
Pore Engineering in Carbon Monoliths Through Soft Templating, In Situ Grown Graphene, and Post-Activation for CO2 Capture, H2 Storage, and Electrochemical Capacitor

 , , , ,
, , , ,  , , and
, , and
Abstract
1. Introduction
2. Experimental Section
2.1. Synthesis of CM and In Situ Incorporation of Graphene
2.2. Characterization of Samples
2.3. Gas Physisorption Experiments
2.4. Electrochemical Characterization
3. Results and Discussions
3.1. Formation of CM with Compositional and Structural Changes
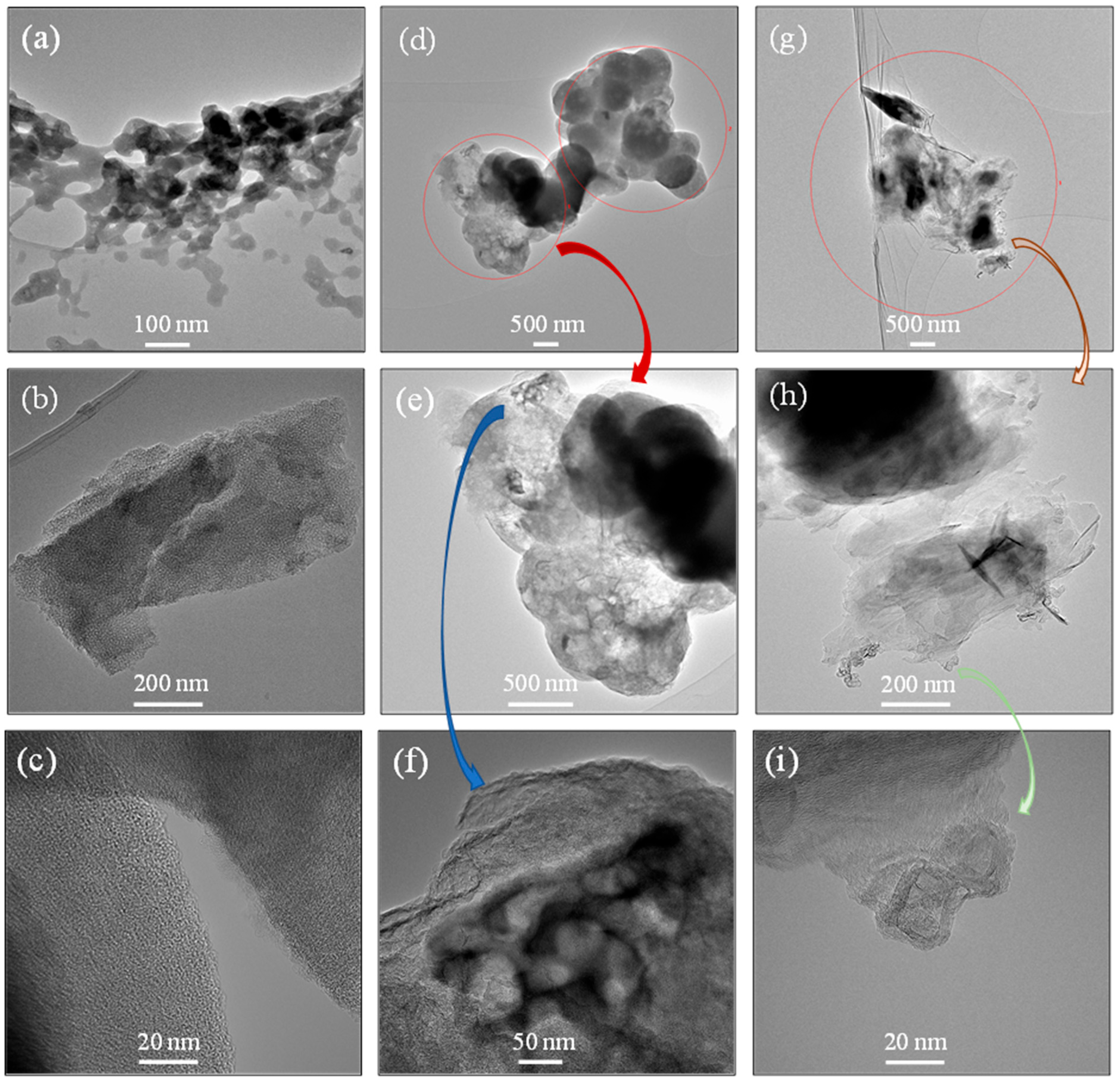
3.2. Morphological Changes and Porosity Creation
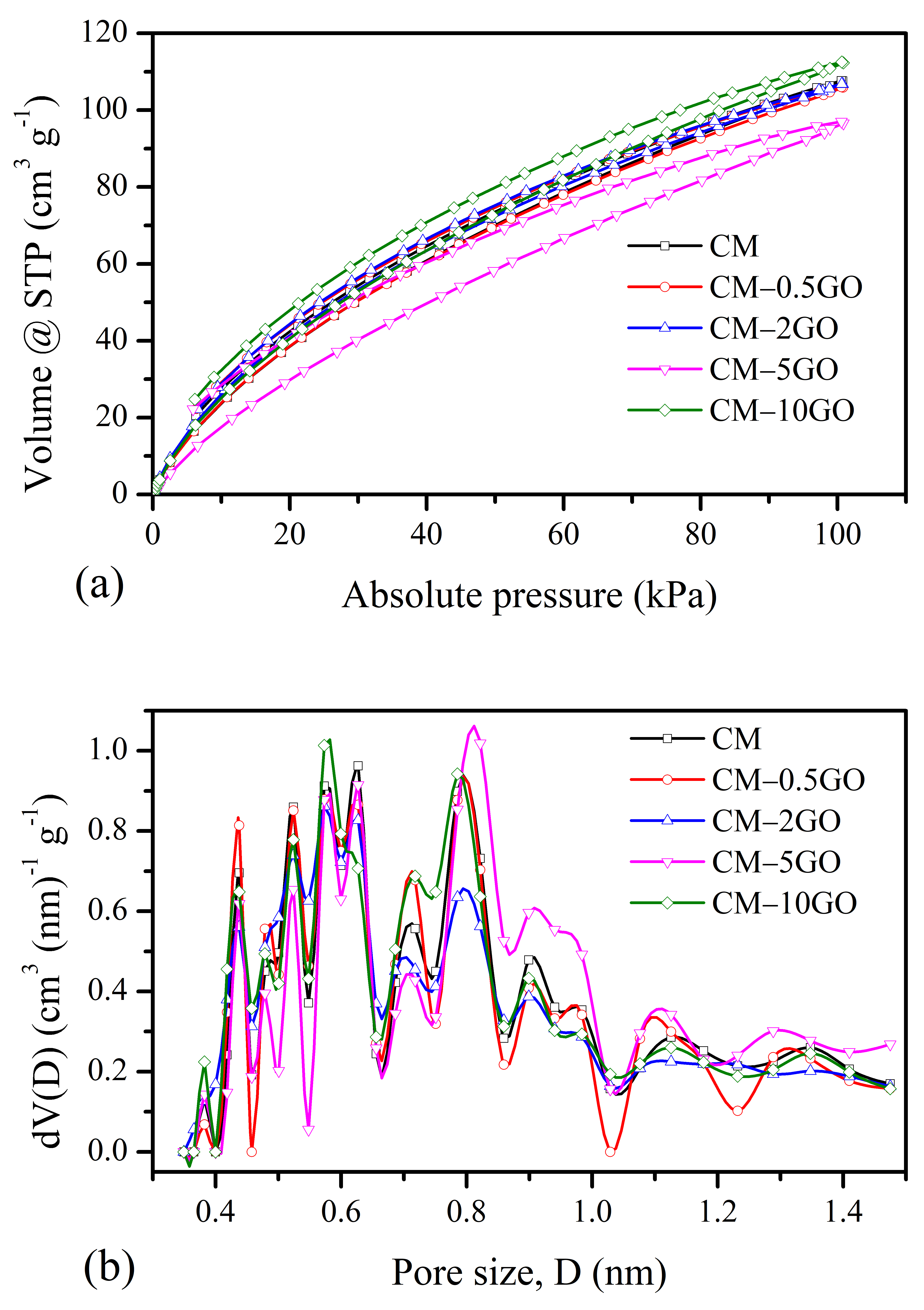
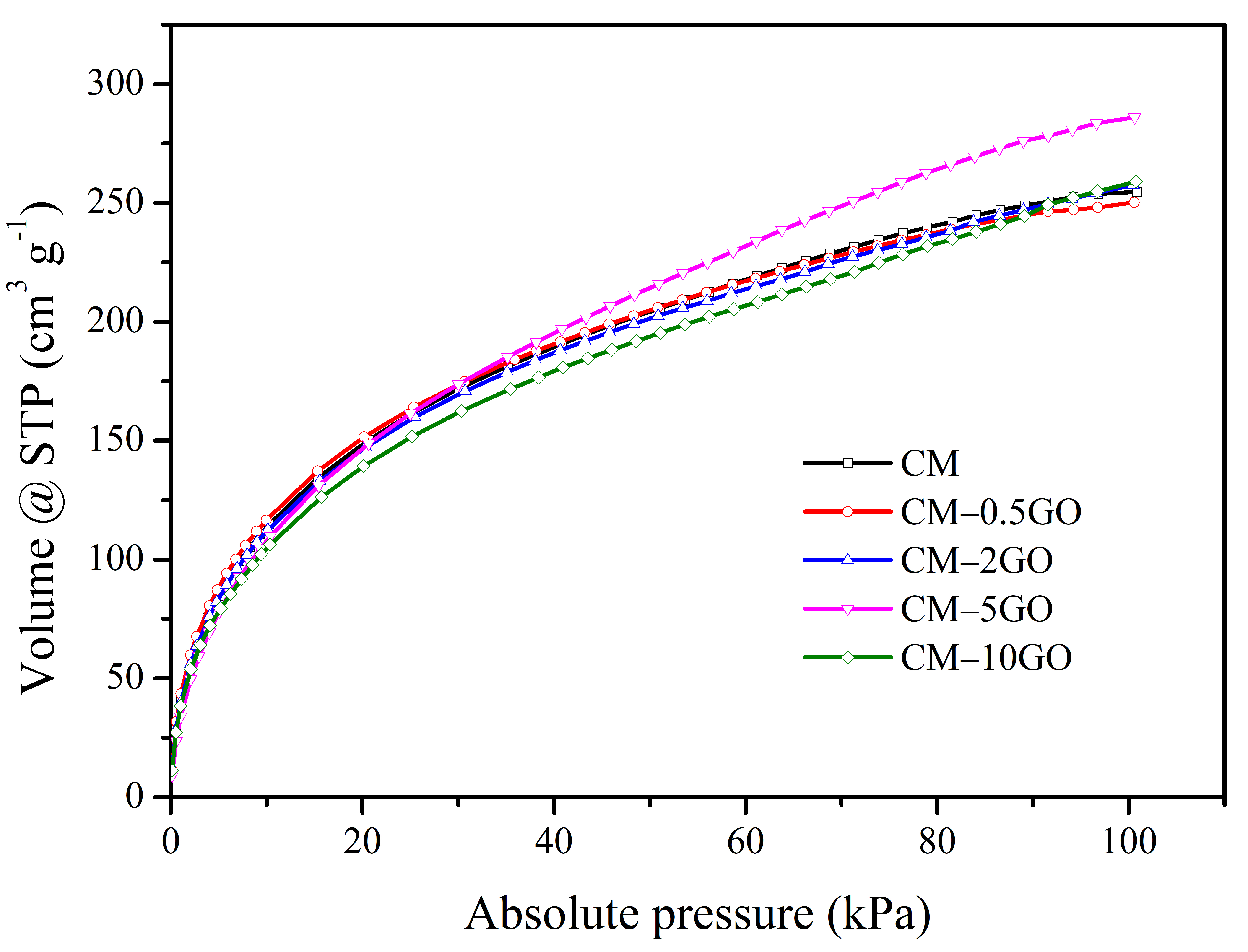
3.3. CO2 and H2 Sorption
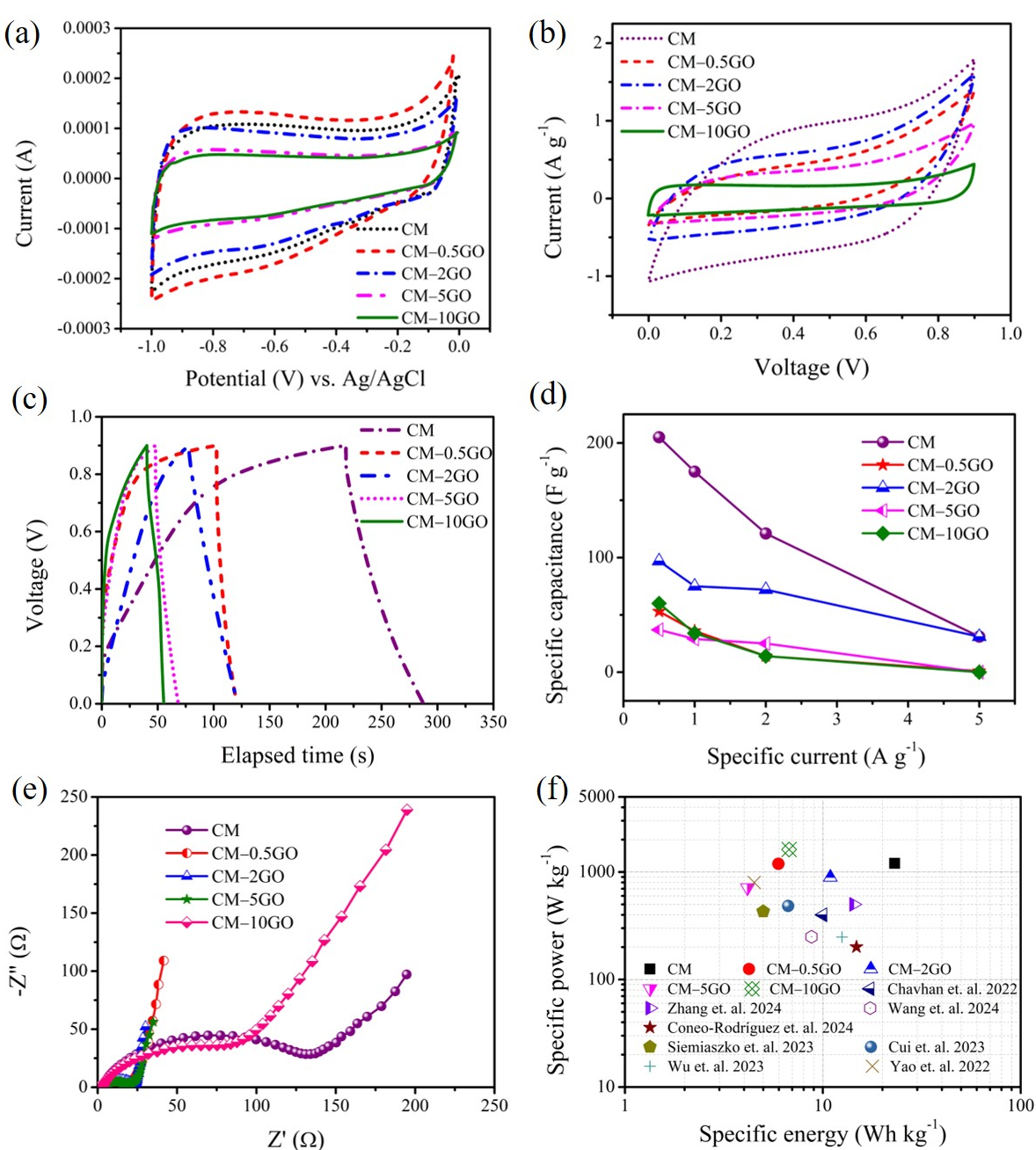
3.4. Electrochemical Performance
4. Conclusions
- For CO2 adsorption, the highest adsorption capacity is the result of the enhanced ultramicroporosity, as well as the wider micropores created in the CM-10GO sample. The increased polymerization rate from the addition of a higher concentration of 10% by weight GO loading leads to highly crosslinked network clusters developing over a smaller space, which, upon the use of pyrolysis protocols, leads to those micropore characteristics that are suitable for more CO2 adsorption. Furthermore, the high content of oxygen present in the structure of the CM-10GO sample additionally provides affinity for such CO2 adsorption.
- In the case of H2 storage, the highest significant value of adsorption capacity of 12.8 mmol g−1 (at −196 ° C and 101 kPa) is possible in the CM sample, derived from the GO with a loading of 5 wt %. The improved micropore volume in the entire pore sizes from 0.75 nm to 2 nm, as well as the significant accessibility provided by mesopores in the 2 to 3 nm sizes compared to other CM samples, are advantageous in achieving the highest adsorption capacity value for H2 storage.
