
First-Principles Investigations of Two-Sided Functionalised MoS2 Monolayer


Abstract
1. Introduction
2. Materials and Methods
2.1. Computational Details
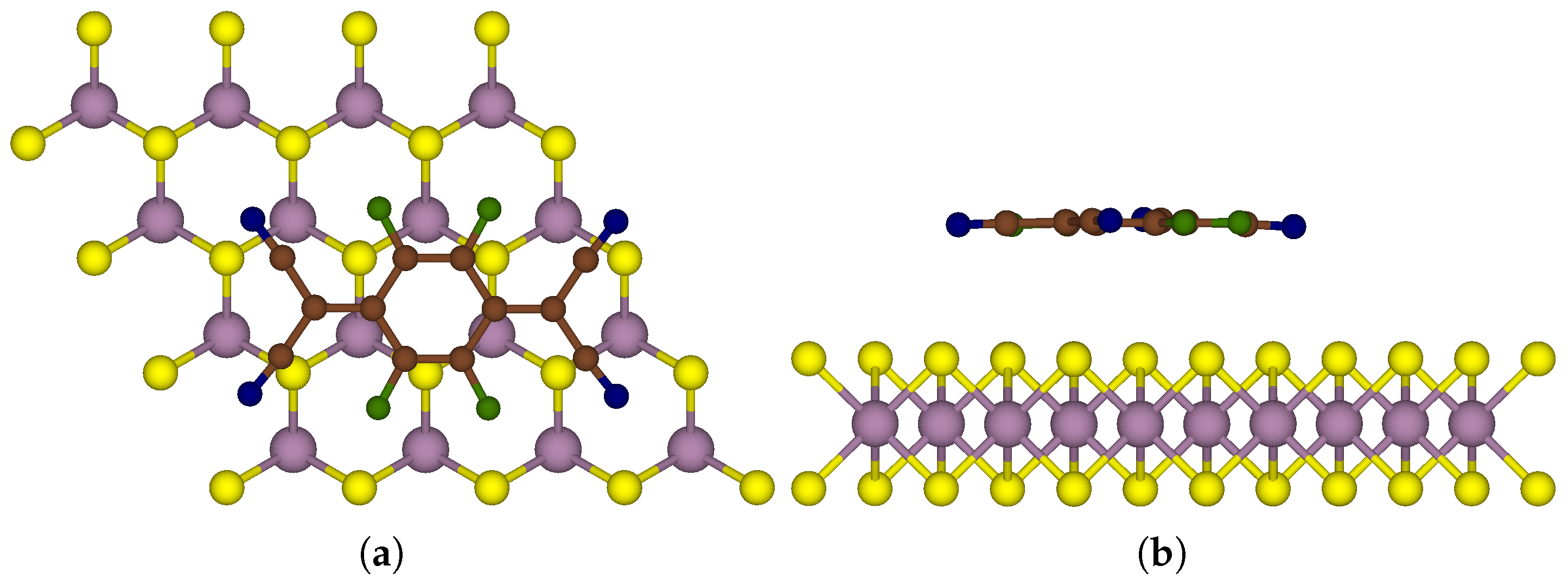
2.2. Model Construction
3. Results and Discussion
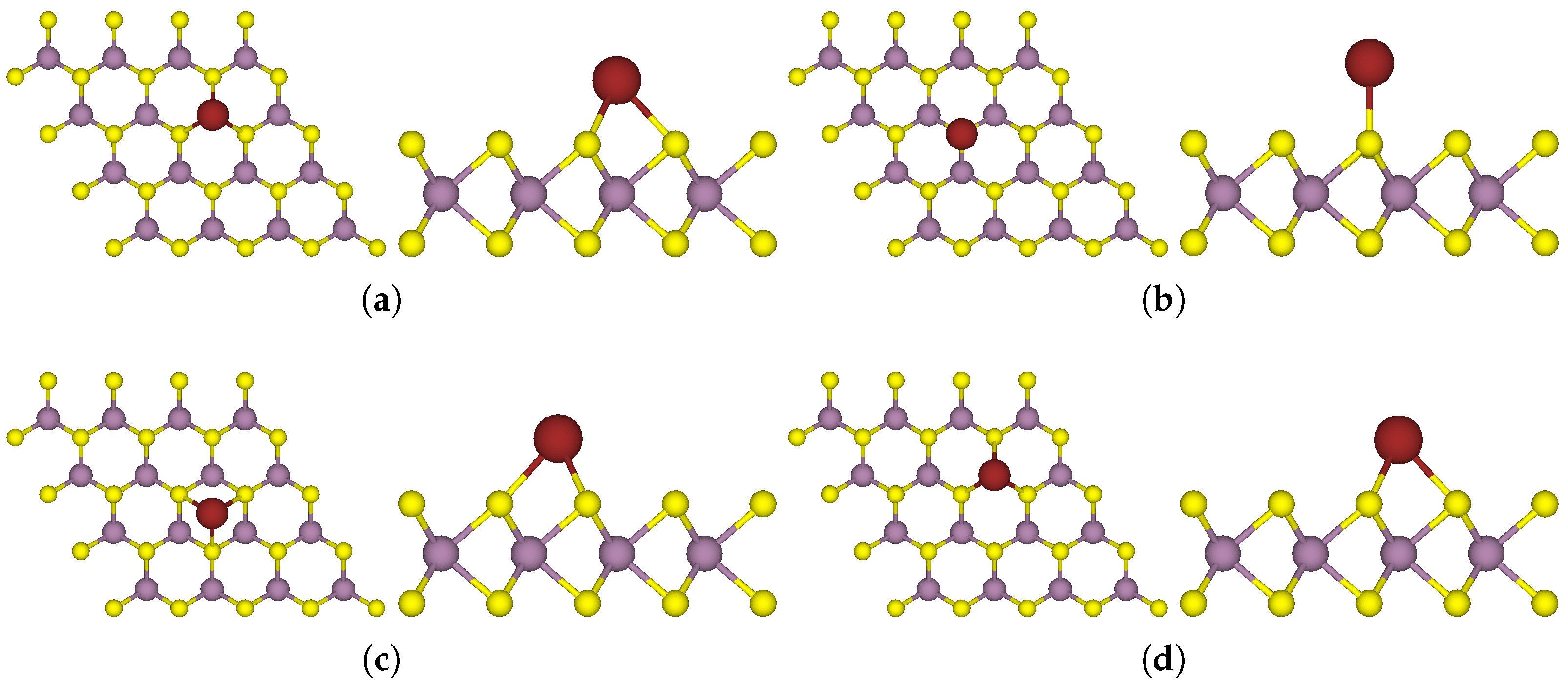
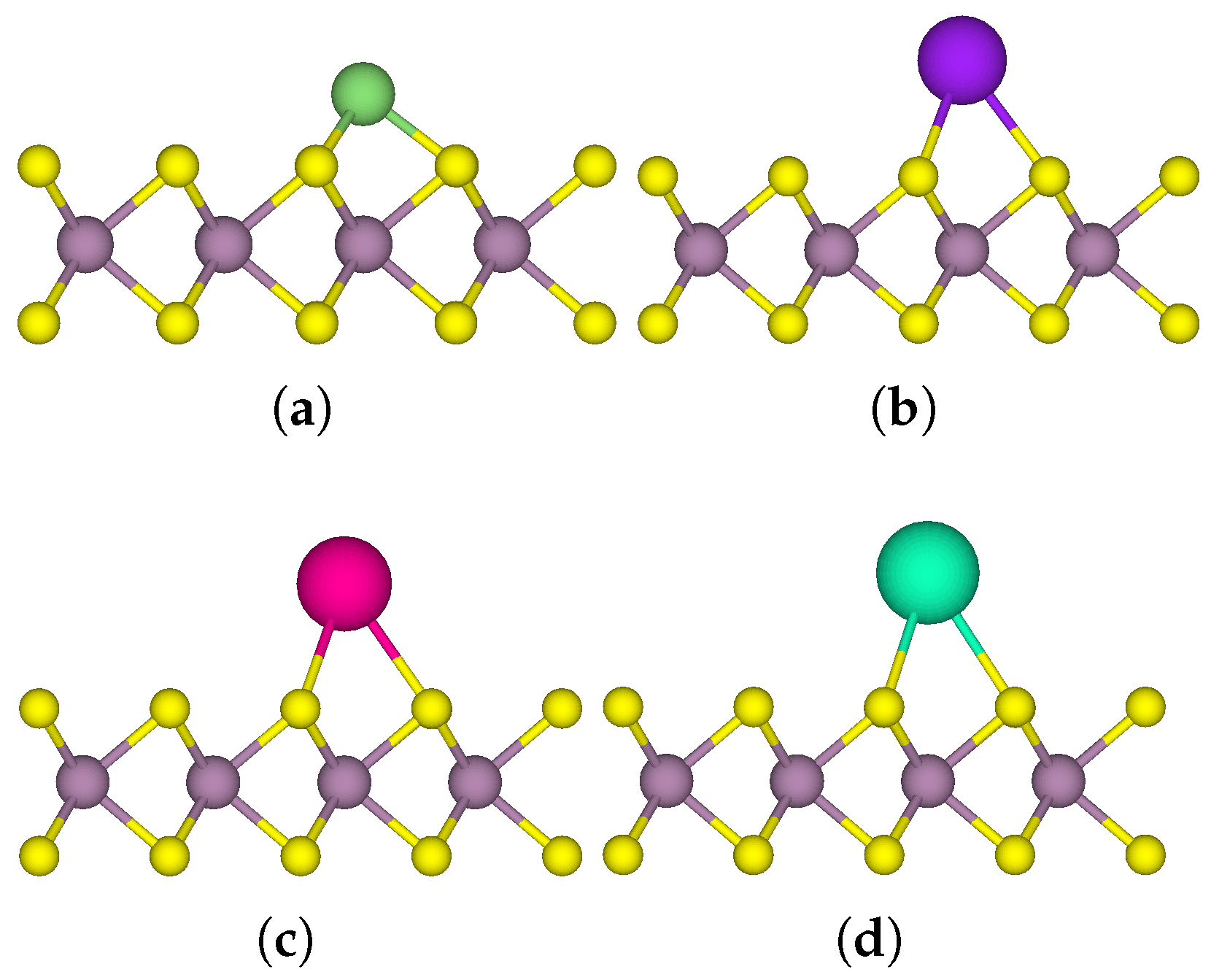
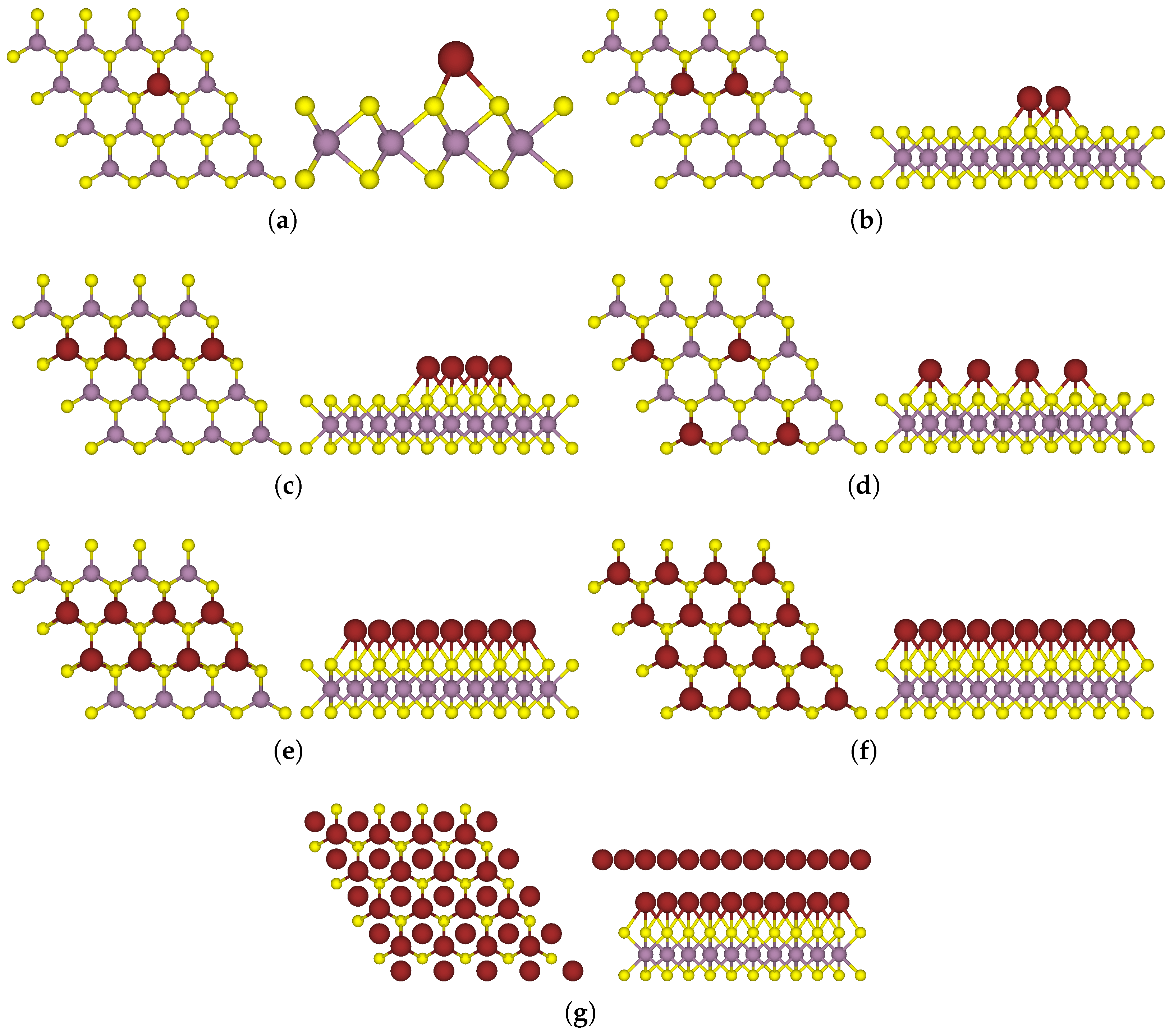
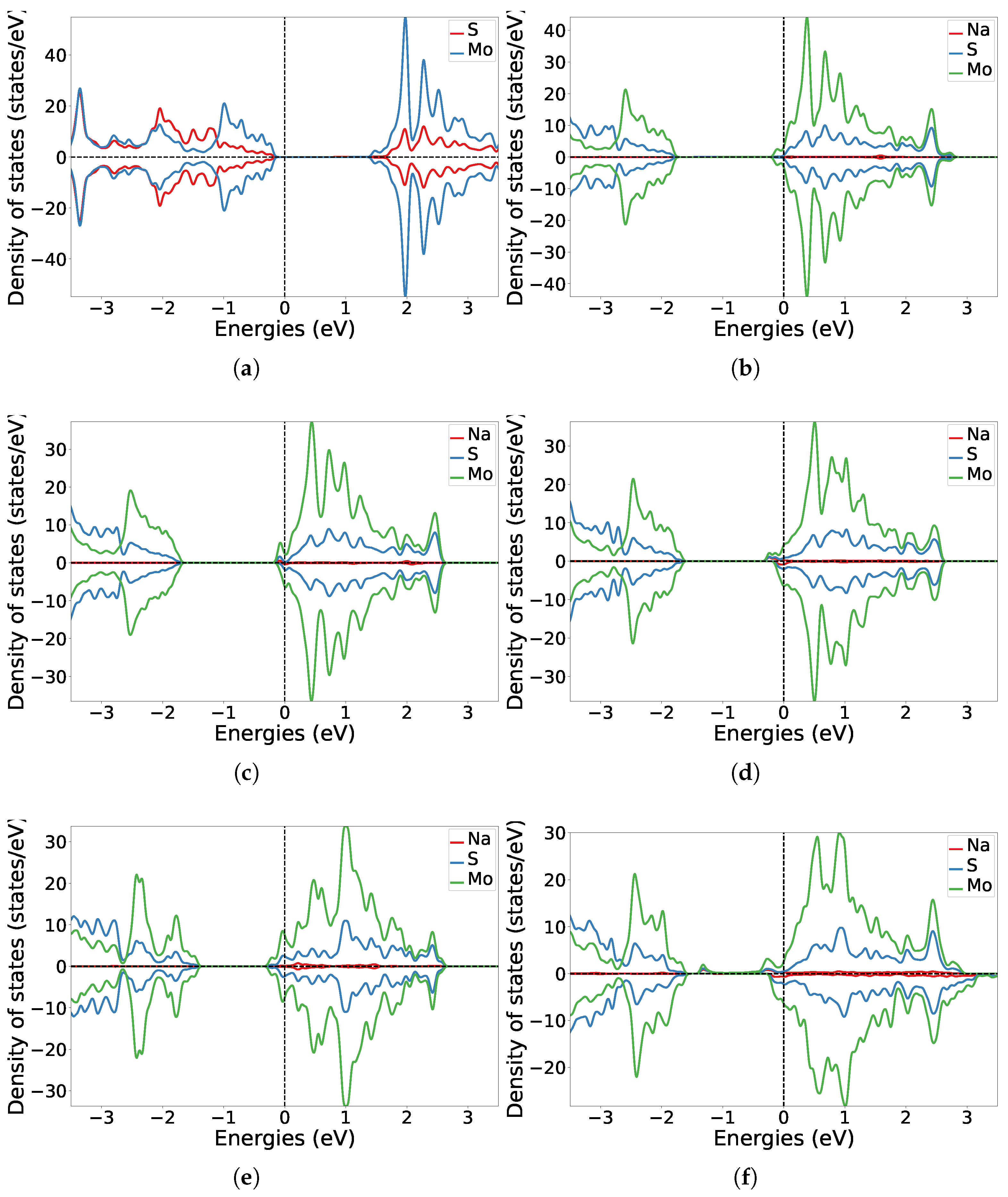
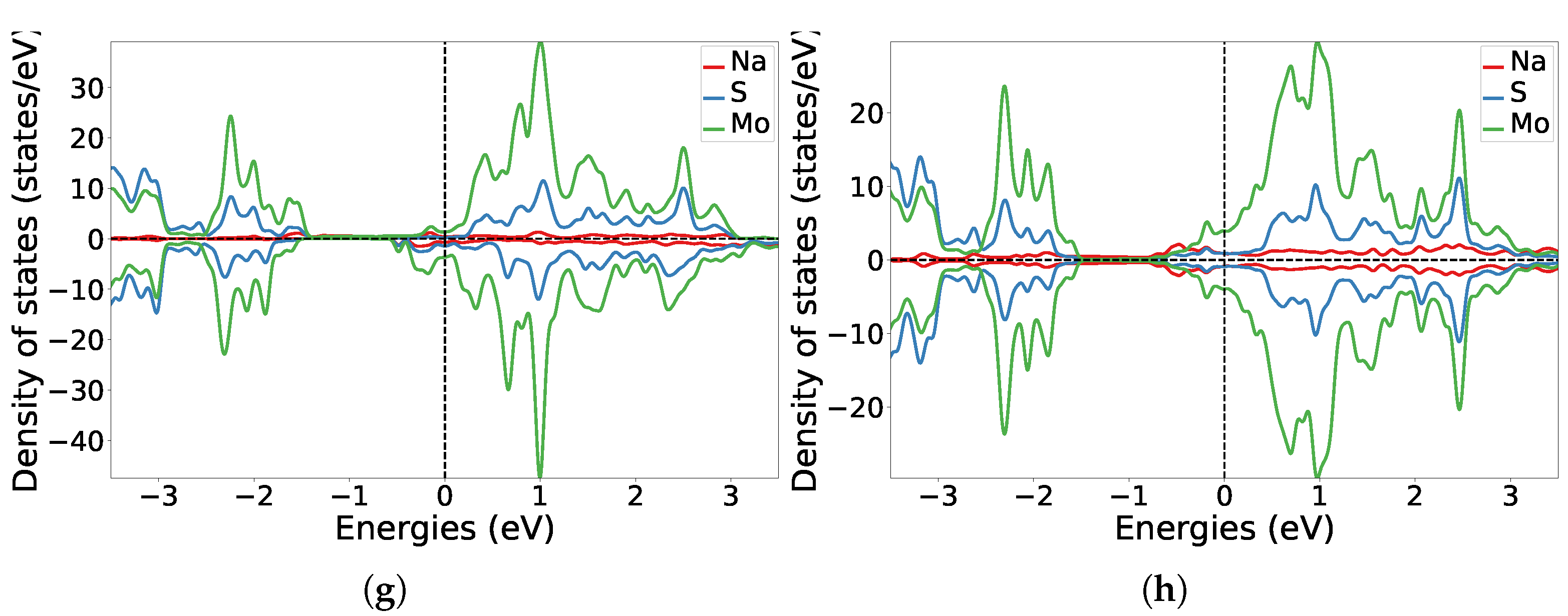
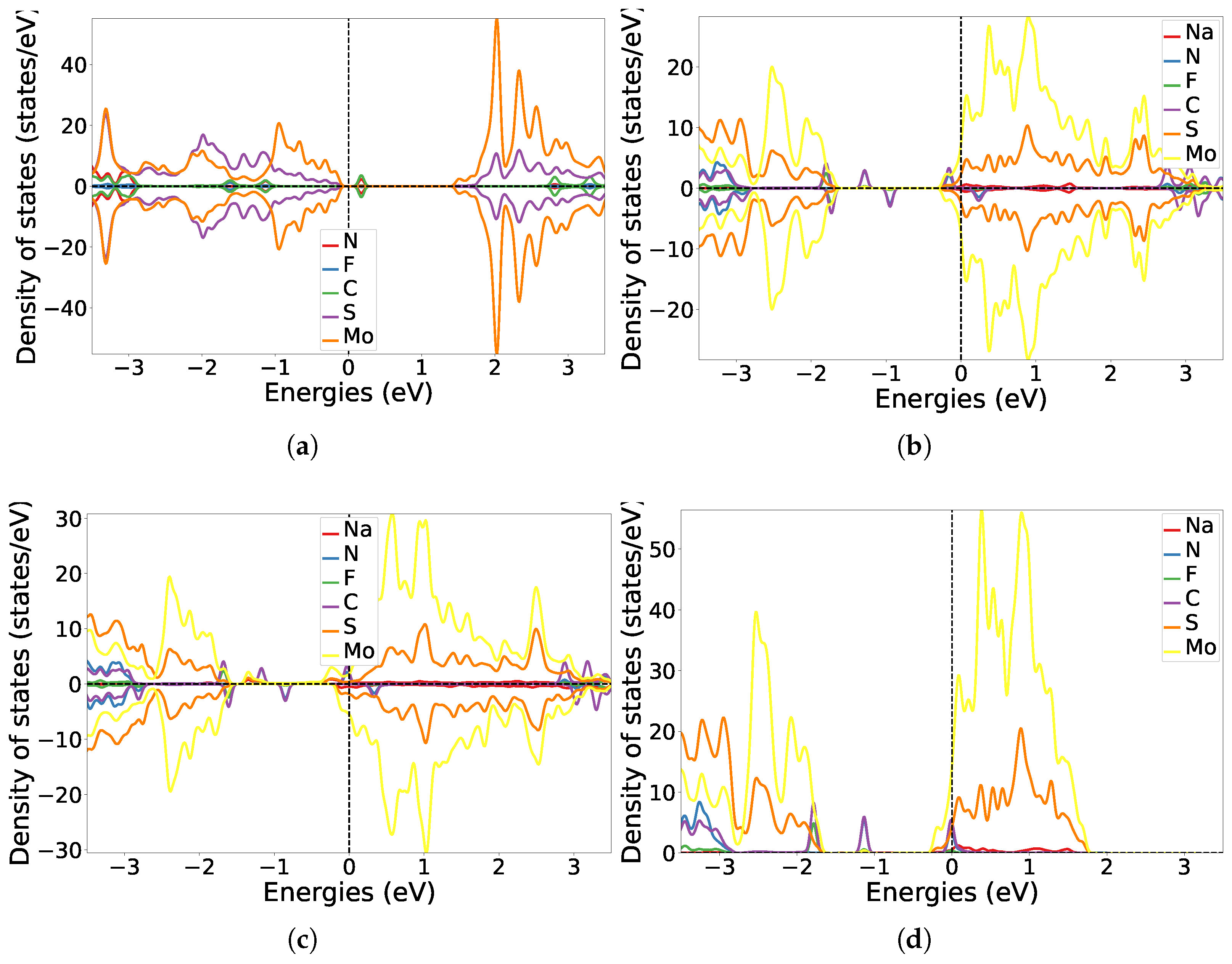
3.1. One-Sided Functionalisation with Only Donors
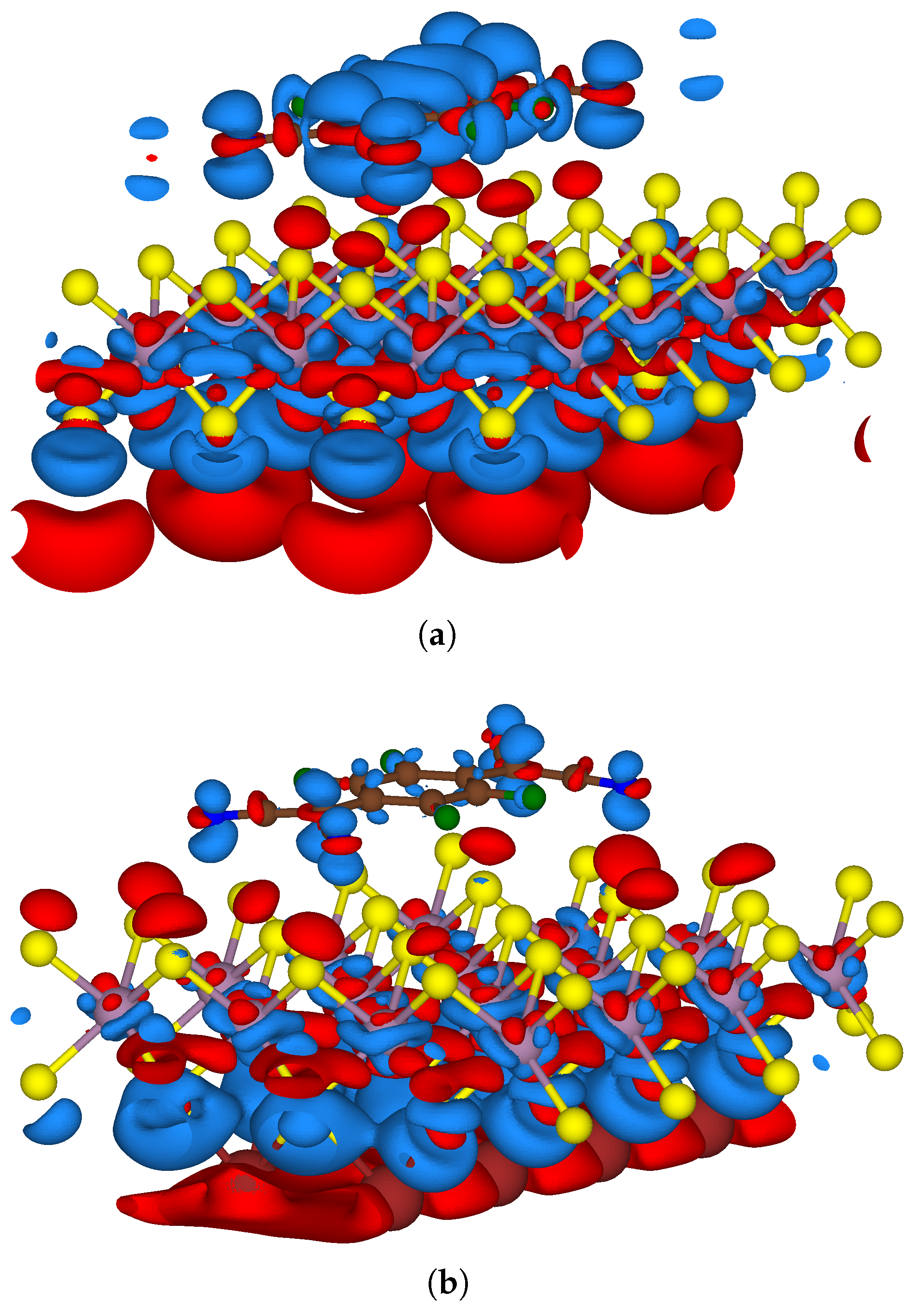
3.2. Two-Sided Functionalisation with Both Donor and Acceptor
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TMD | transition metal dichalcogenide |
| 2DM | two-dimensional material |
| GIC | graphite intercalation compound |
| DOS | density of states |
| DFT | density functional theory |
| GGA | generalised gradient approximations |
| PBE | Perdew-Burke-Ernzerhof |
| HSE | Heyd–Scuseria–Ernzerhof |
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Jiang, D.; Schedin, F.; Booth, T.; Khotkevich, V.; Morozov, S.; Geim, A.K. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451–10453. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 2012, 7, 699–712. [Google Scholar] [CrossRef]
- Wilson, J.; Yoffe, A. The transition metal dichalcogenides discussion and interpretation of the observed optical, electrical and structural properties. Adv. Phys. 1969, 18, 193–335. [Google Scholar] [CrossRef]
