
In Situ Incorporation of Atomically Precise Au Nanoclusters within Zeolites for Ambient Temperature CO Oxidation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
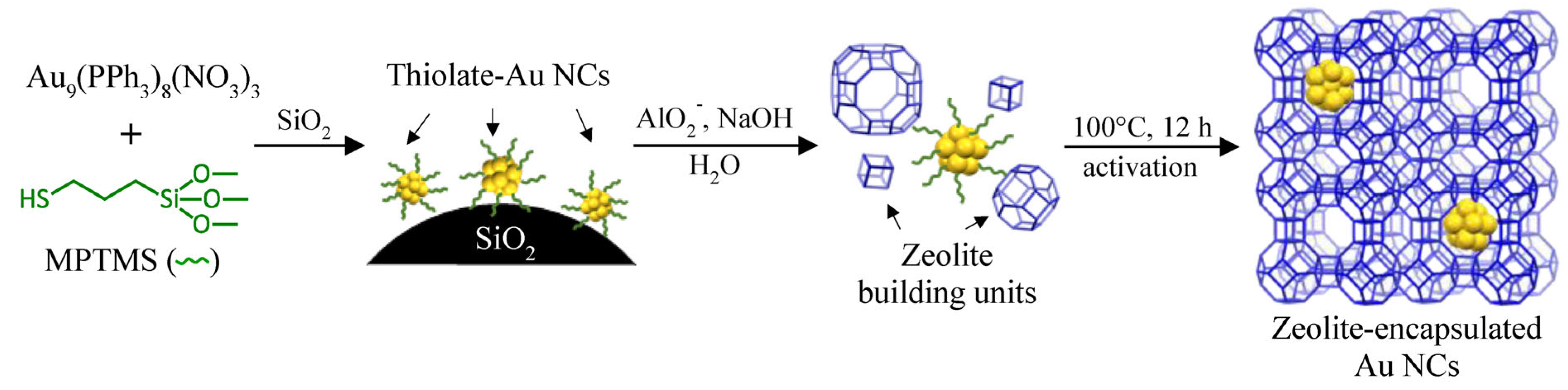
2.1. Materials Synthesis
2.1.1. Synthesis of Au9(PPh3)8(NO3)3 Nanoclusters
2.1.2. Ligand Exchange of Au9(PPh3)8(NO3)3 with (3-mercaptopropyl) Trimethoxysilane
2.1.3. Pre-Mixing of SiO2 with the Au9-MPTMS
2.1.4. Incorporation of Au9-MPTMS within Na-LTA Zeolite
2.1.5. Incorporation of Au9-MPTMS within Na-FAU Zeolite
2.2. Catalyst Activation
2.3. Ambient Temperature CO Oxidation for Testing of Activity and Encapsulation Efficiency
3. Results and Discussion
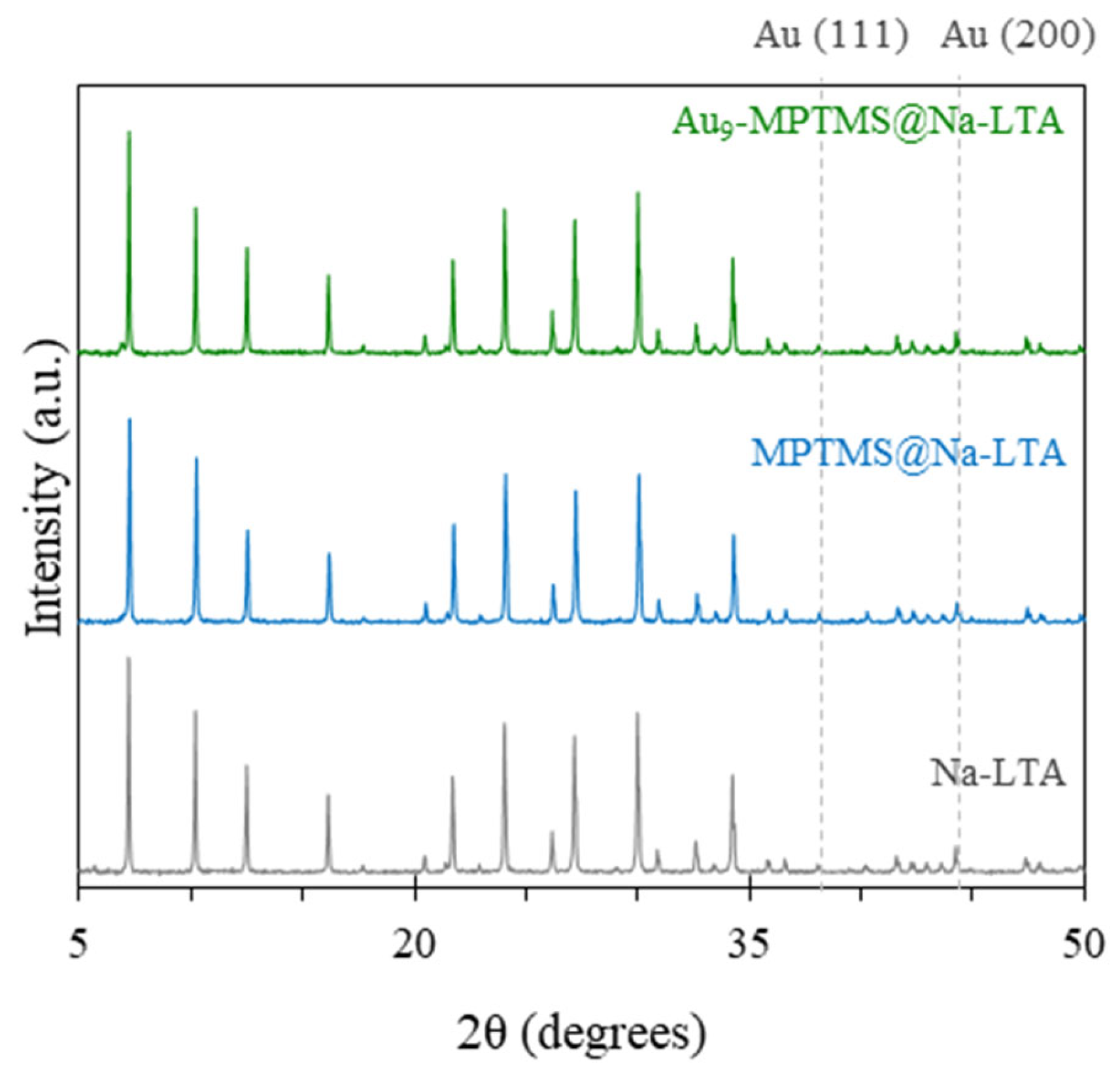
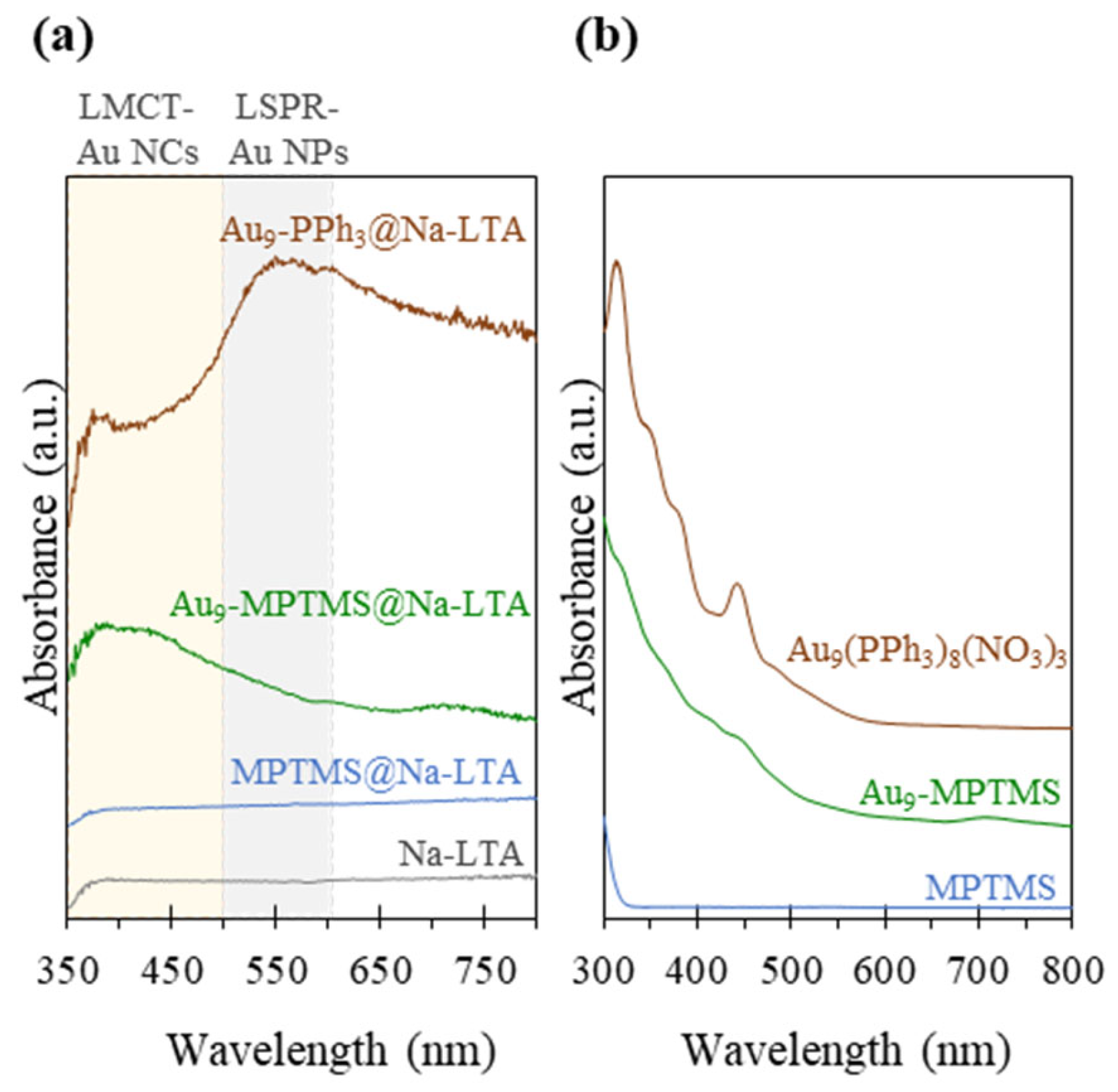
3.1. Synthesis and Characterizations of LTA Zeolite-Encapsulated Au9 Nanoclusters
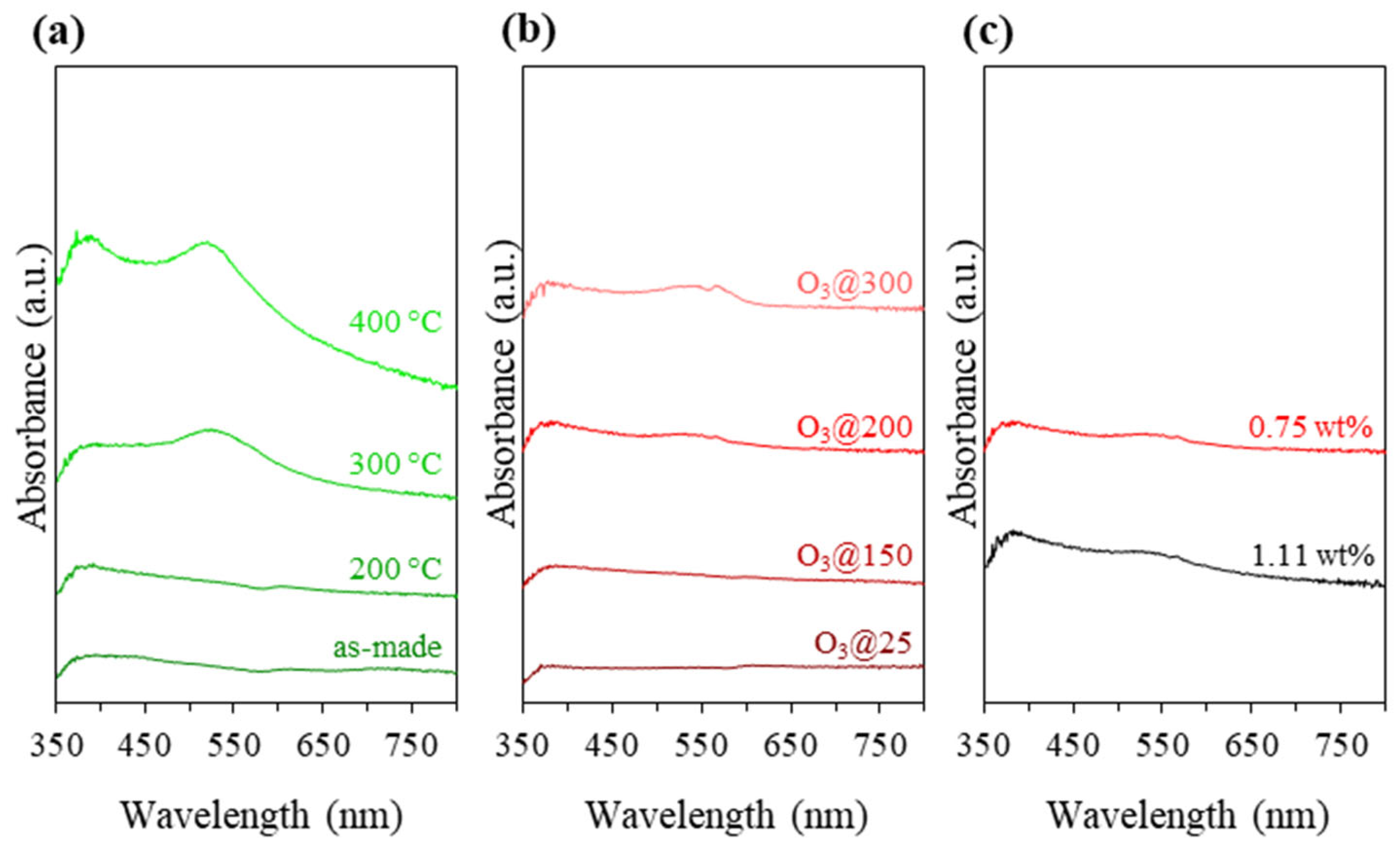
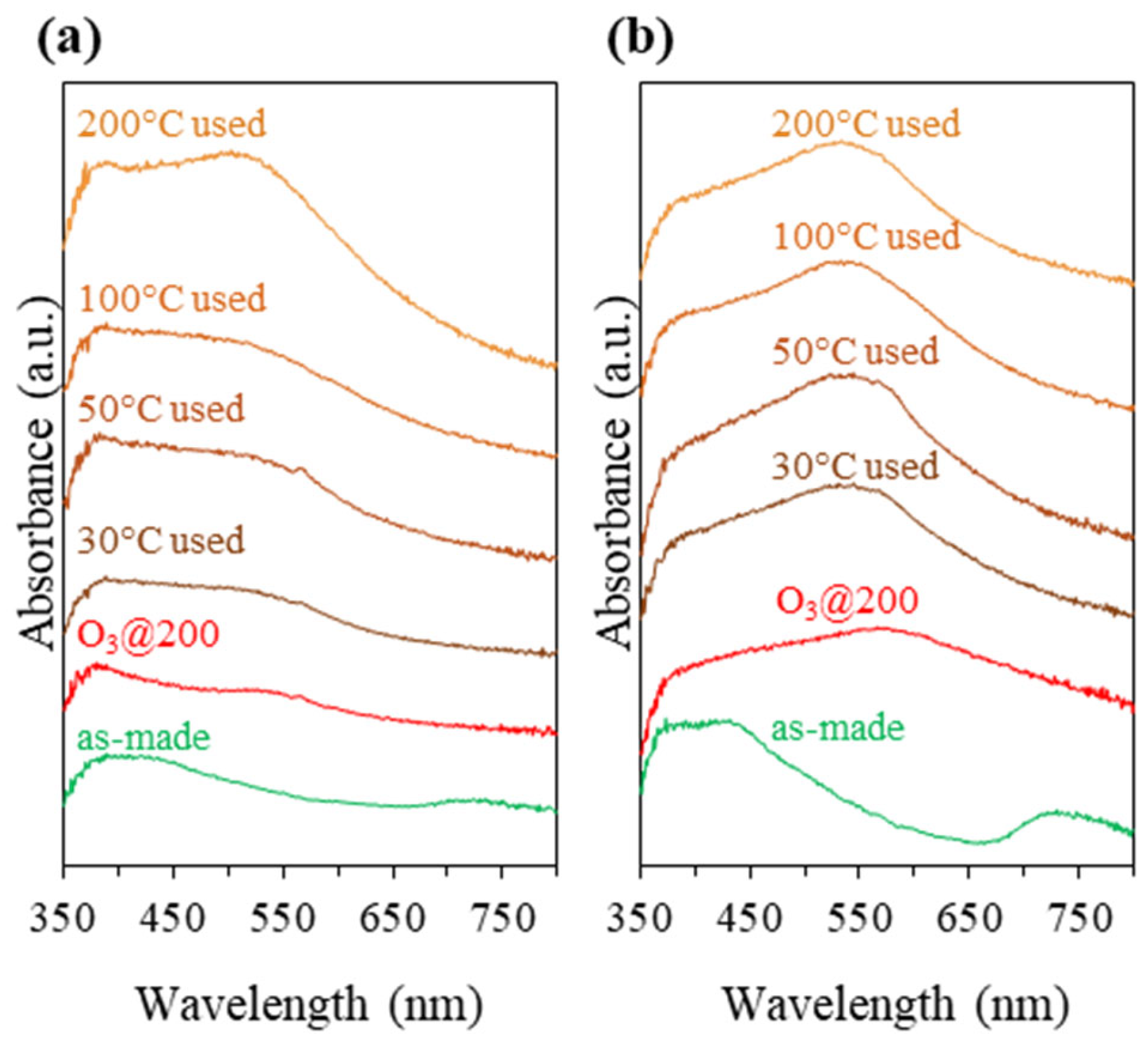
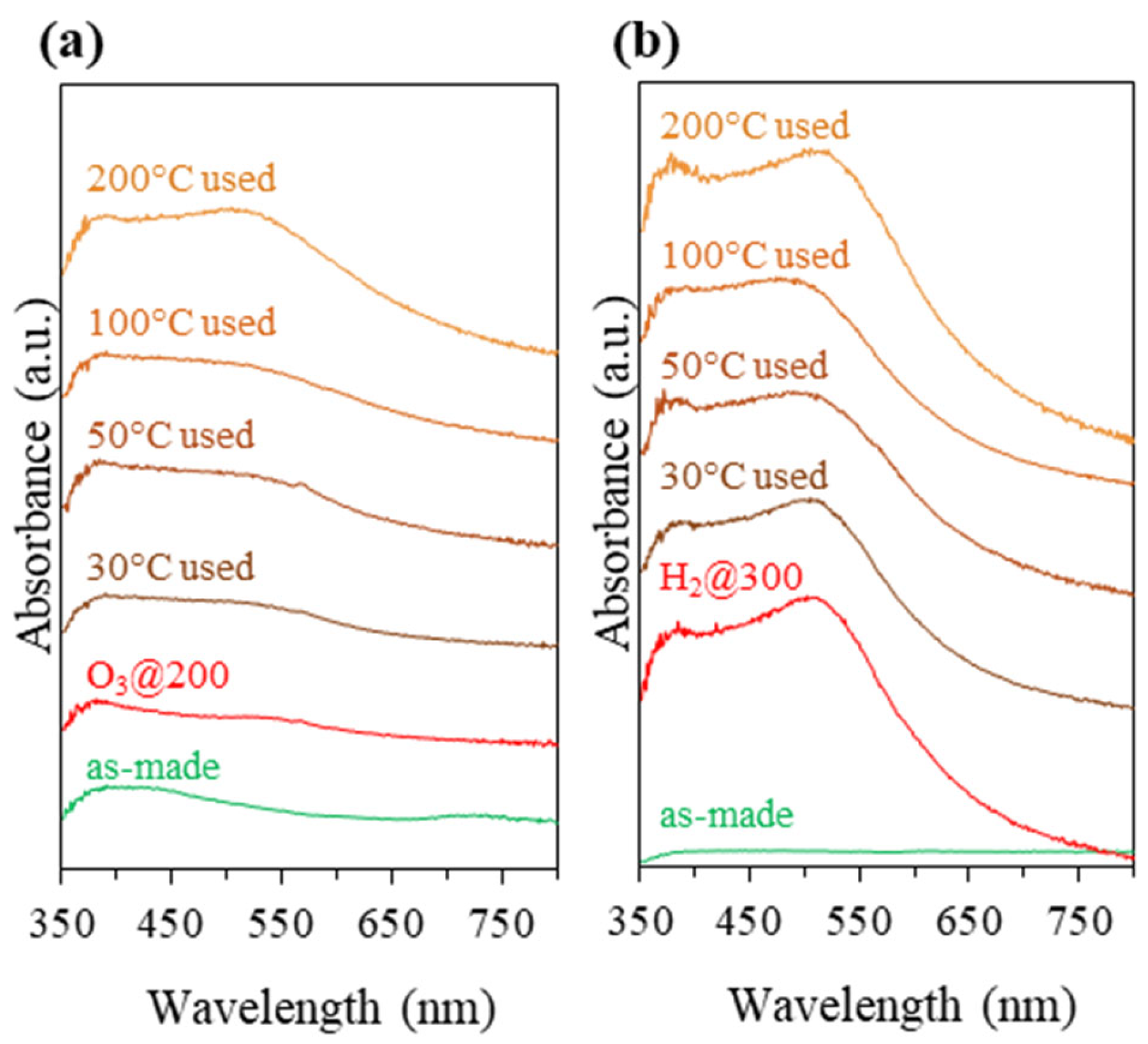
3.2. Catalysts Activation
3.3. Catalytic CO Oxidation
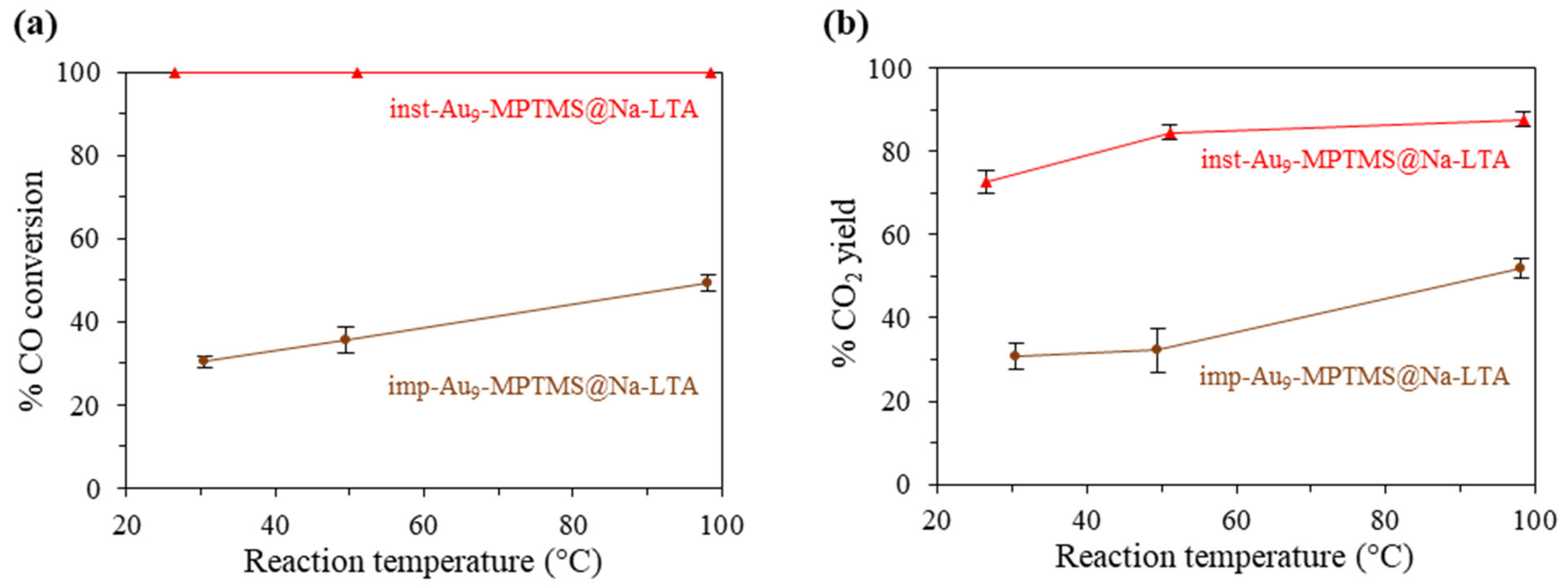
3.3.1. Effect of the Incorporation Approach—In Situ vs. Post-Incorporation
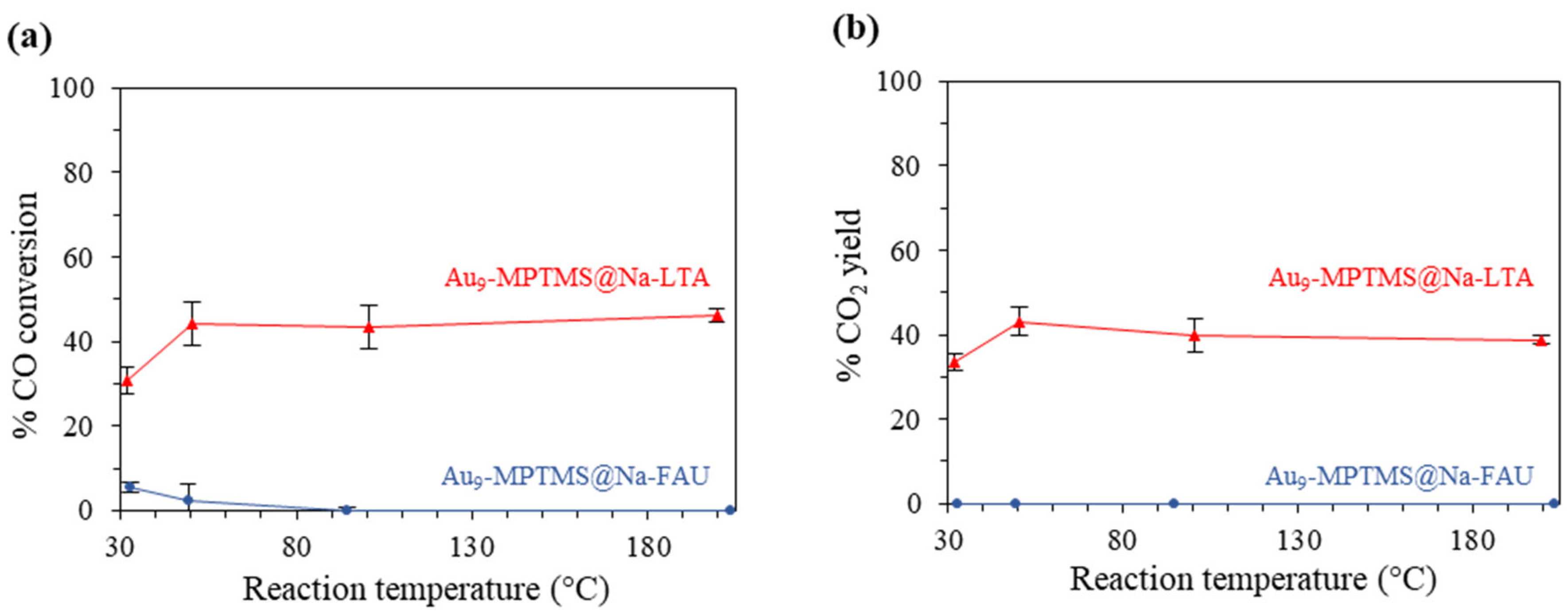
3.3.2. Effect of Zeolite Framework—LTA vs. FAU
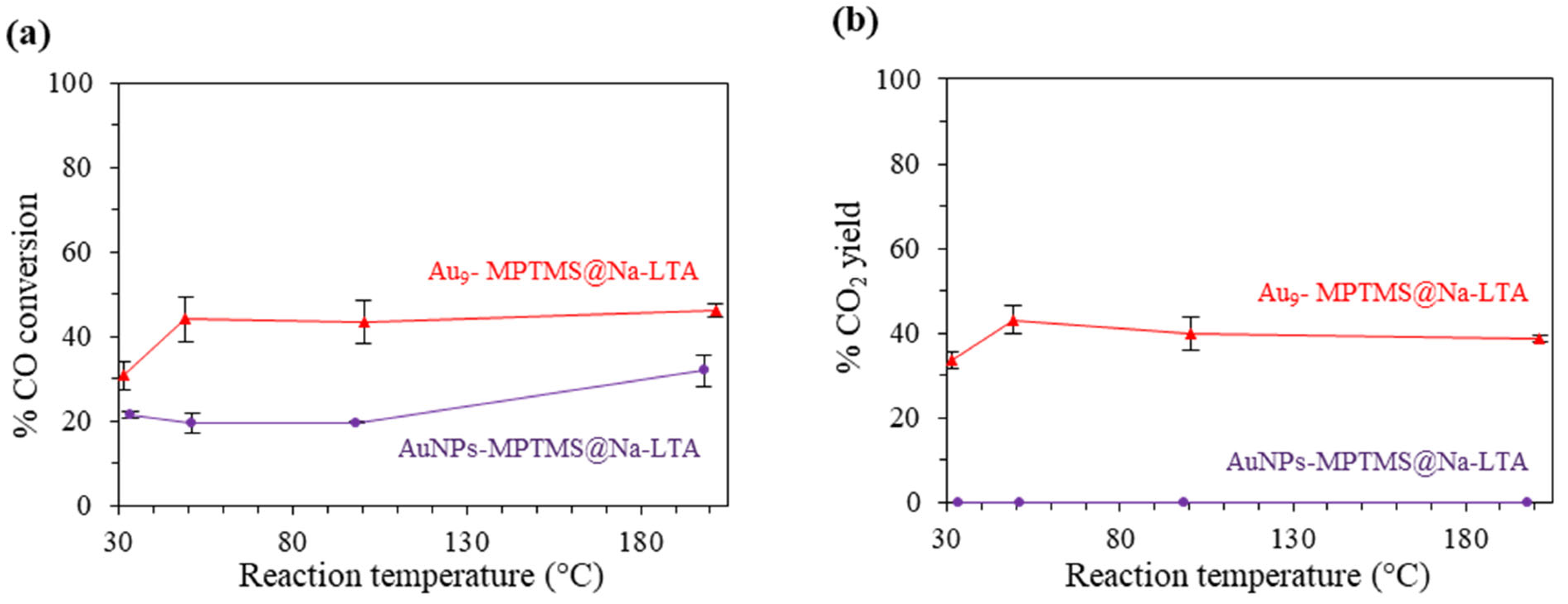
3.3.3. Effect of Incorporated Au Species–Au NCs vs. Au NPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takano, S.; Tsukuda, T. Chapter 2—Controlled synthesis: Size control. In Frontiers of Nanoscience; Tsukuda, T., Häkkinen, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 9, pp. 9–38. [Google Scholar]
- Jin, R.; Zeng, C.; Zhou, M.; Chen, Y. Atomically precise colloidal metal nanoclusters and nanoparticles: Fundamentals and opportunities. Chem. Rev. 2016, 116, 10346–10413. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Meira, D.M.; Arenal, R.; Concepcion, P.; Puga, A.V.; Corma, A. Determination of the evolution of heterogeneous single metal atoms and nanoclusters under reaction conditions: Which are the working catalytic sites? ACS Catal. 2019, 9, 10626–10639. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Li, Y. Removal and utilization of capping agents in nanocatalysis. Chem. Mater. 2014, 26, 72–83. [Google Scholar] [CrossRef]
- Li, Z.; Ji, S.; Liu, Y.; Cao, X.; Tian, S.; Chen, Y.; Niu, Z.; Li, Y. Well-defined materials for heterogeneous catalysis: From nanoparticles to isolated single-atom sites. Chem. Rev. 2020, 120, 623–682. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Corma, A. Evolution of isolated atoms and clusters in catalysis. Trends Chem. 2020, 2, 383–400. [Google Scholar] [CrossRef]
- Fukamori, Y.; König, M.; Yoon, B.; Wang, B.; Esch, F.; Heiz, U.; Landman, U. Fundamental Insight into the Substrate-Dependent Ripening of Monodisperse Clusters. ChemCatChem 2013, 5, 3330–3341. [Google Scholar] [CrossRef]