- For CM electrodes, significant access for solvated ions provided by mesopores in the 4 to 6 nm sizes toward enhanced micropores improves the charge storage in the double layer. Such mesopore characteristics are created from the lower amount of GO loadings (0.5 to 2 wt %), where the relatively lower rate of polymerization leads to the formation of larger and fewer network clusters during polymerization compared to high GO loadings, which benefits CMs for the relatively higher values of electrochemical performance. Although more branching in the pore network at the lowest level of the pore hierarchy in graphene-incorporated CM electrodes offers more leakage resistance, the rate capability improves because of enhanced conduction from the incorporation of graphene.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xia, Y.; Yang, Z.; Zhu, Y. Porous Carbon-Based Materials for Hydrogen Storage: Advancement and Challenge. Mater. Chem. A 2013, 1, 9365–9381. [Google Scholar] [CrossRef]
- Blankenship, T.S.; Balahmar, N.; Mokaya, R. Oxygen-Rich Microporous Carbons with Exceptional Hydrogen Storage Capacity. Nat. Commun. 2017, 8, 1545. [Google Scholar] [CrossRef] [PubMed]
- Cabria, I.; López, M.J.; Alonso, J.A. The Optimum Average Nanopore Size for Hydrogen Storage in Carbon Nanoporous Materials. Carbon 2007, 45, 2649–2658. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Portet, C.; Osswald, S.; Simmons, J.M.; Yildirim, T.; Laudisio, G.; Fischer, J.E. Importance of Pore Size in High-Pressure Hydrogen Storage by Porous Carbons. Int. J. Hydrogen Energy 2009, 34, 6314–6319. [Google Scholar] [CrossRef]
- Zelenková, G.; Zelenka, T.; Almáši, M.; Soldánová, M. Graphene as a Promising Additive to Hierarchically Porous Carbon Monoliths for Enhanced H2and CO2sorption. J. CO2 Util. 2023, 68, 102371. [Google Scholar] [CrossRef]
- Rehman, A.; Park, S.J. Tunable Nitrogen-Doped Microporous Carbons: Delineating the Role of Optimum Pore Size for Enhanced CO2 Adsorption. Chem. Eng. J. 2019, 362, 731–742. [Google Scholar] [CrossRef]
- Chavhan, M.P.; Ganguly, S. Drying Stresses in Precursor Gel: Effect on Pore Connectivity in Carbonized Form, and Resulting Performance in a Supercapacitor Electrode. J. Sol-Gel Sci. Technol. 2018, 88, 395–406. [Google Scholar] [CrossRef]
- Xia, K.; Xiong, R.; Chen, Y.; Liu, D.; Tian, Q.; Gao, Q.; Han, B.; Zhou, C. Tuning the Pore Structure and Surface Chemistry of Porous Graphene for CO2 Capture and H2 Storage. Colloids Surf. A Physicochem. Eng. Asp. 2021, 622, 126640. [Google Scholar] [CrossRef]
