
Controlling the Morphology of Poly(ethylene glycol)-b-poly(lactide) Self-Assemblies in Solution: Interplay of Homopolymer Additives and Kinetic Traps

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Methods
2.1. Materials
2.2. Fabrication of BCP Self-Assembled Structures
2.3. TEM Characterization
2.4. Reverse Shape Transition
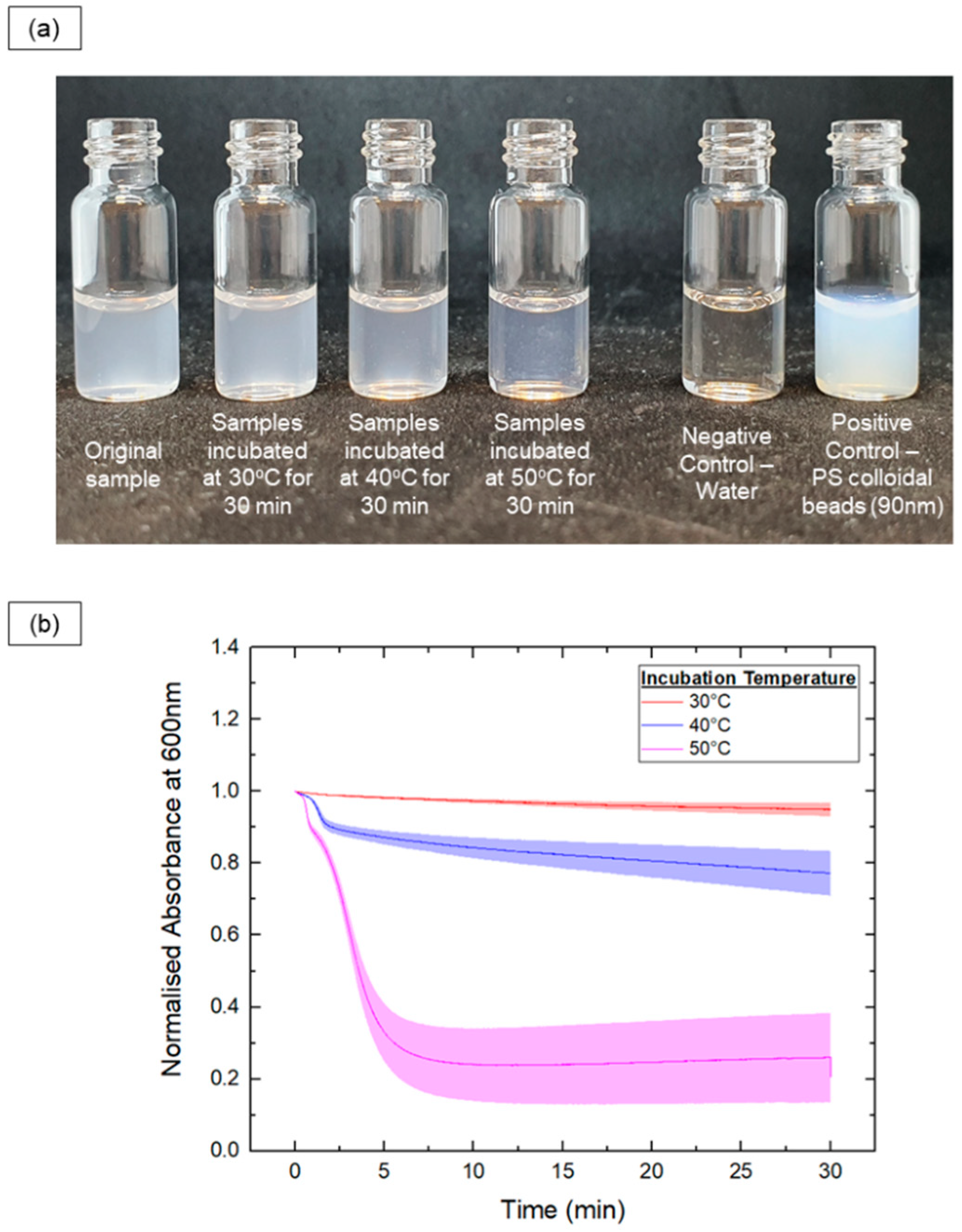
2.5. UV-Vis Characterization
2.6. Cargo Release from Vesicle Samples
2.7. Statistical Analysis
3. Results
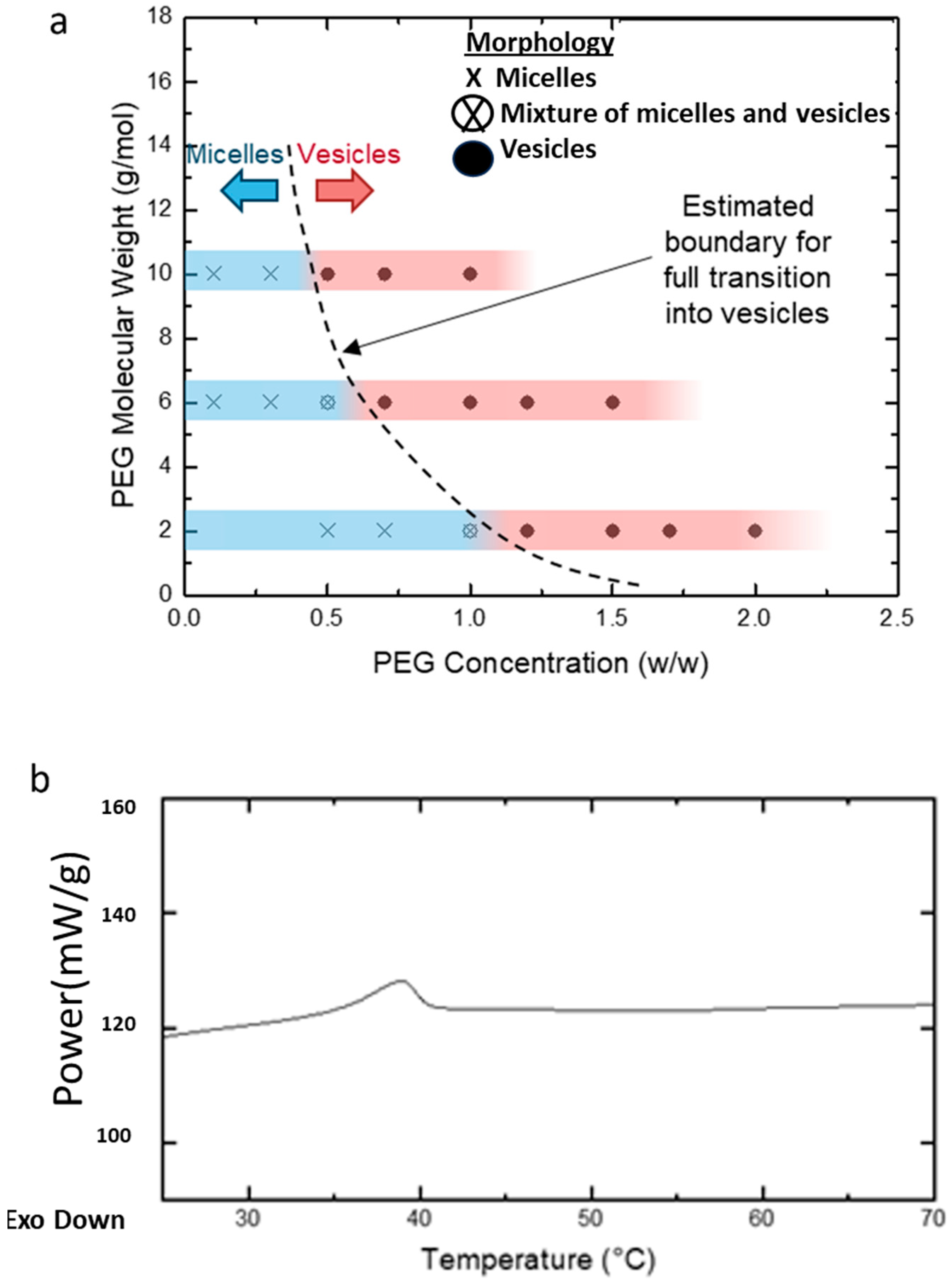
3.1. Presence of Free Homopolymer Causes the Micelle-to-Vesicle Transition
3.2. Shape Transition Is Reversible upon the Homopolymers’ Removal and Is Time- and Temperature-Dependent
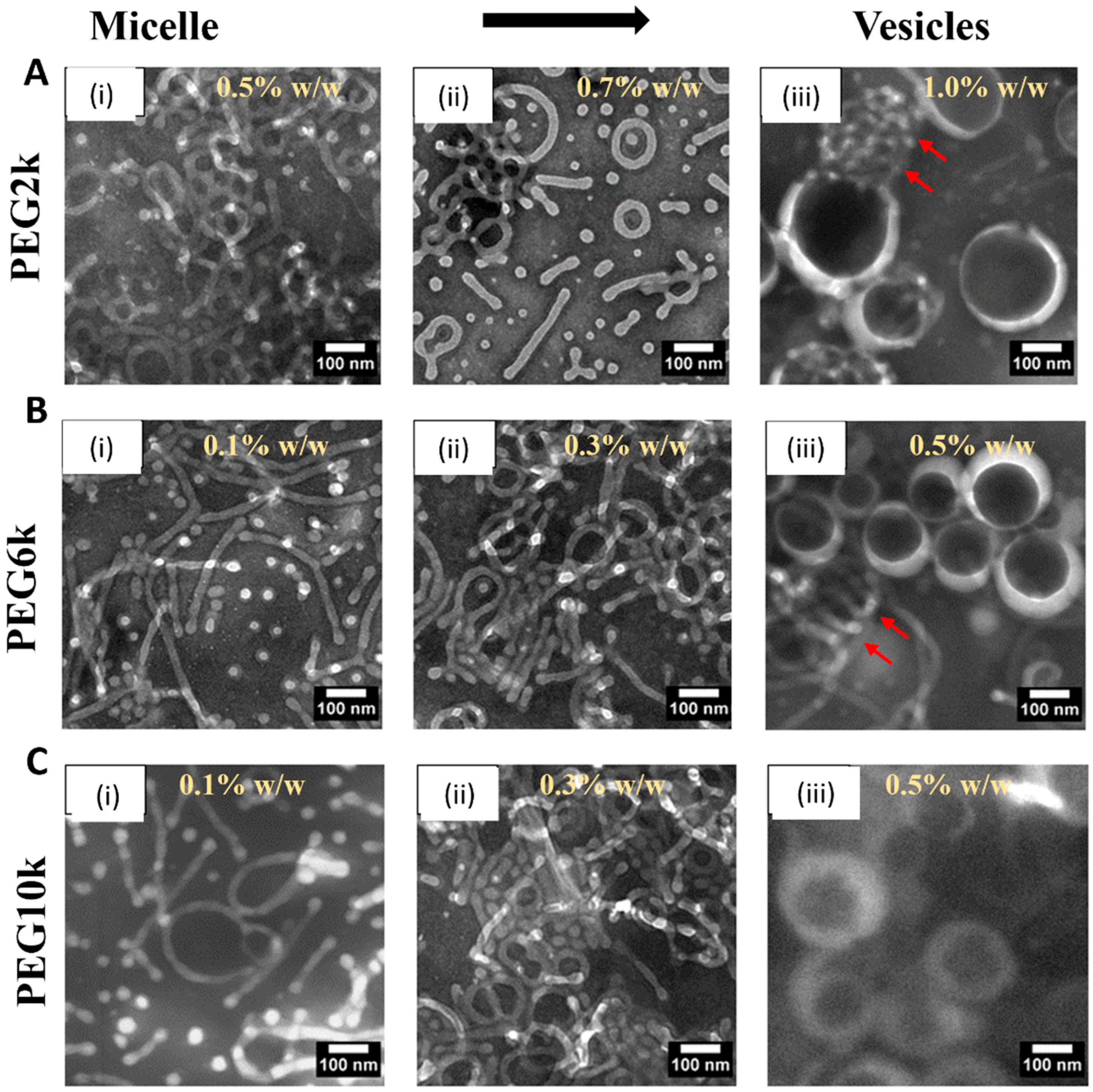
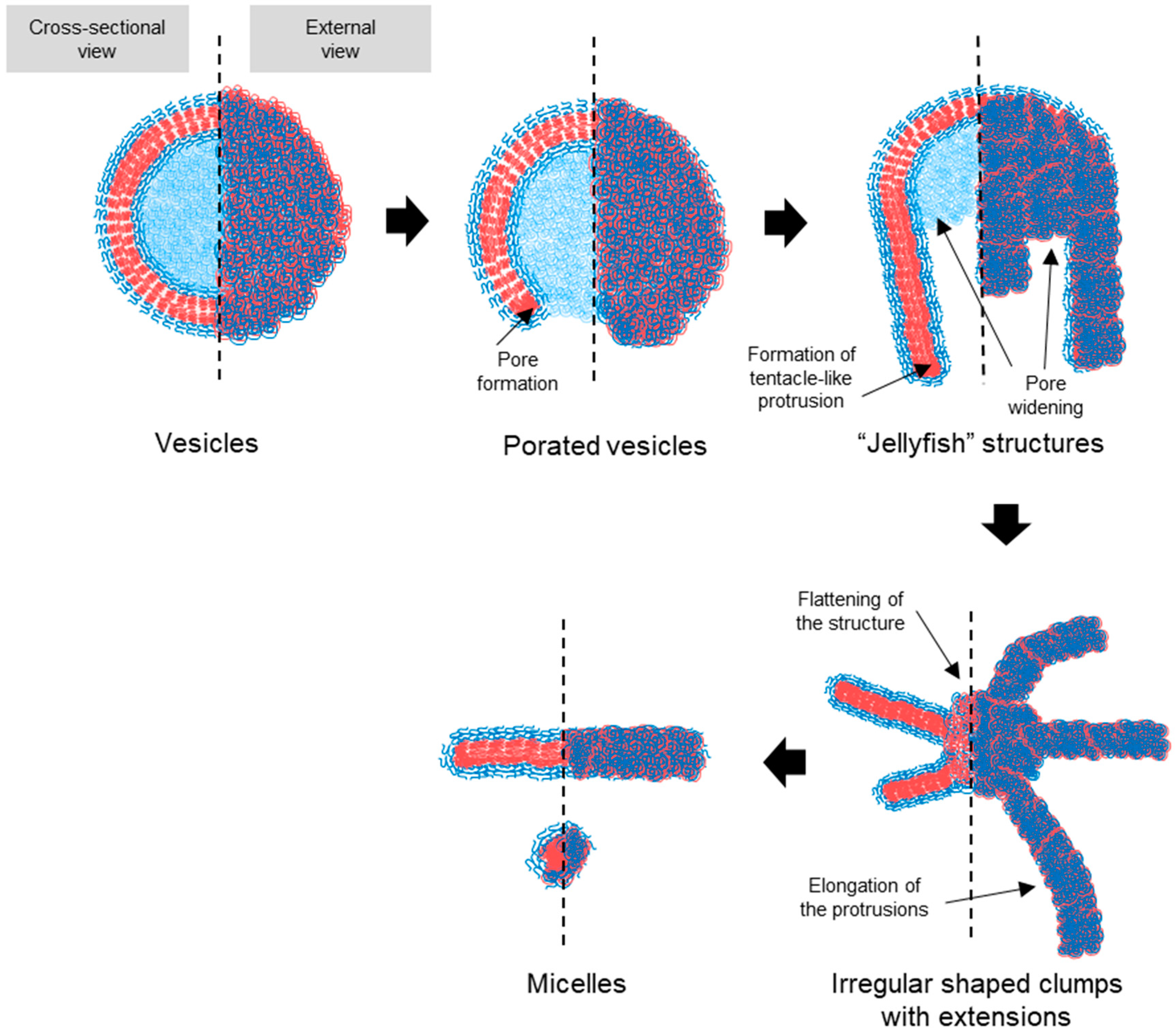
3.3. Transition Intermediates During Vesicle-to-Micelle Conversion and Their Relation to Packing Geometry
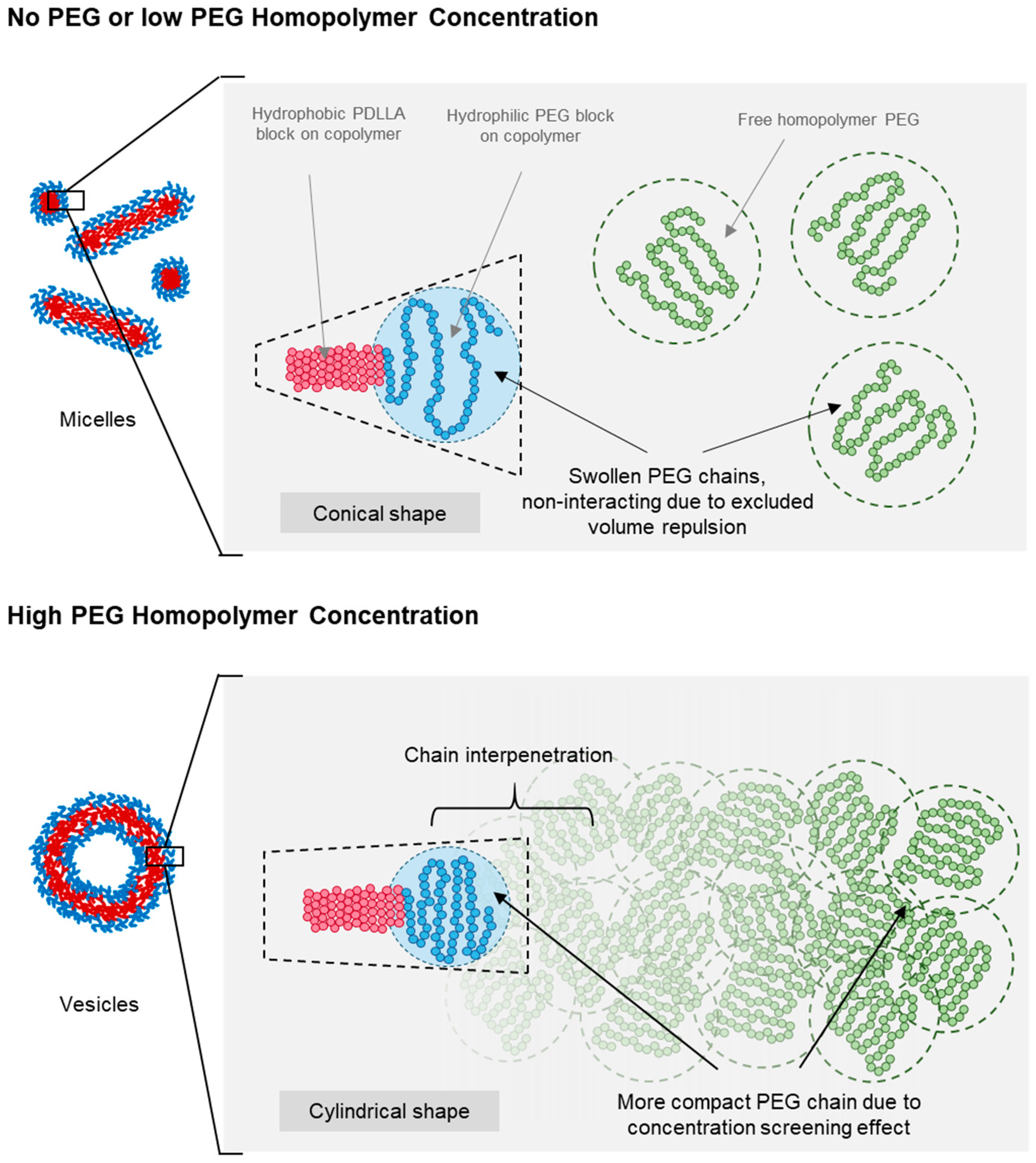
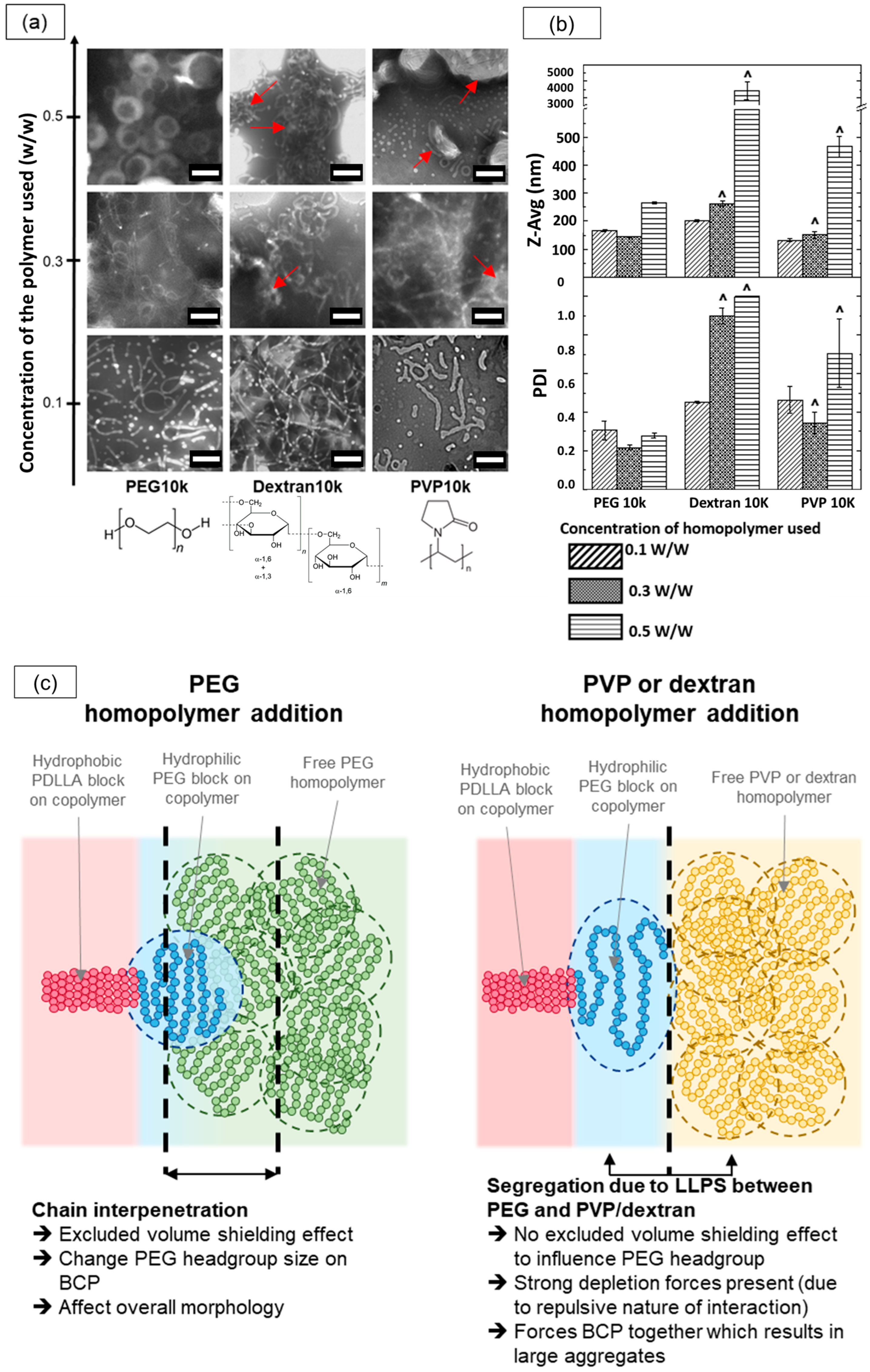
3.4. Effect of PEG Homopolymers on PEG-PLA Self-Assembly
3.5. Shape Transition in PEG-PLA BCPs Induced by PEG Homopolymers
3.6. The Shape Transition Effect Was Not Observed with Other Non-Interacting Hydrophilic Homopolymers
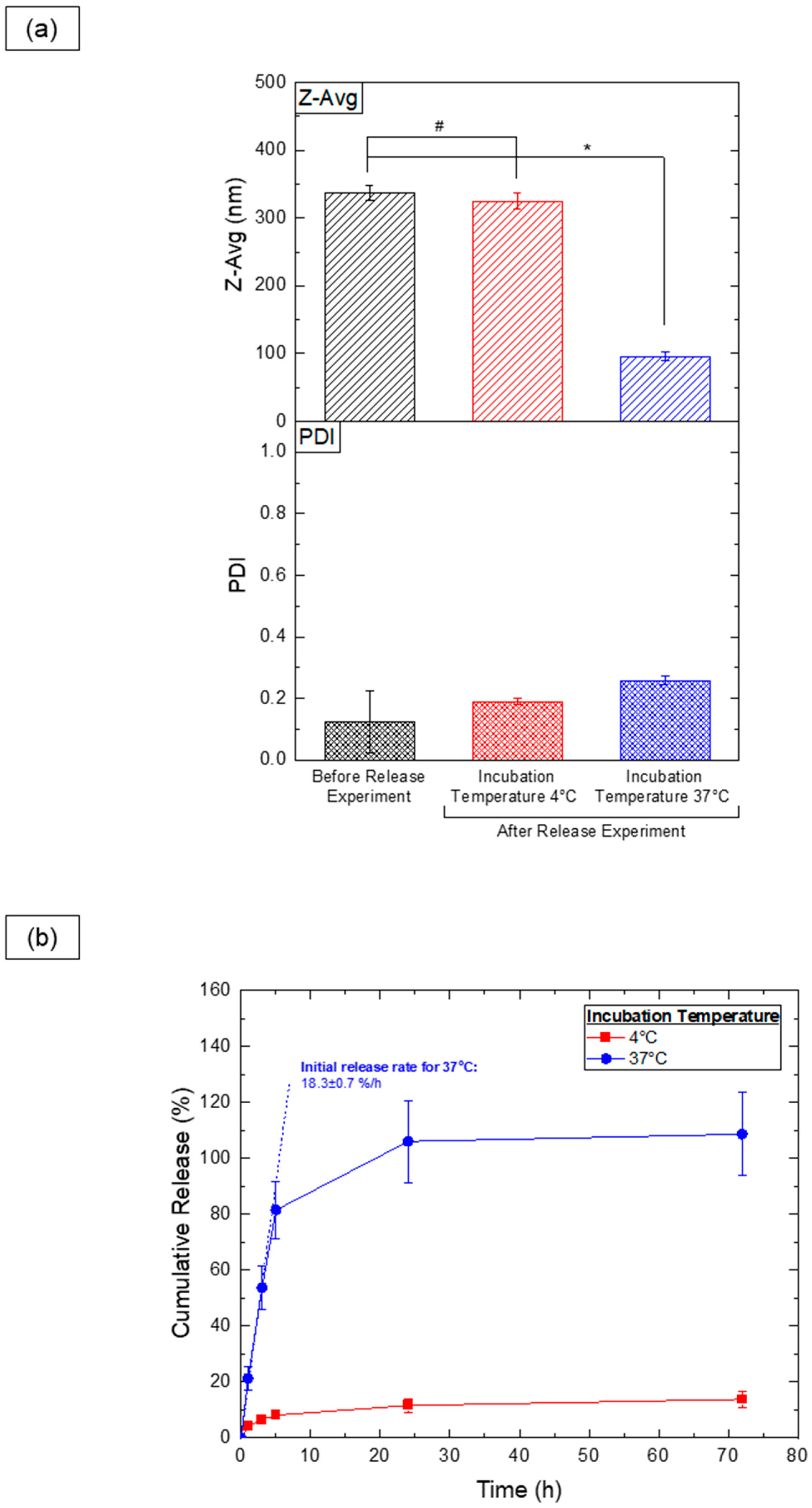
3.7. Thermoresponsive Cargo Delivery Systems