- Krishnan, G.; Al Qahtani, H.S.; Li, J.; Yin, Y.; Eom, N.; Golovko, V.B.; Metha, G.F.; Andersson, G.G. Investigation of Ligand-Stabilized Gold Clusters on Defect-Rich Titania. J. Phys. Chem. C 2017, 121, 28007–28016. [Google Scholar] [CrossRef]
- Mousavi, H.; Yin, Y.; Howard-Fabretto, L.; Sharma, S.K.; Golovko, V.; Andersson, G.G.; Shearer, C.J.; Metha, G.F. Au101–rGO nanocomposite: Immobilization of phosphine-protected gold nanoclusters on reduced graphene oxide without aggregation. Nanoscale Adv. 2021, 3, 1422–1430. [Google Scholar] [CrossRef]
- Al Qahtani, H.S.; Metha, G.F.; Walsh, R.B.; Golovko, V.B.; Andersson, G.G.; Nakayama, T. Aggregation Behavior of Ligand-Protected Au9 Clusters on Sputtered Atomic Layer Deposition TiO2. J. Phys. Chem. C 2017, 121, 10781–10789. [Google Scholar] [CrossRef]
- Al Qahtani, H.S.; Higuchi, R.; Sasaki, T.; Alvino, J.F.; Metha, G.F.; Golovko, V.B.; Adnan, R.; Andersson, G.G.; Nakayama, T. Grouping and aggregation of ligand protected Au9 clusters on TiO2 nanosheets. RSC Adv. 2016, 6, 110765–110774. [Google Scholar] [CrossRef]
- Ruzicka, J.-Y.; Abu Bakar, F.; Hoeck, C.; Adnan, R.; McNicoll, C.; Kemmitt, T.; Cowie, B.C.; Metha, G.F.; Andersson, G.G.; Golovko, V.B. Toward Control of Gold Cluster Aggregation on TiO2 via Surface Treatments. J. Phys. Chem. C 2015, 119, 24465–24474. [Google Scholar] [CrossRef]
- Anderson, D.P.; Alvino, J.F.; Gentleman, A.; Qahtani, H.A.; Thomsen, L.; Polson, M.I.J.; Metha, G.F.; Golovko, V.B.; Andersson, G.G. Chemically-synthesised, atomically-precise gold clusters deposited and activated on titania. Phys. Chem. Chem. Phys. 2013, 15, 3917–3929. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.P.; Adnan, R.H.; Alvino, J.F.; Shipper, O.; Donoeva, B.; Ruzicka, J.-Y.; Al Qahtani, H.; Harris, H.H.; Cowie, B.; Aitken, J.B.; et al. Chemically synthesised atomically precise gold clusters deposited and activated on titania. Part II. Phys. Chem. Chem. Phys. 2013, 15, 14806–14813. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T.; Parker, S.C.; Starr, D.E. The effect of size-dependent nanoparticle energetics on catalyst sintering. Science 2002, 298, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, R.; Liu, J.-X.; Li, W.-X. Atomistic theory of Ostwald Ripening and disintegration of supported metal particles under reaction conditions. J. Am. Chem. Soc. 2013, 135, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Goodman, E.D.; Schwalbe, J.A.; Cargnello, M. Mechanistic understanding and the rational design of sinter-resistant heterogeneous catalysts. ACS Catal. 2017, 7, 7156–7173. [Google Scholar] [CrossRef]