- Xia, K.; Tian, X.; Fei, S.; You, K. Hierarchical Porous Graphene-Based Carbons Prepared by Carbon Dioxide Activation and Their Gas Adsorption Properties. Int. J. Hydrogen Energy 2014, 39, 11047–11054. [Google Scholar] [CrossRef]
- Zhang, N.; Gao, N.; Fu, C.; Liu, D.; Li, S.; Jiang, L.; Zhou, H.; Kuang, Y. Hierarchical Porous Carbon Spheres/Graphene Composite for Supercapacitor with Both Aqueous Solution and Ionic Liquid. Electrochim. Acta 2017, 235, 340–347. [Google Scholar] [CrossRef]
- Chang, J.; Gao, Z.; Liu, X.; Wu, D.; Xu, F.; Guo, Y.; Guo, Y.; Jiang, K. Hierarchically Porous Carbons with Graphene Incorporation for Efficient Supercapacitors. Electrochim. Acta 2016, 213, 382–392. [Google Scholar] [CrossRef]
- Yang, Z.; Ren, J.; Zhang, Z.; Chen, X.; Guan, G.; Qiu, L.; Zhang, Y.; Peng, H. Recent Advancement of Nanostructured Carbon for Energy Applications. Chem. Rev. 2015, 115, 5159–5223. [Google Scholar] [CrossRef] [PubMed]
- Emenike, E.C.; Odimayomi, K.P.; Iwuozor, K.O.; Ndagi, M.; Adeniyi, A.G. Synthesis of Activated Carbon Monolith from Lignocellulosic Material: Evaluation of Product Quality. MRS Adv. 2023, 8, 816–822. [Google Scholar] [CrossRef]
- Roberts, A.D.; Li, X.; Zhang, H. Porous Carbon Spheres and Monoliths: Morphology Control, Pore Size Tuning and Their Applications as Li-Ion Battery Anode Materials. Chem. Soc. Rev. 2014, 43, 4341–4356. [Google Scholar] [CrossRef]
- Hosseini, S.; Moghaddas, H.; Masoudi Soltani, S.; Kheawhom, S. Technological Applications of Honeycomb Monoliths in Environmental Processes: A Review. Process Saf. Environ. Prot. 2020, 133, 286–300. [Google Scholar] [CrossRef]
- Chavhan, M.P.; Musielak, M.; Slovák, V. Mesoporosity Enhancement in Carbon Monoliths through Incorporation of a Nonpolar Solvent with a Templating Source for Fast Adsorption of Organic Dyes. Microporous Mesoporous Mater. 2024, 366, 112919. [Google Scholar] [CrossRef]
- Chavhan, M.P.; Slovak, V.; Zelenkova, G.; Dominko, D. Revisiting the Effect of Pyrolysis Temperature and Type of Activation on the Performance of Carbon Electrodes in an Electrochemical Capacitor. Materials 2022, 15, 2431. [Google Scholar] [CrossRef]