- Yoffe, A. Layer compounds. Annu. Rev. Mater. Sci. 1973, 3, 147–170. [Google Scholar] [CrossRef]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef]
- Bratschitsch, R. Monolayer diodes light up. Nat. Nanotechnol. 2014, 9, 247–248. [Google Scholar] [CrossRef]
- Jena, D. Tunneling transistors based on graphene and 2-D crystals. Proc. IEEE 2013, 101, 1585–1602. [Google Scholar] [CrossRef]
- Wang, H.; Yu, L.; Lee, Y.H.; Shi, Y.; Hsu, A.; Chin, M.L.; Li, L.J.; Dubey, M.; Kong, J.; Palacios, T. Integrated circuits based on bilayer MoS2 transistors. Nano Lett. 2012, 12, 4674–4680. [Google Scholar] [CrossRef]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef]
- Bhimanapati, G.R.; Lin, Z.; Meunier, V.; Jung, Y.; Cha, J.; Das, S.; Xiao, D.; Son, Y.; Strano, M.S.; Cooper, V.R.; et al. Recent Advances in Two-Dimensional Materials beyond Graphene. ACS Nano 2015, 9, 11509–11539. [Google Scholar] [CrossRef] [PubMed]
- Voiry, D.; Goswami, A.; Kappera, R.; Silva, C.D.C.C.E.; Kaplan, D.; Fujita, T.; Chen, M.; Asefa, T.; Chhowalla, M. Covalent functionalization of monolayered transition metal dichalcogenides by phase engineering. Nat. Chem. 2014, 7, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Tareen, A.K.; Aslam, M.; Wang, R.; Zhang, Y.; Mahmood, A.; Ouyang, Z.; Zhang, H.; Guo, Z. Recent developments in emerging two-dimensional materials and their applications. J. Mater. Chem. C 2020, 8, 387–440. [Google Scholar] [CrossRef]