- Babucci, M.; Guntida, A.; Gates, B.C. Atomically dispersed metals on well-defined supports including zeolites and metal–organic frameworks: Structure, bonding, reactivity, and catalysis. Chem. Rev. 2020, 120, 11956–11985. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Confining isolated atoms and clusters in crystalline porous materials for catalysis. Nat. Rev. Mater. 2021, 6, 244–263. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Xiao, F.-S. Metal@zeolite hybrid materials for catalysis. ACS Cent. Sci. 2020, 6, 1685–1697. [Google Scholar] [CrossRef]
- Kosinov, N.; Liu, C.; Hensen, E.J.M.; Pidko, E.A. Engineering of transition metal catalysts confined in zeolites. Chem. Mater. 2018, 30, 3177–3198. [Google Scholar] [CrossRef] [PubMed]
- Pagis, C.; Morgado Prates, A.R.; Farrusseng, D.; Bats, N.; Tuel, A. Hollow zeolite structures: An overview of synthesis methods. Chem. Mater. 2016, 28, 5205–5223. [Google Scholar] [CrossRef]
- Ou, Z.; Li, Y.; Wu, W.; Bi, Y.; Xing, E.; Yu, T.; Chen, Q. Encapsulating subnanometric metal clusters in zeolites for catalysis and their challenges. Chem. Eng. J. 2022, 430, 132925. [Google Scholar] [CrossRef]
- Liu, L.; Lopez-Haro, M.; Calvino, J.J.; Corma, A. Tutorial: Structural characterization of isolated metal atoms and subnanometric metal clusters in zeolites. Nat. Protoc. 2021, 16, 1871–1906. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Davies, T.E.; Nasrallah, A.; Sainna, M.A.; Howe, A.G.R.; Lewis, R.J.; Quesne, M.; Catlow, C.R.A.; Willock, D.J.; He, Q.; et al. Au-ZSM-5 catalyses the selective oxidation of CH4 to CH3OH and CH3COOH using O2. Nat. Catal. 2022, 5, 45–54. [Google Scholar] [CrossRef]
- De Graaf, J.; van Dillen, A.J.; de Jong, K.P.; Koningsberger, D.C. Preparation of highly dispersed Pt particles in zeolite Y with a narrow particle size distribution: Characterization by hydrogen chemisorption, TEM, EXAFS spectroscopy, and particle modeling. J. Catal. 2001, 203, 307–321. [Google Scholar] [CrossRef]
- Zečević, J.; van der Eerden, A.M.J.; Friedrich, H.; de Jongh, P.E.; de Jong, K.P. Heterogeneities of the nanostructure of platinum/zeolite Y catalysts revealed by electron tomography. ACS Nano 2013, 7, 3698–3705. [Google Scholar] [CrossRef]
- Serna, P.; Gates, B.C. Molecular metal catalysts on supports: Organometallic chemistry meets surface science. Acc. Chem. Res. 2014, 47, 2612–2620. [Google Scholar] [CrossRef]
- Zeng, S.; Ding, S.; Li, S.; Wang, R.; Zhang, Z. Controlled growth of gold nanoparticles in zeolite L via ion-exchange reactions and thermal reduction processes. Inorg. Chem. Commun. 2014, 47, 63–66. [Google Scholar] [CrossRef]
- Gu, J.; Zhang, Z.; Hu, P.; Ding, L.; Xue, N.; Peng, L.; Guo, X.; Lin, M.; Ding, W. Platinum nanoparticles encapsulated in MFI zeolite crystals by a two-step dry gel conversion method as a highly selective hydrogenation catalyst. ACS Catal. 2015, 5, 6893–6901. [Google Scholar] [CrossRef]