- Rodríguez, N.; Agámez-Pertuz, Y.Y.; Romero, E.; Díaz-Velásquez, J.d.J.; Odriozola, J.A.; Centeno, M.Á. Effect of Starch as Binder in Carbon Aerogel and Carbon Xerogel Preparation. J. Non-Cryst. Solids 2019, 522, 119554. [Google Scholar] [CrossRef]
- Okolo, G.N.; Neomagus, H.W.J.P.; Everson, R.C.; Roberts, M.J.; Bunt, J.R.; Sakurovs, R.; Mathews, J.P. Chemical-Structural Properties of South African Bituminous Coals: Insights from Wide Angle XRD-Carbon Fraction Analysis, ATR-FTIR, Solid State 13C NMR, and HRTEM Techniques. Fuel 2015, 158, 779–792. [Google Scholar] [CrossRef]
- Collins, J.; Zheng, D.; Ngo, T.; Qu, D.; Foster, M. Partial Graphitization of Activated Carbon by Surface Acidification. Carbon 2014, 79, 500–517. [Google Scholar] [CrossRef]
- Lei, Q.; Song, H.; Chen, X.; Li, M.; Li, A.; Tang, B.; Zhou, D. Effects of Graphene Oxide Addition on the Synthesis and Supercapacitor Performance of Carbon Aerogel Particles. RSC Adv. 2016, 6, 40683–40690. [Google Scholar] [CrossRef]
- Hasegawa, G.; Yano, T.; Akamatsu, H.; Hayashi, K.; Nakanishi, K. Variation of Meso- and Macroporous Morphologies in Resorcinol–Formaldehyde (RF) Gels Tailored via a Sol–Gel Process Combined with Soft-Templating and Phase Separation. J. Sol-Gel Sci. Technol. 2020, 95, 801–812. [Google Scholar] [CrossRef]
- Szczęśniak, B.; Choma, J.; Jaroniec, M. Effect of Graphene Oxide on the Adsorption Properties of Ordered Mesoporous Carbons toward H2, C6H6, CH4 and CO2. Microporous Mesoporous Mater. 2018, 261, 105–110. [Google Scholar] [CrossRef]
- Kouketsu, Y.; Mizukami, T.; Mori, H.; Endo, S.; Aoya, M.; Hara, H.; Nakamura, D.; Wallis, S. A New Approach to Develop the Raman Aman Carbonaceous Material Geothermometer for Low-grade Metamorphism Using Peak Width. Isl. Arc 2014, 23, 33–50. [Google Scholar] [CrossRef]
- Wang, Y.; Alsmeyer, D.C.; McCreery, R.L. Raman Spectroscopy of Carbon Materials: Structural Basis of Observed Spectra. Chem. Mater. 1990, 2, 557–563. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman Spectra of Disordered and Amorphous Carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef]