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mai, Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Q.; Kaneti, Y.V.; Hou, D.; Yamauchi, Y.; Mai, Y. Self-assembly of block copolymers towards mesoporous materials for energy storage and conversion systems. Chem. Soc. Rev. 2020, 49, 4681–4736. [Google Scholar] [CrossRef] [PubMed]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.; Singh, S.; Horechyy, A.; Fery, A.; Nandan, B. Block copolymer template-directed catalytic systems: Recent progress and perspectives. Membranes 2011, 11, 318. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Du, W.; Liu, C.; Li, Z.; Zhang, H.; Wei, L.; Yu, M. Self-Assembled Micellar Nanosensor toward pH with high photo-stability and its application in living cells. Sens. Actuators B Chem. 2018, 273, 927–934. [Google Scholar] [CrossRef]
- Chen, Q.; Schönherr, H.; Vancso, G.J. Block-Copolymer Vesicles as Nanoreactors for Enzymatic Reactions. Small 2009, 5, 1436–1445. [Google Scholar] [CrossRef]
- Lombardo, D.; Kiselev, M.A.; Magazù, S.; Calandra, P. Amphiphiles self-assembly: Basic concepts and future perspectives of supramolecular approaches. Adv. Condens. Matter. Phys. 2015, 2015, 151683. [Google Scholar] [CrossRef]
- Yu, K.; Eisenberg, A. Multiple morphologies in aqueous solutions of aggregates of polystyrene-block-poly (ethylene oxide) diblock copolymers. Macromolecules 1966, 29, 6359–6361. [Google Scholar] [CrossRef]
- Van Hest, J.C.M.; Delnoye, D.A.P.; Baars, M.; Van Genderen, M.H.P.; Meijer, E.W. Polystyrene-dendrimer amphiphilic block copolymers with a generation-dependent aggregation. Science 1995, 268, 1592–1595. [Google Scholar] [CrossRef]