- Liu, L.; Zakharov, D.N.; Arenal, R.; Concepcion, P.; Stach, E.A.; Corma, A. Evolution and stabilization of subnanometric metal species in confined space by in situ TEM. Nat. Commun. 2018, 9, 574. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Wu, Z.; Zones, S.I.; Iglesia, E. Synthesis and catalytic properties of metal clusters encapsulated within small-pore (SOD, GIS, ANA) zeolites. J. Am. Chem. Soc. 2012, 134, 17688–17695. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Goel, S.; Choi, M.; Iglesia, E. Hydrothermal synthesis of LTA-encapsulated metal clusters and consequences for catalyst stability, reactivity, and selectivity. J. Catal. 2014, 311, 458–468. [Google Scholar] [CrossRef]
- Otto, T.; Zones, S.I.; Hong, Y.; Iglesia, E. Synthesis of highly dispersed cobalt oxide clusters encapsulated within LTA zeolites. J. Catal. 2017, 356, 173–185. [Google Scholar] [CrossRef]
- Otto, T.; Zones, S.I.; Iglesia, E. Synthetic strategies for the encapsulation of nanoparticles of Ni, Co, and Fe oxides within crystalline microporous aluminosilicates. Microporous Mesoporous Mater. 2018, 270, 10–23. [Google Scholar] [CrossRef]
- Wang, N.; Sun, Q.; Bai, R.; Li, X.; Guo, G.; Yu, J. In situ confinement of ultrasmall Pd clusters within nanosized Silicalite-1 zeolite for highly efficient catalysis of hydrogen generation. J. Am. Chem. Soc. 2016, 138, 7484–7487. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, N.; Bing, Q.; Si, R.; Liu, J.; Bai, R.; Zhang, P.; Jia, M.; Yu, J. Subnanometric hybrid Pd-M(OH)2, M = Ni, Co clusters in zeolites as highly efficient nanocatalysts for hydrogen generation. Chem 2017, 3, 477–493. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, M.; Zhang, C.; Ren, K.; Xin, Y.; Zhao, M.; Xing, E. Selectivity control on hydrogenation of substituted nitroarenes through end-on adsorption of reactants in zeolite-encapsulated platinum nanoparticles. Chem. Asian J. 2018, 13, 2077–2084. [Google Scholar] [CrossRef]
- Shan, Y.; Sui, Z.; Zhu, Y.; Zhou, J.; Zhou, X.; Chen, D. Boosting size-selective hydrogen combustion in the presence of propene using controllable metal clusters encapsulated in zeolite. Angew. Chem. Int. Ed. 2018, 57, 9770–9774. [Google Scholar] [CrossRef]
- Liu, L.; Lopez-Haro, M.; Lopes, C.W.; Li, C.; Concepcion, P.; Simonelli, L.; Calvino, J.J.; Corma, A. Regioselective generation and reactivity control of subnanometric platinum clusters in zeolites for high-temperature catalysis. Nat. Mater. 2019, 18, 866–873. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Yu, Q.; Chen, Y.; Chai, Z.; Zhao, G.; Liu, S.; Cheong, W.-C.; Pan, Y.; Zhang, Q.; et al. A general strategy for fabricating isolated single metal atomic site catalysts in Y zeolite. J. Am. Chem. Soc. 2019, 141, 9305–9311. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, N.; Zhang, T.; Bai, R.; Mayoral, A.; Zhang, P.; Zhang, Q.; Terasaki, O.; Yu, J. Zeolite-encaged single-atom rhodium catalysts: Highly-efficient hydrogen generation and shape-selective tandem hydrogenation of nitroarenes. Angew. Chem. Int. Ed. 2019, 58, 18570–18576. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Wu, Z.; Iglesia, E. Mercaptosilane-assisted synthesis of metal clusters within zeolites and catalytic consequences of encapsulation. J. Am. Chem. Soc. 2010, 132, 9129–9137. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Zones, S.I.; Iglesia, E. Challenges and strategies in the encapsulation and stabilization of monodisperse Au clusters within zeolites. J. Catal. 2016, 339, 195–208. [Google Scholar] [CrossRef]
- Otto, T.; Ramallo-López, J.M.; Giovanetti, L.J.; Requejo, F.G.; Zones, S.I.; Iglesia, E. Synthesis of stable monodisperse AuPd, AuPt, and PdPt bimetallic clusters encapsulated within LTA-zeolites. J. Catal. 2016, 342, 125–137. [Google Scholar] [CrossRef]
- Lee, S.; Lee, K.; Im, J.; Kim, H.; Choi, M. Revisiting hydrogen spillover in Pt/LTA: Effects of physical diluents having different acid site distributions. J. Catal. 2015, 325, 26–34. [Google Scholar] [CrossRef]
- Moliner, M.; Gabay, J.E.; Kliewer, C.E.; Carr, R.T.; Guzman, J.; Casty, G.L.; Serna, P.; Corma, A. Reversible transformation of Pt nanoparticles into single atoms inside high-silica Chabazite zeolite. J. Am. Chem. Soc. 2016, 138, 15743–15750. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Han, W.; Lyu, J.; Zhang, Q.; Guo, L.; Li, X. In situ encapsulation of platinum clusters within H-ZSM-5 zeolite for highly stable benzene methylation catalysis. Catal. Sci. Technol. 2017, 7, 6140–6150. [Google Scholar] [CrossRef]
- Li, S.; Tuel, A.; Laprune, D.; Meunier, F.; Farrusseng, D. Transition-metal nanoparticles in hollow zeolite single crystals as bifunctional and size-selective hydrogenation catalysts. Chem. Mater. 2015, 27, 276–282. [Google Scholar] [CrossRef]
- Ingham, B.; Lim, T.H.; Dotzler, C.J.; Henning, A.; Toney, M.F.; Tilley, R.D. How nanoparticles coalesce: An in situ study of Au nanoparticle aggregation and grain growth. Chem. Mater. 2011, 23, 3312–3317. [Google Scholar] [CrossRef]
- Woehrle, G.H.; Hutchison, J.E. Thiol-functionalized undecagold clusters by ligand exchange: Synthesis, mechanism, and properties. Inorg. Chem. 2005, 44, 6149–6158. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.W.; Franklin, K.C. Chapter 55—LTA Linde Type A Si(50), Al(50). In Verified Syntheses of Zeolitic Materials; Robson, H., Lillerud, K.P., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; pp. 179–181. [Google Scholar]
- Liu, L.; Díaz, U.; Arenal, R.; Agostini, G.; Concepción, P.; Corma, A. Generation of subnanometric platinum with high stability during transformation of a 2D zeolite into 3D. Nat. Mater. 2017, 16, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Holder, C.F.; Schaak, R.E. Tutorial on powder X-ray diffraction for characterizing nanoscale materials. ACS Nano 2019, 13, 7359–7365. [Google Scholar] [CrossRef] [PubMed]