- Katagiri, G.; Ishida, H.; Ishitani, A. Raman Spectra of Graphite Edge Planes. Carbon 1988, 26, 565–571. [Google Scholar] [CrossRef]
- Livneh, T.; Bar-Ziv, E.; Senneca, O.; Salatino, P. Evolution of Reactivity of Highly Porous Chars from Raman Microscopy. Combust. Sci. Technol. 2000, 153, 65–82. [Google Scholar] [CrossRef]
- Zaida, A.; Bar-Ziv, E.; Radovic, L.R.; Lee, Y.J. Further Development of Raman Microprobe Spectroscopy for Characterization of Char Reactivity. Proc. Combust. Inst. 2007, 31, 1881–1887. [Google Scholar] [CrossRef]
- Bar-Ziv, E.; Zaida, A.; Salatino, P.; Senneca, O. Diagnostics of Carbon Gasification by Raman Microprobe Spectroscopy. Proc. Combust. Inst. 2000, 28, 2369–2374. [Google Scholar] [CrossRef]
- Raman, N.F.; Dippel, S.B.; Heintzenberg, J. Soot Characterization in Atmospheric Particles From Different Sources by NIR FT Raman spectroscopy. J. Aerosol Sci. 1999, 30, 907–908. [Google Scholar]
- Cuesta, A.; Dhamelincourt, P.; Laureyns, J.; Martínez-Alonso, A.; Tascón, J.M.D. Raman Microprobe Studies on Carbon Materials. Carbon 1994, 32, 1523–1532. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution ( IUPAC Technical Report ). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Rehman, A.; Park, S.J. Comparative Study of Activation Methods to Design Nitrogen-Doped Ultra-Microporous Carbons as Efficient Contenders for CO2 Capture. Chem. Eng. J. 2018, 352, 539–548. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, J.; Tan, J.; Ma, C.H.; Luo, S.; Li, W.; Liu, S. Hierarchical Porous Graphene Oxide/Carbon Foam Nanocomposites Derived from Larch for Enhanced CO2 Capture and Energy Storage Performance. J. CO2 Util. 2021, 52, 101666. [Google Scholar] [CrossRef]
- Hasegawa, G.; Kanamori, K.; Nakanishi, K.; Abe, T. New Insights into the Relationship between Micropore Properties, Ionic Sizes, and Electric Double-Layer Capacitance in Monolithic Carbon Electrodes. J. Phys. Chem. C 2012, 116, 26197–26203. [Google Scholar] [CrossRef]