- Sarkar, D.; Xie, X.; Kang, J.; Zhang, H.; Liu, W.; Navarrete, J.; Moskovits, M.; Banerjee, K. Functionalization of Transition Metal Dichalcogenides with Metallic Nanoparticles: Implications for Doping and Gas-Sensing. Nano Lett. 2015, 15, 2852–2862. [Google Scholar] [CrossRef]
- Knirsch, K.C.; Berner, N.C.; Nerl, H.C.; Cucinotta, C.S.; Gholamvand, Z.; McEvoy, N.; Wang, Z.; Abramovic, I.; Vecera, P.; Halik, M.; et al. Basal-Plane Functionalization of Chemically Exfoliated Molybdenum Disulfide by Diazonium Salts. ACS Nano 2015, 9, 6018–6030. [Google Scholar] [CrossRef]
- Chen, X.; Berner, N.C.; Backes, C.; Duesberg, G.S.; McDonald, A.R. Functionalization of Two-Dimensional MoS2: On the Reaction Between MoS2 and Organic Thiols. Angew. Chem. Int. Ed. 2016, 55, 5803–5808. [Google Scholar] [CrossRef]
- Backes, C.; Berner, N.C.; Chen, X.; Lafargue, P.; LaPlace, P.; Freeley, M.; Duesberg, G.S.; Coleman, J.N.; McDonald, A.R. Functionalization of Liquid-Exfoliated Two-Dimensional 2H-MoS2. Angew. Chem. Int. Ed. 2015, 54, 2638–2642. [Google Scholar] [CrossRef]
- Wang, K.; Paulus, B. Tuning the binding energy of excitons in the MoS2 monolayer by molecular functionalization and defective engineering. Phys. Chem. Chem. Phys. 2020, 22, 11936–11942. [Google Scholar] [CrossRef]