- Motl, N.E.; Smith, A.F.; DeSantis, C.J.; Skrabalak, S.E. Engineering plasmonic metal colloids through composition and structural design. Chem. Soc. Rev. 2014, 43, 3823–3834. [Google Scholar] [CrossRef] [PubMed]
- Eustis, S.; El-Sayed, M.A. Why gold nanoparticles are more precious than pretty gold: Noble metal surface plasmon resonance and its enhancement of the radiative and nonradiative properties of nanocrystals of different shapes. Chem. Soc. Rev. 2006, 35, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Aikens, C.M. Chapter 9—Optical properties and chirality. In Frontiers of Nanoscience; Tsukuda, T., Häkkinen, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 9, pp. 223–261. [Google Scholar]
- Häkkinen, H. The gold–sulfur interface at the nanoscale. Nat. Chem. 2012, 4, 443–455. [Google Scholar] [CrossRef]
- Tai, Y.; Yamaguchi, W.; Okada, M.; Ohashi, F.; Shimizu, K.-i.; Satsuma, A.; Tajiri, K.; Kageyama, H. Depletion of CO oxidation activity of supported Au catalysts prepared from thiol-capped Au nanoparticles by sulfates formed at Au–titania boundaries: Effects of heat treatment conditions on catalytic activity. J. Catal. 2010, 270, 234–241. [Google Scholar] [CrossRef]
- Ma, G.; Binder, A.; Chi, M.; Liu, C.; Jin, R.; Jiang, D.-e.; Fan, J.; Dai, S. Stabilizing gold clusters by heterostructured transition-metal oxide–mesoporous silica supports for enhanced catalytic activities for CO oxidation. Chem. Commun. 2012, 48, 11413–11415. [Google Scholar] [CrossRef]
- Gaur, S.; Wu, H.; Stanley, G.G.; More, K.; Kumar, C.S.S.R.; Spivey, J.J. CO oxidation studies over cluster-derived Au/TiO2 and AUROlite™ Au/TiO2 catalysts using DRIFTS. Catal. Today 2013, 208, 72–81. [Google Scholar] [CrossRef]
- Nie, X.; Qian, H.; Ge, Q.; Xu, H.; Jin, R. CO oxidation catalyzed by oxide-supported Au25(SR)18 nanoclusters and identification of perimeter sites as active centers. ACS Nano 2012, 6, 6014–6022. [Google Scholar] [CrossRef]
- Nie, X.; Zeng, C.; Ma, X.; Qian, H.; Ge, Q.; Xu, H.; Jin, R. CeO2-supported Au38(SR)24 nanocluster catalysts for CO oxidation: A comparison of ligand-on and -off catalysts. Nanoscale 2013, 5, 5912–5918. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, R.G.; Fusco, F.A.; Allara, D.L. Spontaneously organized molecular assemblies—Preparation and properties of solution adsorbed monolayers of organic disulfides on gold surfaces. J. Am. Chem. Soc. 1987, 109, 2358–2368. [Google Scholar] [CrossRef]
- Toulhoat, H.; Raybaud, P.; Kasztelan, S.; Kresse, G.; Hafner, J. Transition metals to sulfur binding energies relationship to catalytic activities in HDS: Back to Sabatier with first principle calculations. Catal. Today 1999, 50, 629–636. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C.; Nieuwenhuys, B.E. Adsorption of small molecules on gold single crystal surfaces. Gold Bull. 2009, 42, 288–301. [Google Scholar] [CrossRef]