- Zhang, M.; Zou, J.; Yan, Y.; Li, W.; Dai, Q.; Li, H.; Shi, Z.; Zhang, Z.; Wang, R.; Qiu, S. Adjustable Nanoarchitectonics of N-Doping Yolk-Shell Carbon Spheres via “Pyrolysis-Capture” Method for High-Performance Supercapacitor. J. Colloid Interface Sci. 2024, 674, 686–694. [Google Scholar] [CrossRef]
- Wang, G.; Hu, G.; Lan, J.; Miao, F.; Zhang, P.; Shao, G. Rational Design of One-Dimensional Skin-Core Multilayer Structure for Electrospun Carbon Nanofibers with Bicontinuous Electron/Ion Transport toward High-Performance Supercapacitors. J. Colloid Interface Sci. 2024, 653, 148–158. [Google Scholar] [CrossRef]
- Coneo-Rodríguez, R.; Tesio, A.Y.; Cometto, F.P.; Morales, G.M.; Planes, G.Á.; Caballero, A. Synergetic Combination of Carbon Xerogels, Graphene Oxide and Nano-ZnO for Aqueous and Organic Supercapacitors. Batter. Supercaps 2024, 8, e202400502. [Google Scholar] [CrossRef]
- Siemiaszko, G.; Breczko, J.; Hryniewicka, A.; Ilnicka, A.; Markiewicz, K.H.; Terzyk, A.P.; Plonska-Brzezinska, M.E. Composites Containing Resins and Carbon Nano-Onions as Efficient Porous Carbon Materials for Supercapacitors. Sci. Rep. 2023, 13, 6606. [Google Scholar] [CrossRef]
- Cui, X.; Jiang, Y.; He, Z.; Liu, Z.; Yang, X.; Wan, J.; Liu, Y.; Ma, F. Preparation of Tank-like Resin-Derived Porous Carbon Sphere for Supercapacitor: The Influence of KOH Activator and Activation Temperature on Structure and Performance. Diam. Relat. Mater. 2023, 136, 110054. [Google Scholar] [CrossRef]
- Wu, H.; Du, J.; Chen, A. N-Doped Hollow Porous Carbon Nanotubes Derived from in Situ Activation Approach for Supercapacitor. J. Mater. Sci. 2023, 58, 5362–5371. [Google Scholar] [CrossRef]
- Yao, H.; Zhang, J.; Du, J.; Li, B.; Zong, S.; Chen, A. Carbon Spheres Prepared by Amino Acid-Catalyzed Resorcinol-Formaldehyde Polymerization for Supercapacitors. J. Alloys Compd. 2022, 926, 166948. [Google Scholar] [CrossRef]
- Chavhan, M.P.; Pankaj; Ganguly, S. Charge Transport in Carbon Electrodes Made by Electrospray of Precursor Sol and Subsequent Carbonization in Situ. J. Solid State Electrochem. 2018, 22, 2149–2157. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Elemental Composition (%) | Structural and Lattice Parameters Calculated from XRD Analyses | |||||