- Deng, Z.; Liu, S. Emerging trends in solution self-assembly of block copolymers. Polymer 2020, 207, 122914. [Google Scholar] [CrossRef]
- Ianiro, A.; Hendrix, M.M.R.M.; Hurst, P.J.; Patterson, J.P.; Vis, M.; Sztucki, M.; Esteves, A.C.C.; Tuinier, R. Solvent selectivity governs the emergence of temperature responsiveness in block copolymer self-assembly. Macromolecules 2021, 54, 2912–2920. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Geng, Y.; Discher, D.E.; Yang, S. Temperature-controlled assembly and release from polymer vesicles of poly (ethylene oxide)-block-poly (N-isopropylacrylamide). Drug Deliv. 2006, 6, 7. [Google Scholar] [CrossRef]
- Choucair, A.; Eisenberg, A. Control of amphiphilic block copolymer morphologies using solution conditions. Eur. Phys. J. E 2003, 10, 37–44. [Google Scholar] [CrossRef]
- Matsen, M.W. Phase behavior of block copolymer/homopolymer blends. Macromolecules 1995, 28, 5765–5773. [Google Scholar] [CrossRef]
- Jeong, U.; Ryu, D.Y.; Kho, D.H.; Lee, D.H.; Kim, J.K.; Russell, T.P. Phase behavior of mixtures of block copolymer and homopolymers in thin films and bulk. Macromolecules 2003, 36, 3626–3634. [Google Scholar] [CrossRef]
- Kim, Y.; Mun, J.; Yu, G.; Char, K. Phase transition of block copolymer/homopolymer binary blends under 2D confinement. Macromol. Res. 2017, 25, 656–661. [Google Scholar] [CrossRef]
- García, A.G.; Ianiro, A.; Tuinier, R. On the colloidal stability of spherical copolymeric micelles. ACS Omega 2018, 3, 17976–17985. [Google Scholar] [CrossRef]