- Angell, C.L.; Schaffer, P.C. Infrared spectroscopic investigations of zeolites and adsorbed molecules. II. Adsorbed carbon monoxide. J. Phys. Chem. 1966, 70, 1413–1418. [Google Scholar] [CrossRef]
- Lopez, N.; Nørskov, J.K. Catalytic CO oxidation by a gold nanoparticle: A density functional study. J. Am. Chem. Soc. 2002, 124, 11262–11263. [Google Scholar] [CrossRef] [PubMed]
- Lopez, N.; Janssens, T.V.W.; Clausen, B.S.; Xu, Y.; Mavrikakis, M.; Bligaard, T.; Nørskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Min, B.K.; Friend, C.M. Heterogeneous gold-based catalysis for green chemistry: Low-temperature CO oxidation and propene oxidation. Chem. Rev. 2007, 107, 2709–2724. [Google Scholar] [CrossRef]
- Janssens, T.V.W.; Clausen, B.S.; Hvolbæk, B.; Falsig, H.; Christensen, C.H.; Bligaard, T.; Nørskov, J.K. Insights into the reactivity of supported Au nanoparticles: Combining theory and experiments. Top. Catal. 2007, 44, 15–26. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, C.-J.; Yamasaki, J.; Terasaki, O.; Schüth, F. Highly active iron oxide supported gold catalysts for CO oxidation: How small must the gold nanoparticles be? Angew. Chem. Int. Ed. 2010, 49, 5771–5775. [Google Scholar] [CrossRef]
- Qian, K.; Luo, L.; Bao, H.; Hua, Q.; Jiang, Z.; Huang, W. Catalytically active structures of SiO2-supported Au nanoparticles in low-temperature CO oxidation. Catal. Sci. Technol. 2013, 3, 679–687. [Google Scholar] [CrossRef]
- Qiao, B.; Liang, J.-X.; Wang, A.; Xu, C.-Q.; Li, J.; Zhang, T.; Liu, J.J. Ultrastable single-atom gold catalysts with strong covalent metal-support interaction (CMSI). Nano Res. 2015, 8, 2913–2924. [Google Scholar] [CrossRef]
- Menard, L.D.; Xu, F.; Nuzzo, R.G.; Yang, J.C. Preparation of TiO2-supported Au nanoparticle catalysts from a Au13 cluster precursor: Ligand removal using ozone exposure versus a rapid thermal treatment. J. Catal. 2006, 243, 64–73. [Google Scholar] [CrossRef]
- Cargnello, M.; Chen, C.; Diroll, B.T.; Doan-Nguyen, V.V.T.; Gorte, R.J.; Murray, C.B. Efficient removal of organic ligands from supported nanocrystals by fast thermal annealing enables catalytic studies on well-defined active phases. J. Am. Chem. Soc. 2015, 137, 6906–6911. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.J.; Delmon, B. Low-temperature oxidation of CO over gold supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. [Google Scholar] [CrossRef]
- Sankar, M.; He, Q.; Engel, R.V.; Sainna, M.A.; Logsdail, A.J.; Roldan, A.; Willock, D.J.; Agarwal, N.; Kiely, C.J.; Hutchings, G.J. Role of the support in gold-containing nanoparticles as heterogeneous catalysts. Chem. Rev. 2020, 120, 3890–3938. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Lemire, C.; Shaikhutdinov, S.K.; Freund, H.J. Surface chemistry of catalysis by gold. Gold Bull. 2004, 37, 72–124. [Google Scholar] [CrossRef]