|---|---|---|---|---|---|---|---|
| C | H | O | (A0) | (A0) | (A0) | ||
| CM | 92.20 | 1.07 | 6.70 | 3.94 | 12.74 | 34.81 | 4.23 |
| CM-0.5GO | 90.97 | 1.00 | 8.03 | 4.05 | 12.34 | 37.00 | 4.05 |
| CM-2GO | 89.23 | 0.96 | 9.67 | 3.94 | 12.12 | 42.80 | 4.07 |
| CM-5GO | 85.49 | 1.11 | 13.26 | 3.89 | 10.51 | 47.11 | 3.70 |
| CM-10GO | 82.95 | 1.63 | 15.36 | 3.93 | 11.19 | 43.03 | 3.85 |
| Sample | D4 | D1 | D3 | G | D2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Center | FWHM | Center | FWHM | Center | FWHM | Center | FWHM | Center | FWHM | |
| CM | 1245 | 355 | 1336 | 112 | 1465 | 294 | 1593 | 68 | 1596 | 333 |
| CM-0.5GO | 1245 | 282 | 1337 | 104 | 1466 | 297 | 1593 | 59 | 1600 | 261 |
| CM-2GO | 1245 | 249 | 1337 | 107 | 1467 | 196 | 1593 | 65 | 1597 | 218 |
| CM-5GO | 1245 | 263 | 1328 | 99 | 1517 | 276 | 1593 | 63 | 1597 | 1137 |
| CM-10GO | 1245 | 301 | 1343 | 127 | 1502 | 534 | 1593 | 62 | 1611 | 695 |
| Sample | QSDFT Pore Volume (cm3 g−1) | QSDFT Surface Area (m2 g−1) | BET area (m2 g−1) | ||
|---|---|---|---|---|---|
| Vmic | Vmeso | Smic | Smeso | ||
| CM | 0.512 | 0.434 | 1483 | 376 | 1751 |
| CM-0.5GO | 0.496 | 0.519 | 1478 | 408 | 1727 |
| CM-2GO | 0.611 | 0.583 | 1448 | 507 | 2154 |
| CM-5GO | 0.646 | 0.397 | 1452 | 464 | 2287 |
| CM-10GO | 0.518 | 0.273 | 1331 | 257 | 1661 |
| Sample | CO2 Adsorption (101 kPa, 0 °C) | Micropore (<1.47 nm) | Ultramicropores (≤0.72 nm) | ||||
|---|---|---|---|---|---|---|---|
| cm3(STP) g−1 | mmol g−1 | wt % | Vmic (cm3 g−1) | Smic (m2 g−1) | Vumic (cm3 g−1) | Sumic (m2 g−1) | |
| CM | 107.6 | 4.80 | 21.13 | 0.424 | 1153 | 0.184 | 666 |
| CM-0.5GO | 105.9 | 4.72 | 20.79 | 0.402 | 1115 | 0.189 | 683 |
| CM-2GO | 106.8 | 4.76 | 20.96 | 0.398 | 1120 | 0.192 | 707 |
| CM-5GO | 96.6 | 4.31 | 18.96 | 0.44 | 1108 | 0.145 | 515 |
| CM-10GO | 112.3 | 5.01 | 22.05 | 0.427 | 1182 | 0.194 | 701 |
| Sample | H2 Adsorption (101 kPa, −196 °C) | ||
|---|---|---|---|
| cm3(STP) g−1 | mmol g−1 | wt % | |
| CM | 255 | 11.4 | 2.3 |
| CM-0.5GO | 250 | 11.2 | 2.3 |
| CM-2GO | 258 | 11.5 | 2.3 |
| CM-5GO | 286 | 12.8 | 2.6 |
| CM-10GO | 259 | 11.6 | 2.3 |