- Abbas, S.; Lodge, T.P. Depletion interactions: Effects of added homopolymer on ordered phases formed by spherical block copolymer micelles. Macromolecules 2008, 41, 8895–8902. [Google Scholar] [CrossRef]
- Watanabe, H.; Kotaka, T. Rheology of Ternary Mixtures of Styrene-Butadiene Diblock Copolymer, Homopolybutadiene, and n-Tetradecane. J. Rheol. 1983, 27, 223–240. [Google Scholar] [CrossRef]
- Malmsten, M.; Lindman, B. Effects of homopolymers on the gel formation in aqueous block copolymer solutions. Macromolecules 1993, 26, 1282–1286. [Google Scholar] [CrossRef]
- Constantinou, A.P.; Provatakis, N.; Li, Q.; Georgiou, T.K. Homopolymer and ABC Triblock Copolymer Mixtures for Thermoresponsive Gel Formulations. Gels 2021, 7, 116. [Google Scholar] [CrossRef]
- García, Á.G.; Ianiro, A.; Beljon, R.; Leermakers, F.A.M.; Tuinier, R. (Homo) polymer-mediated colloidal stability of micellar solutions. Soft Matter 2020, 16, 1560–1571. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Cowman, C.D.; Lodge, T.P. Effect of added homopolymer on the hexagonal phase formed by cylindrical block copolymer micelles in a selective solvent. Macromol. Rapid Commun. 2009, 30, 352–361. [Google Scholar] [CrossRef]
- Yang, T.; Lei, Z.; Yang, S.; Chen, E.-Q. Depletion driven self-assembly of block copolymer solutions by homopolymers. Phys. Chem. Chem. Phys. 2019, 21, 2121–2127. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Samal, S.K.; Biswal, M.; Mohanty, S.; Nayak, S.K. Preparation and characterization of poly (lactic acid)/poly (ethylene oxide) blend film: Effects of poly (ethylene oxide) and poly (ethylene glycol) on the properties. Polym. Int. 2019, 68, 164–172. [Google Scholar] [CrossRef]
