
Electrospun PLGA Membranes with Incorporated Moxifloxacin-Loaded Silica-Based Mesoporous Nanocarriers for Periodontal Regeneration

 ,
,  , ,
, ,  ,
, 
 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Synthesis of MSNs
2.2. Preparation of Solutions for Electrospinning Process
2.3. Electrospinning Process
2.4. Characterization
2.4.1. Scanning Electron Microscopy and Energy Dispersive Spectroscopic Analysis (SEM-EDS)
- yo = offset
- = center
- w = width
- A = area
2.4.2. Attenuated Total Reflectance Spectroscopy (ATR)
2.4.3. X-ray Diffraction (XRD)
2.4.4. Mechanical Properties
2.4.5. Equilibrium Swelling Performance and In Vitro Degradation
2.4.6. Drug Loading and Drug Content Quantification
2.4.7. In Vitro Drug Release
2.4.8. Hemocompatibility Assay
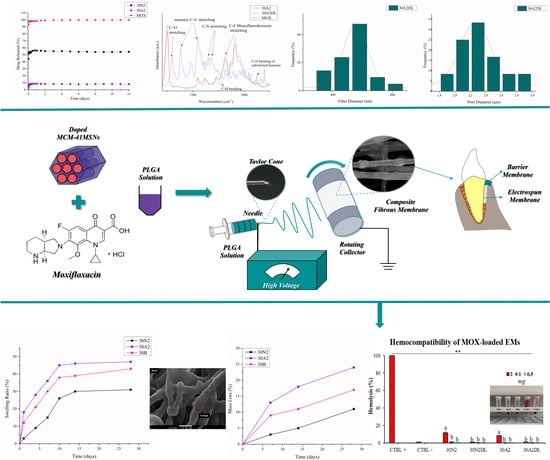
3. Results
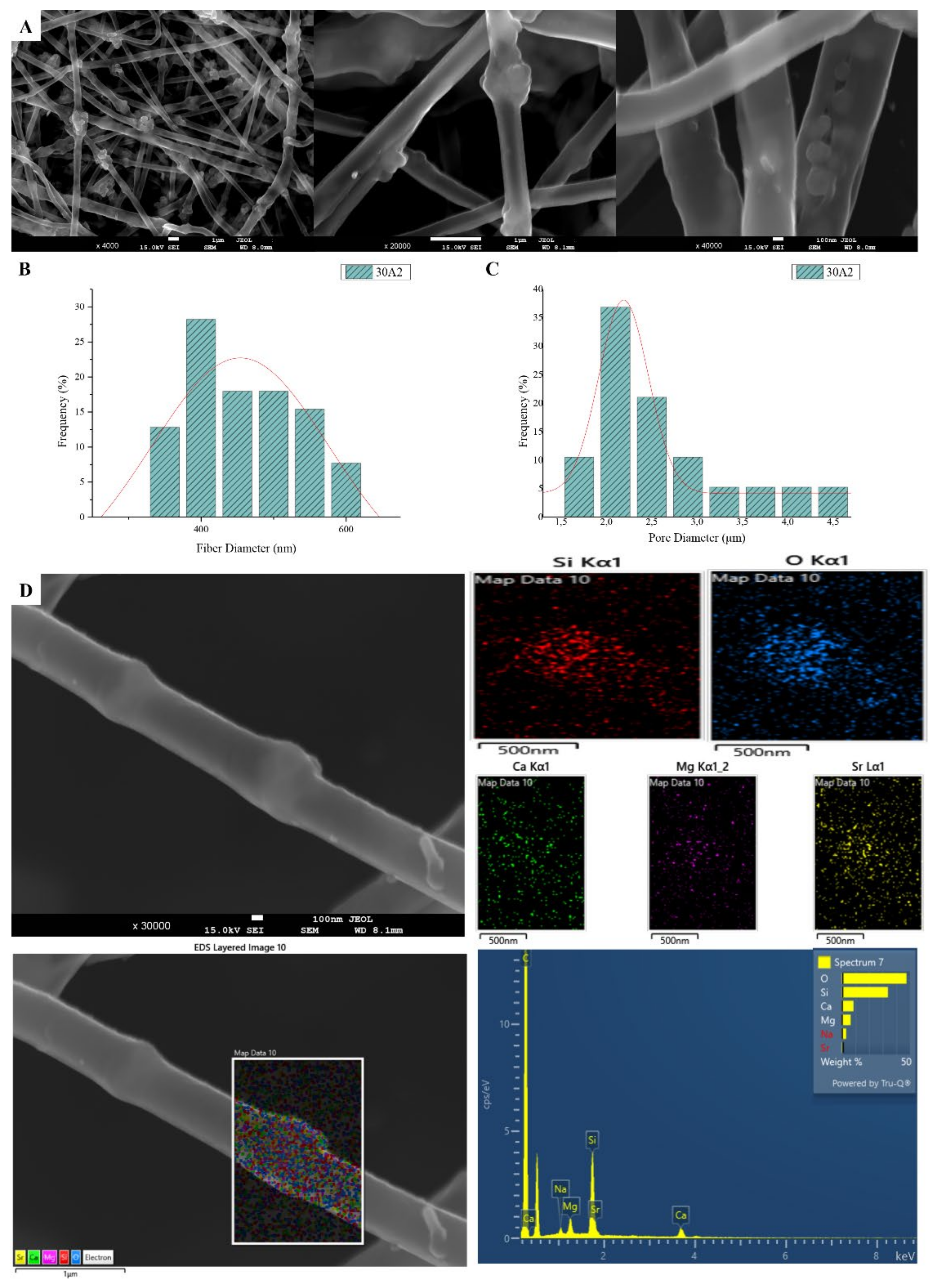
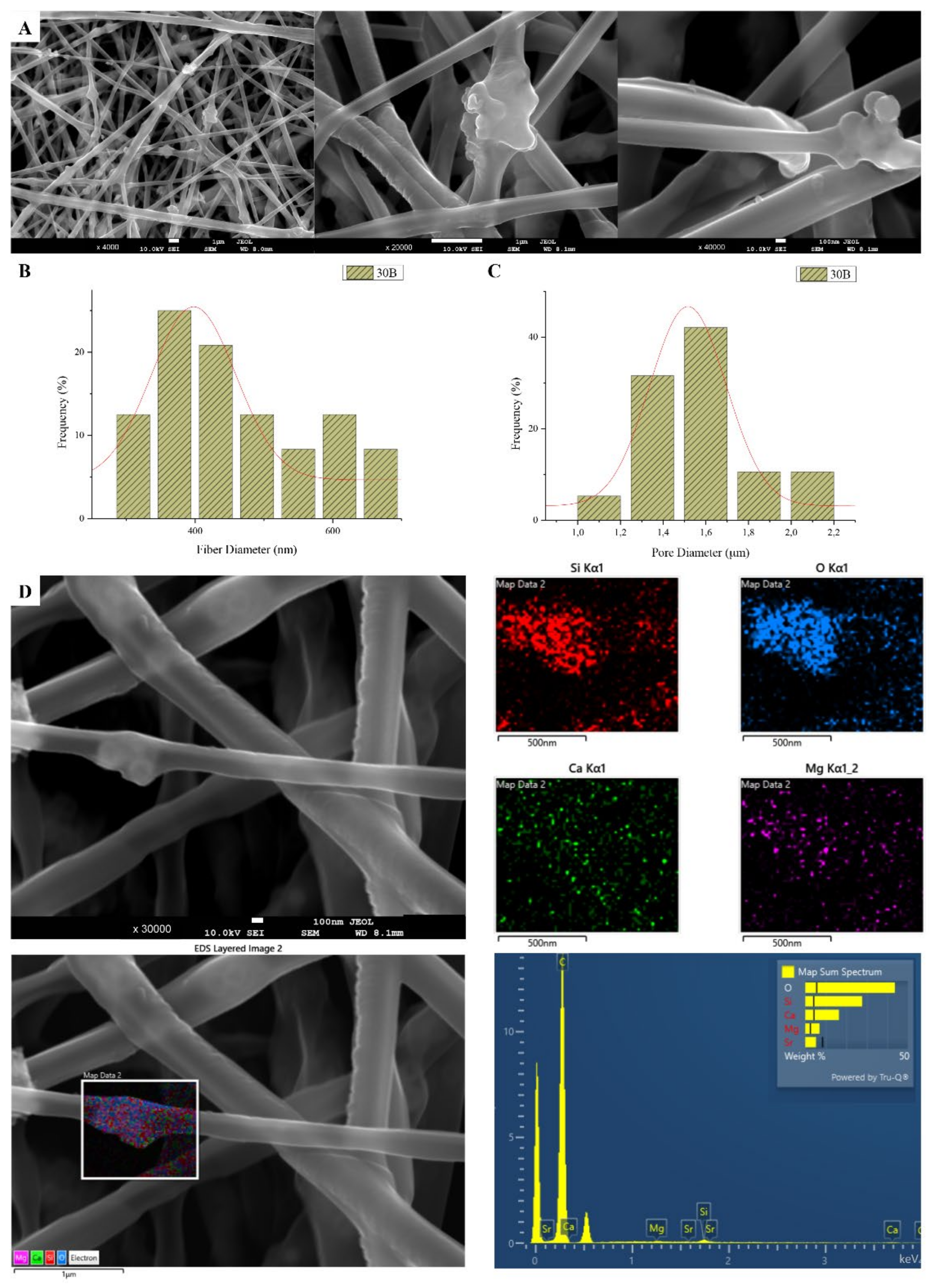