- Wang, K.; Paulus, B. Toward a Comprehensive Understanding of Oxygen on MoS2: From Reaction to Optical Properties. J. Phys. Chem. C 2021, 125, 19544–19550. [Google Scholar] [CrossRef]
- Rangger, G.M.; Hofmann, O.T.; Romaner, L.; Heimel, G.; Bröker, B.; Blum, R.P.; Johnson, R.L.; Koch, N.; Zojer, E. F4TCNQ on Cu, Ag, and Au as prototypical example for a strong organic acceptor on coinage metals. Phys. Rev. B 2009, 79, 165306. [Google Scholar] [CrossRef]
- Hofmann, O.T.; Egger, D.A.; Zojer, E. Work-Function Modification beyond Pinning: When Do Molecular Dipoles Count? Nano Lett. 2010, 10, 4369–4374. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, O.T.; Atalla, V.; Moll, N.; Rinke, P.; Scheffler, M. Interface dipoles of organic molecules on Ag(111) in hybrid density-functional theory. New J. Phys. 2013, 15, 123028. [Google Scholar] [CrossRef]
- Pingel, P.; Neher, D. Comprehensive picture of p-type doping of P3HT with the molecular acceptor F4TCNQ. Phys. Rev. B 2013, 87, 115209. [Google Scholar] [CrossRef]
- Duong, D.T.; Wang, C.; Antono, E.; Toney, M.F.; Salleo, A. The chemical and structural origin of efficient p-type doping in P3HT. Org. Electron. 2013, 14, 1330–1336. [Google Scholar] [CrossRef]
- Cochran, J.E.; Junk, M.J.N.; Glaudell, A.M.; Miller, P.L.; Cowart, J.S.; Toney, M.F.; Hawker, C.J.; Chmelka, B.F.; Chabinyc, M.L. Molecular Interactions and Ordering in Electrically Doped Polymers: Blends of PBTTT and F4TCNQ. Macromolecules 2014, 47, 6836–6846. [Google Scholar] [CrossRef]