- Ahmed, F.; Discher, D.E. Self-porating polymersomes of PEG–PLA and PEG–PCL: Hydrolysis-triggered controlled release vesicles. J. Control. Release 2004, 96, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Podgornik, R. Book Review: Polymer Physics; Rubinshtein, M., Colby, R.H., Eds.; Oxford University Press: London, UK, 2004. [Google Scholar]
- Taylor, W.; Jones, R.A.L. Producing high-density high-molecular-weight polymer brushes by a ‘grafting to’ method from a concentrated homopolymer solution. Langmuir 2010, 26, 13954–13958. [Google Scholar] [CrossRef] [PubMed]
- Apolinário, A.C.; Magoń, M.S.; Pessoa, A., Jr.; De Oliveira Rangel-Yagui, C. Challenges for the self-assembly of poly (ethylene glycol)–poly (lactic acid) (PEG-PLA) into polymersomes: Beyond the theoretical paradigms. Nanomaterials 2018, 8, 373. [Google Scholar] [CrossRef]
- Gurnev, P.A.; Stanley, C.B.; Aksoyoglu, M.A.; Hong, K.; Parsegian, V.A.; Bezrukov, S.M. Poly (ethylene glycol) s in Semidilute Regime: Radius of Gyration in the Bulk and Partitioning into a Nanopore. Macromolecules 2017, 50, 2477–2483. [Google Scholar] [CrossRef]
- Israelachvili, J. The different faces of poly (ethylene glycol). Proc. Natl. Acad. Sci. USA 1997, 94, 8378–8379. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Lodge, T.P. Depletion Interactions: A New Control Parameter for the Self-Assembly<? format?> of Diblock Copolymer Micelles. Phys. Rev. Lett. 2007, 99, 137802. [Google Scholar]