- Ginter, D. Chapter 46—FAU Linde Type Y Si(71), Al(29). In Verified Syntheses of Zeolitic Materials; Robson, H., Lillerud, K.P., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; pp. 156–158. [Google Scholar]
- Haiss, W.; Thanh, N.T.K.; Aveyard, J.; Fernig, D.G. Determination of size and concentration of gold nanoparticles from UV−vis spectra. Anal. Chem. 2007, 79, 4215–4221. [Google Scholar] [CrossRef]
- Higaki, T.; Zhou, M.; Lambright, K.J.; Kirschbaum, K.; Sfeir, M.Y.; Jin, R. Sharp transition from nonmetallic Au246 to metallic Au279 with nascent surface plasmon resonance. J. Am. Chem. Soc. 2018, 140, 5691–5695. [Google Scholar] [CrossRef]
- Lee, S.; Fan, C.; Wu, T.; Anderson, S.L. CO oxidation on Aun/TiO2 catalysts produced by size-selected cluster deposition. J. Am. Chem. Soc. 2004, 126, 5682–5683. [Google Scholar] [CrossRef]
- Li, L.; Gao, Y.; Li, H.; Zhao, Y.; Pei, Y.; Chen, Z.; Zeng, X.C. CO oxidation on TiO2 (110) supported subnanometer gold clusters: Size and shape effects. J. Am. Chem. Soc. 2013, 135, 19336–19346. [Google Scholar] [CrossRef]
- Lopez-Acevedo, O.; Kacprzak, K.A.; Akola, J.; Häkkinen, H. Quantum size effects in ambient CO oxidation catalysed by ligand-protected gold clusters. Nat. Chem. 2010, 2, 329–334. [Google Scholar] [CrossRef]
- Haruta, M. Spiers Memorial Lecture Role of perimeter interfaces in catalysis by gold nanoparticles. Faraday Discuss. 2011, 152, 11–32. [Google Scholar] [CrossRef]
- Glemser, O.; Sauer, H. Copper, silver, gold. In Handbook of Preparative Inorganic Chemistry, 2nd ed.; Brauer, G., Ed.; Academic Press inc.: London, UK, 1963; Volume 1. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tesana, S.; Kennedy, J.V.; Yip, A.C.K.; Golovko, V.B. In Situ Incorporation of Atomically Precise Au Nanoclusters within Zeolites for Ambient Temperature CO Oxidation. Nanomaterials 2023, 13, 3120. https://doi.org/10.3390/nano13243120
Tesana S, Kennedy JV, Yip ACK, Golovko VB. In Situ Incorporation of Atomically Precise Au Nanoclusters within Zeolites for Ambient Temperature CO Oxidation. Nanomaterials. 2023; 13(24):3120. https://doi.org/10.3390/nano13243120
Chicago/Turabian StyleTesana, Siriluck, John V. Kennedy, Alex C. K. Yip, and Vladimir B. Golovko. 2023. "In Situ Incorporation of Atomically Precise Au Nanoclusters within Zeolites for Ambient Temperature CO Oxidation" Nanomaterials 13, no. 24: 3120. https://doi.org/10.3390/nano13243120
APA StyleTesana, S., Kennedy, J. V., Yip, A. C. K., & Golovko, V. B. (2023). In Situ Incorporation of Atomically Precise Au Nanoclusters within Zeolites for Ambient Temperature CO Oxidation. Nanomaterials, 13(24), 3120. https://doi.org/10.3390/nano13243120