- Wang, C.; Duong, D.T.; Vandewal, K.; Rivnay, J.; Salleo, A. Optical measurement of doping efficiency in poly(3-hexylthiophene) solutions and thin films. Phys. Rev. B 2015, 91, 085205. [Google Scholar] [CrossRef]
- Chen, W.; Chen, S.; Qi, D.C.; Gao, X.Y.; Wee, A.T.S. Surface Transfer p-Type Doping of Epitaxial Graphene. J. Am. Chem. Soc. 2007, 129, 10418–10422. [Google Scholar] [CrossRef]
- Coletti, C.; Riedl, C.; Lee, D.S.; Krauss, B.; Patthey, L.; von Klitzing, K.; Smet, J.H.; Starke, U. Charge neutrality and band-gap tuning of epitaxial graphene on SiC by molecular doping. Phys. Rev. B 2010, 81, 235401. [Google Scholar] [CrossRef]
- Wang, X.; Xu, J.B.; Xie, W.; Du, J. Quantitative Analysis of Graphene Doping by Organic Molecular Charge Transfer. J. Phys. Chem. C 2011, 115, 7596–7602. [Google Scholar] [CrossRef]
- Song, J.; Kam, F.Y.; Png, R.Q.; Seah, W.L.; Zhuo, J.M.; Lim, G.K.; Ho, P.K.H.; Chua, L.L. A general method for transferring graphene onto soft surfaces. Nat. Nanotechnol. 2013, 8, 356–362. [Google Scholar] [CrossRef]
- Kumar, A.; Banerjee, K.; Dvorak, M.; Schulz, F.; Harju, A.; Rinke, P.; Liljeroth, P. Charge-Transfer-Driven Nonplanar Adsorption of F4TCNQ Molecules on Epitaxial Graphene. ACS Nano 2017, 11, 4960–4968. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xia, F.; Shao, Z.; Zhao, J.; Jie, J. Surface Charge Transfer Doping of Monolayer Phosphorene via Molecular Adsorption. J. Phys. Chem. Lett. 2015, 6, 4701–4710. [Google Scholar] [CrossRef] [PubMed]