- Kourti, T. Turbidimetry in particle size analysis. In Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Shen, H.; Zhang, L.; Eisenberg, A. Multiple pH-induced morphological changes in aggregates of polystyrene-block-poly (4-vinylpyridine) in DMF/H2O mixtures. J. Am. Chem. Soc. 1999, 121, 2728–2740. [Google Scholar] [CrossRef]
- Zhang, L.; Eisenberg, A. Crew-cut aggregates from self-assembly of blends of polystyrene-b-poly (acrylic acid) block copolymers and homopolystyrene in solution. J. Polym. Sci. B Polym. Phys. 1999, 37, 1469–1484. [Google Scholar] [CrossRef]
- Liu, C.; Hong, C.-Y.; Pan, C.-Y. Polymerization techniques in polymerization-induced self-assembly (PISA). Polym. Chem. 2020, 11, 3673–3689. [Google Scholar] [CrossRef]
- Penfold, N.J.W.; Yeow, J.; Boyer, C.; Armes, S.P. Emerging trends in polymerization-induced self-assembly. ACS Macro Lett. 2019, 8, 1029–1054. [Google Scholar] [CrossRef] [PubMed]
- Huo, F.; Li, S.; He, X.; Shah, S.A.; Li, Q.; Zhang, W. Disassembly of block copolymer vesicles into nanospheres through vesicle mediated RAFT polymerization. Macromolecules 2014, 47, 8262–8269. [Google Scholar] [CrossRef]
- d’Agosto, F.; Rieger, J.; Lansalot, M. RAFT-mediated polymerization-induced self-assembly. Angew. Chem. Int. Ed. 2020, 59, 8368–8392. [Google Scholar] [CrossRef]
