.; Liu, C. Exploration of the Electronic and Catalytic Properties of [Co5MS8(PEt3)5]1+ Nanoclusters: A Computational Study. Nanomaterials 2026, 16, 587. https://doi.org/10.3390/nano16100587
Havenridge S, Miller AG, Liu C. Exploration of the Electronic and Catalytic Properties of [Co5MS8(PEt3)5]1+ Nanoclusters: A Computational Study. Nanomaterials. 2026; 16(10):587. https://doi.org/10.3390/nano16100587
Chicago/Turabian StyleHavenridge, Shana, Audrey Grace Miller, and Cong Liu. 2026. "Exploration of the Electronic and Catalytic Properties of [Co5MS8(PEt3)5]1+ Nanoclusters: A Computational Study" Nanomaterials 16, no. 10: 587. https://doi.org/10.3390/nano16100587
APA StyleHavenridge, S., Miller, A. G., & Liu, C. (2026). Exploration of the Electronic and Catalytic Properties of [Co5MS8(PEt3)5]1+ Nanoclusters: A Computational Study. Nanomaterials, 16(10), 587. https://doi.org/10.3390/nano16100587