- Le, O.K.; Chihaia, V.; Van On, V.; Son, D.N. N-type and p-type molecular doping on monolayer MoS2. RSC Adv. 2021, 11, 8033–8041. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Schultz, T.; Xu, X.; Wegner, B.; Aljarb, A.; Han, A.; Li, L.J.; Tung, V.C.; Amsalem, P.; Koch, N. Demonstration of the key substrate-dependent charge transfer mechanisms between monolayer MoS2 and molecular dopants. Commun. Phys. 2019, 2, 109. [Google Scholar] [CrossRef]
- Park, S.; Wang, H.; Schultz, T.; Shin, D.; Ovsyannikov, R.; Zacharias, M.; Maksimov, D.; Meissner, M.; Hasegawa, Y.; Yamaguchi, T.; et al. Temperature-Dependent Electronic Ground-State Charge Transfer in van der Waals Heterostructures. Adv. Mater. 2021, 33, 2008677. [Google Scholar] [CrossRef]
- Dresselhaus, M.S. Intercalation in layered materials. MRS Bull. 1987, 12, 24–28. [Google Scholar] [CrossRef]
- Nobuhara, K.; Nakayama, H.; Nose, M.; Nakanishi, S.; Iba, H. First-principles study of alkali metal-graphite intercalation compounds. J. Power Sources 2013, 243, 585–587. [Google Scholar] [CrossRef]
- Rüdorff, W. Graphite intercalation compounds. In Advances in Inorganic Chemistry and Radiochemistry; Elsevier: Amsterdam, The Netherlands, 1959; Volume 1, pp. 223–266. [Google Scholar]