- Tiarks, F.; Frechen, T.; Kirsch, S.; Leuninger, J.; Melan, M.; Pfau, A.; Richter, F.; Schuler, B.; Zhao, C.-L. Formulation effects on the distribution of pigment particles in paints. Prog. Org. Coat. 2003, 48, 140–152. [Google Scholar] [CrossRef]
- Fernández-Peña, L.; Guzmán, E. Physicochemical aspects of the performance of hair-conditioning formulations. Cosmetics 2020, 7, 26. [Google Scholar] [CrossRef]
- Van Zee, N.J.; Hillmyer, M.A.; Lodge, T.P. Role of polymer excipients in the kinetic stabilization of drug-rich nanoparticles. ACS Appl. Bio Mater. 2020, 3, 7243–7254. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Minton, A.P. Join the crowd. Nature 2003, 425, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Donth, C.; Weiss, M. Quantitative assessment of the spatial crowding heterogeneity in cellular fluids. Phys. Rev. E 2019, 99, 052415. [Google Scholar] [CrossRef]
- Fu, X.; Hosta-Rigau, L.; Chandrawati, R.; Cui, J. Multi-stimuli-responsive polymer particles, films, and hydrogels for drug delivery. Chem 2018, 4, 2084–2107. [Google Scholar] [CrossRef]
- Pijpers, I.A.B.; Meng, F.; van Hest, J.C.M.; Abdelmohsen, L.K.E.A. Investigating the self-assembly and shape transformation of poly (ethylene glycol)-b-poly (d, l-lactide) (PEG-PDLLA) polymersomes by tailoring solvent-polymer interactions. Polym. Chem. 2020, 275–280. [Google Scholar] [CrossRef]
- Ghasemi, S.; Ahmadi, L.; Farjadian, F. Thermo-responsive PNIPAAm-b-PLA amphiphilic block copolymer micelle as nanoplatform for docetaxel drug release. J. Mater. Sci. 2022, 57, 17433–17447. [Google Scholar] [CrossRef]