| Equivalent Electric Circuit (Ladder Model) | 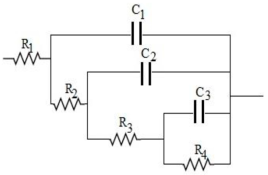 | |||||||
|---|---|---|---|---|---|---|---|---|
| Type of Electrode | R1 (Ω) | C1 (F) | R2 (Ω) | C2 (F) | R3 (Ω) | C3 (F) | R4 (Ω) | Chi2 |
| CM | 3.8 | 1.9 × 10−4 | 41 | 6.4 × 10−4 | 84.1 | 0.075 | 125 | 1.2 × 10−2 |
| CM-0.5GO | 3.2 | 2.0 × 10−4 | 14 | 0.019 | 11.6 | 0.110 | 486 | 1.4 × 10−2 |
| CM-2GO | 3.2 | 2.2 × 10−4 | 12 | 0.003 | 7.9 | 0.279 | 307 | 4.7 × 10−2 |
| CM-5GO | 4.3 | 1.3 × 10−4 | 12 | 0.014 | 9.2 | 0.184 | 196 | 2.5 × 10−2 |
| CM-10GO | 3.4 | 1.2 × 10−4 | 36 | 5.8 × 10−4 | 78 | 0.014 | 1231 | 4.1 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chavhan, M.P.; Marzouki, M.; Jaouadi, M.; Ghodbane, O.; Zelenková, G.; Almasi, M.; Maříková, M.; Bezdicka, P.; Tolasz, J.; Murafa, N. Pore Engineering in Carbon Monoliths Through Soft Templating, In Situ Grown Graphene, and Post-Activation for CO2 Capture, H2 Storage, and Electrochemical Capacitor. Nanomaterials 2025, 15, 900. https://doi.org/10.3390/nano15120900
Chavhan MP, Marzouki M, Jaouadi M, Ghodbane O, Zelenková G, Almasi M, Maříková M, Bezdicka P, Tolasz J, Murafa N. Pore Engineering in Carbon Monoliths Through Soft Templating, In Situ Grown Graphene, and Post-Activation for CO2 Capture, H2 Storage, and Electrochemical Capacitor. Nanomaterials. 2025; 15(12):900. https://doi.org/10.3390/nano15120900
Chicago/Turabian StyleChavhan, Madhav P., Moomen Marzouki, Mouna Jaouadi, Ouassim Ghodbane, Gabriela Zelenková, Miroslav Almasi, Monika Maříková, Petr Bezdicka, Jakub Tolasz, and Natalija Murafa. 2025. "Pore Engineering in Carbon Monoliths Through Soft Templating, In Situ Grown Graphene, and Post-Activation for CO2 Capture, H2 Storage, and Electrochemical Capacitor" Nanomaterials 15, no. 12: 900. https://doi.org/10.3390/nano15120900
APA StyleChavhan, M. P., Marzouki, M., Jaouadi, M., Ghodbane, O., Zelenková, G., Almasi, M., Maříková, M., Bezdicka, P., Tolasz, J., & Murafa, N. (2025). Pore Engineering in Carbon Monoliths Through Soft Templating, In Situ Grown Graphene, and Post-Activation for CO2 Capture, H2 Storage, and Electrochemical Capacitor. Nanomaterials, 15(12), 900. https://doi.org/10.3390/nano15120900