- Besenhard, J. The electrochemical preparation and properties of ionic alkali metal-and NR4-graphite intercalation compounds in organic electrolytes. Carbon 1976, 14, 111–115. [Google Scholar] [CrossRef]
- Dresselhaus, M.; Dresselhaus, G. Intercalation compounds of graphite. Adv. Phys. 1981, 30, 139–326. [Google Scholar] [CrossRef]
- Dolui, K.; Rungger, I.; Das Pemmaraju, C.; Sanvito, S. Possible doping strategies for MoS2monolayers: Anab initiostudy. Phys. Rev. B 2013, 88, 075420. [Google Scholar] [CrossRef]
- Rastogi, P.; Kumar, S.; Bhowmick, S.; Agarwal, A.; Chauhan, Y.S. Doping Strategies for Monolayer MoS2 via Surface Adsorption: A Systematic Study. J. Phys. Chem. C 2014, 118, 30309–30314. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes forab initiototal-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initioparametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Wigner, E.; Seitz, F. On the Constitution of Metallic Sodium. Phys. Rev. 1933, 43, 804–810. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Adsorption Site | Ebind (eV) | d(metal-ML) (Å) | Charge Transfer (e) |
|---|---|---|---|---|
| 1Na-MoS2 | M | −1.46 | 2.73 | 0.85 |
| 1Na-MoS2 | S | −1.05 | 2.60 | 0.79 |
| 1Na-MoS2 | H | −1.44 | 2.74 | 0.84 |
| 1Na-MoS2 | B | −1.46 | 2.74 | 0.84 |
| 1Li-MoS2 | M | −1.97 | 2.35 | 0.87 |
| 1K-MoS2 | M | −1.84 | 3.08 | 0.89 |
| 1Rb-MoS2 | M | −1.87 | 3.23 | 0.90 |