- Cerritelli, S.; Velluto, D.; Hubbell, J.A. PEG-SS-PPS: Reduction-sensitive disulfide block copolymer vesicles for intracellular drug delivery. Biomacromolecules 2007, 8, 1966–1972. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, P.Q.; Vaibavi, S.R.; Parikh, A.N.; Venkatraman, S.; Czarny, B. Controlling the Morphology of Poly(ethylene glycol)-b-poly(lactide) Self-Assemblies in Solution: Interplay of Homopolymer Additives and Kinetic Traps. Nanomaterials 2024, 14, 2015. https://doi.org/10.3390/nano14242015
Lim PQ, Vaibavi SR, Parikh AN, Venkatraman S, Czarny B. Controlling the Morphology of Poly(ethylene glycol)-b-poly(lactide) Self-Assemblies in Solution: Interplay of Homopolymer Additives and Kinetic Traps. Nanomaterials. 2024; 14(24):2015. https://doi.org/10.3390/nano14242015
Chicago/Turabian StyleLim, Pei Qi, Srirangam Ramanujam Vaibavi, Atul N. Parikh, Subbu Venkatraman, and Bertrand Czarny. 2024. "Controlling the Morphology of Poly(ethylene glycol)-b-poly(lactide) Self-Assemblies in Solution: Interplay of Homopolymer Additives and Kinetic Traps" Nanomaterials 14, no. 24: 2015. https://doi.org/10.3390/nano14242015
APA StyleLim, P. Q., Vaibavi, S. R., Parikh, A. N., Venkatraman, S., & Czarny, B. (2024). Controlling the Morphology of Poly(ethylene glycol)-b-poly(lactide) Self-Assemblies in Solution: Interplay of Homopolymer Additives and Kinetic Traps. Nanomaterials, 14(24), 2015. https://doi.org/10.3390/nano14242015