| 1Cs-MoS2 | M | −1.90 | 3.40 | 0.90 |
| System | Ebind (eV) | Ebind/Na (eV) | d(Na-ML) (Å) | Charge Transfer (e) |
|---|---|---|---|---|
| 1Na-MoS2 | −1.46 | −1.46 | 2.73 | 0.85 |
| 2Na-MoS2 | −2.62 | −1.31 | 2.74 | 0.67 |
| 4Na-MoS2(a) * | −4.60 | −1.15 | 2.80 | 0.51 |
| 4Na-MoS2(b) † | −4.92 | −1.23 | 2.67 | 0.79 |
| 8Na-MoS2 | −10.24 | −1.28 | 2.82 | 0.46 |
| 16Na-MoS2 | −20.64 | −1.29 | 2.93 | 0.36 |
| 32Na-MoS2 | −39.36 | −1.23 | 2.88 | 0.18 |
| System | Ebind (eV) | d(F4TCNQ-ML) (Å) | Charge Transfer (e) |
|---|---|---|---|
| F4TCNQ-MoS2 | −1.14 | 3.35 | −1.07 |
| F4TCNQ-MoS2-4Na | −2.05 | 3.24 | −1.22 |
| F4TCNQ-MoS2-8Na | −1.77 | 3.21 | −1.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ray, S.; Paulus, B. First-Principles Investigations of Two-Sided Functionalised MoS2 Monolayer. Nanomaterials 2025, 15, 193. https://doi.org/10.3390/nano15030193
Ray S, Paulus B. First-Principles Investigations of Two-Sided Functionalised MoS2 Monolayer. Nanomaterials. 2025; 15(3):193. https://doi.org/10.3390/nano15030193
Chicago/Turabian StyleRay, Sreejita, and Beate Paulus. 2025. "First-Principles Investigations of Two-Sided Functionalised MoS2 Monolayer" Nanomaterials 15, no. 3: 193. https://doi.org/10.3390/nano15030193
APA StyleRay, S., & Paulus, B. (2025). First-Principles Investigations of Two-Sided Functionalised MoS2 Monolayer. Nanomaterials, 15(3), 193. https://doi.org/10.3390/nano15030193