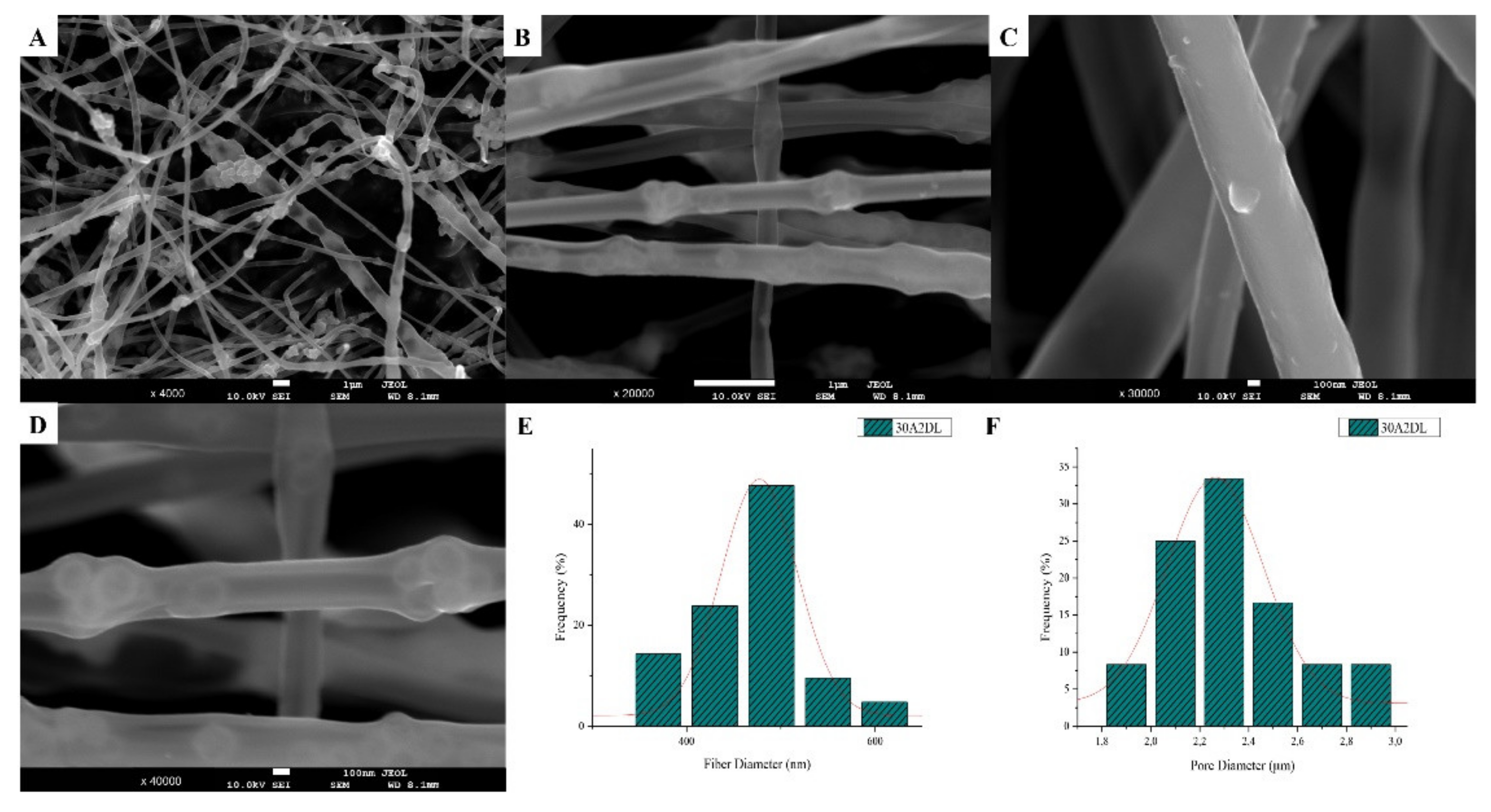
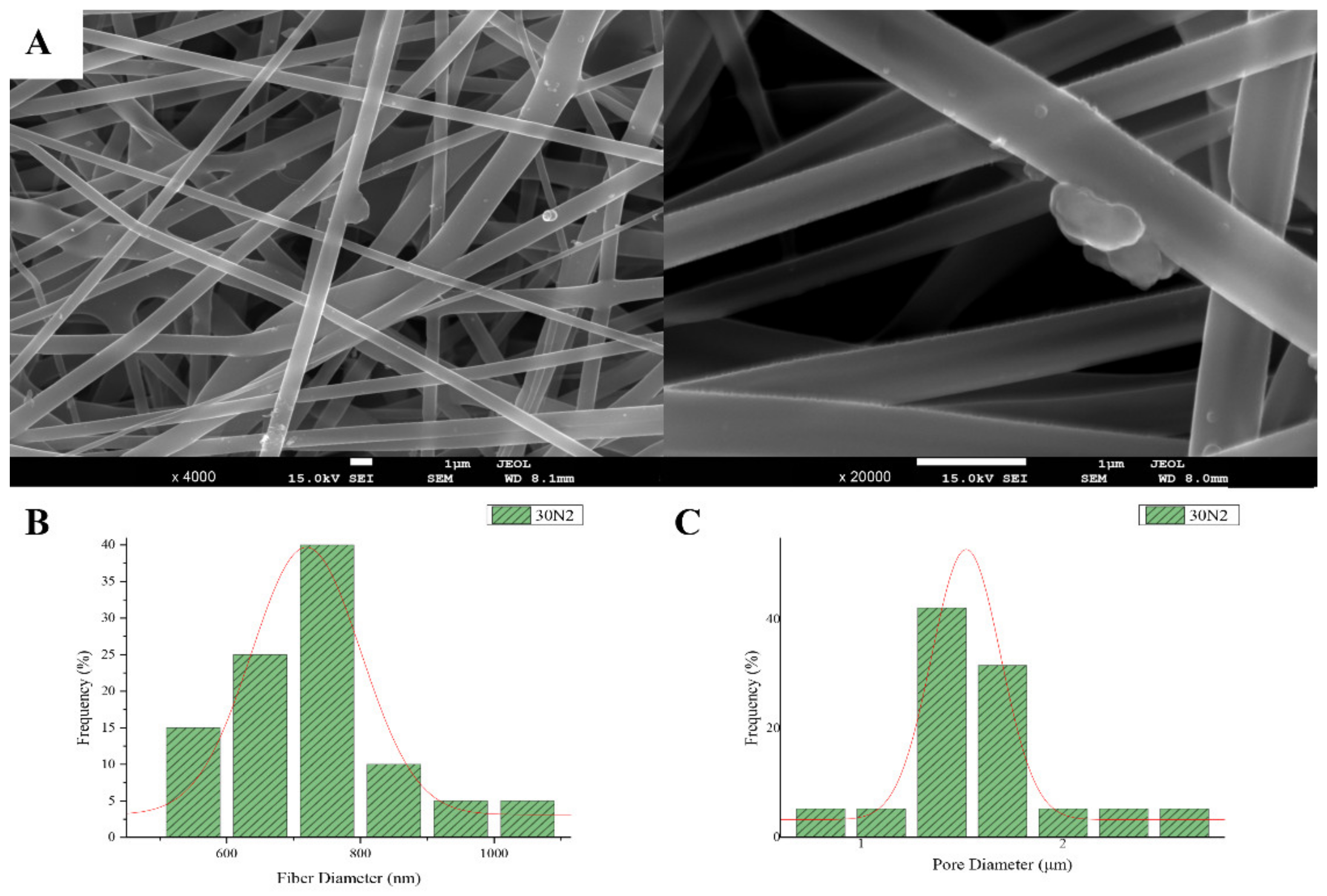
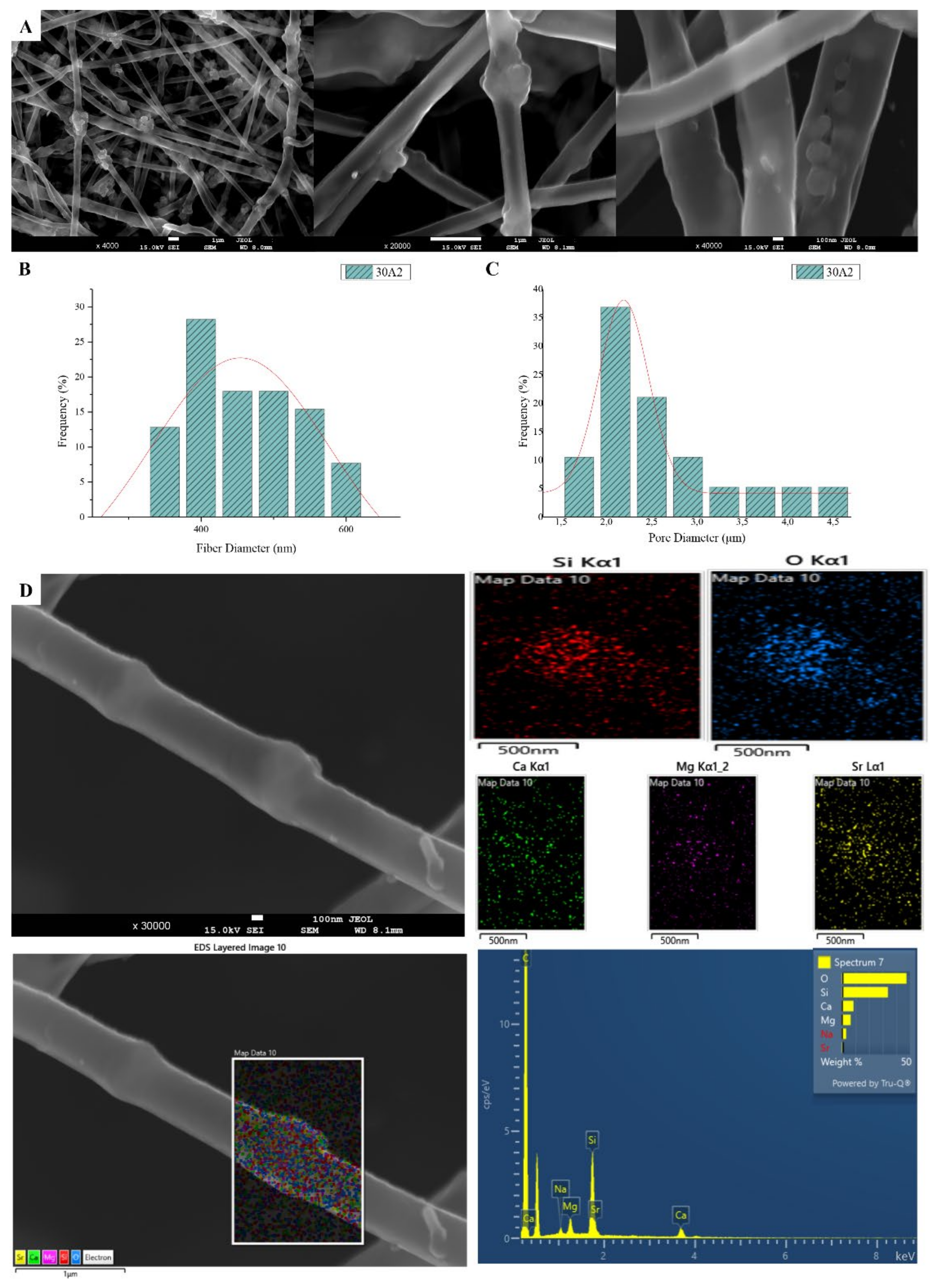
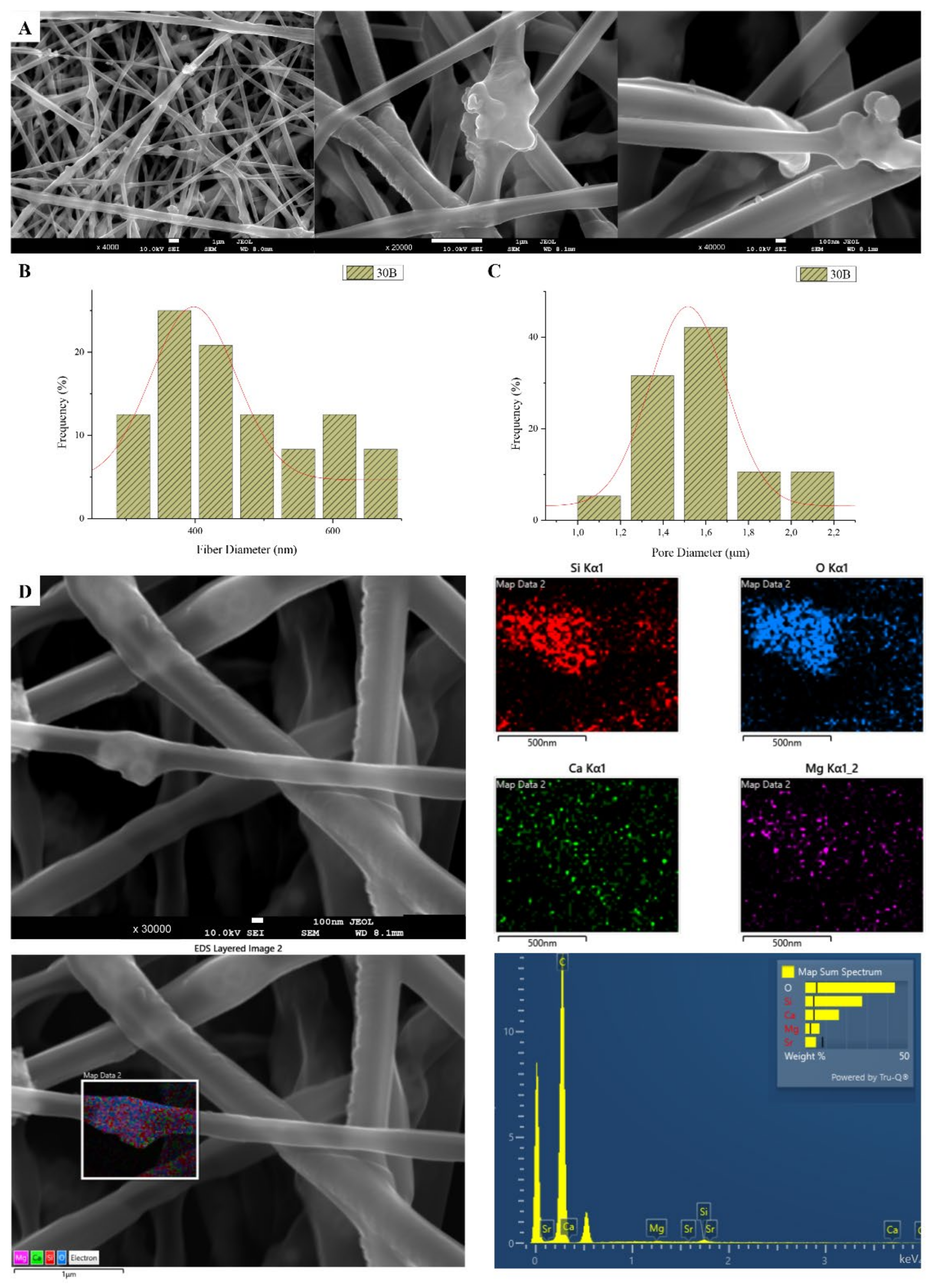
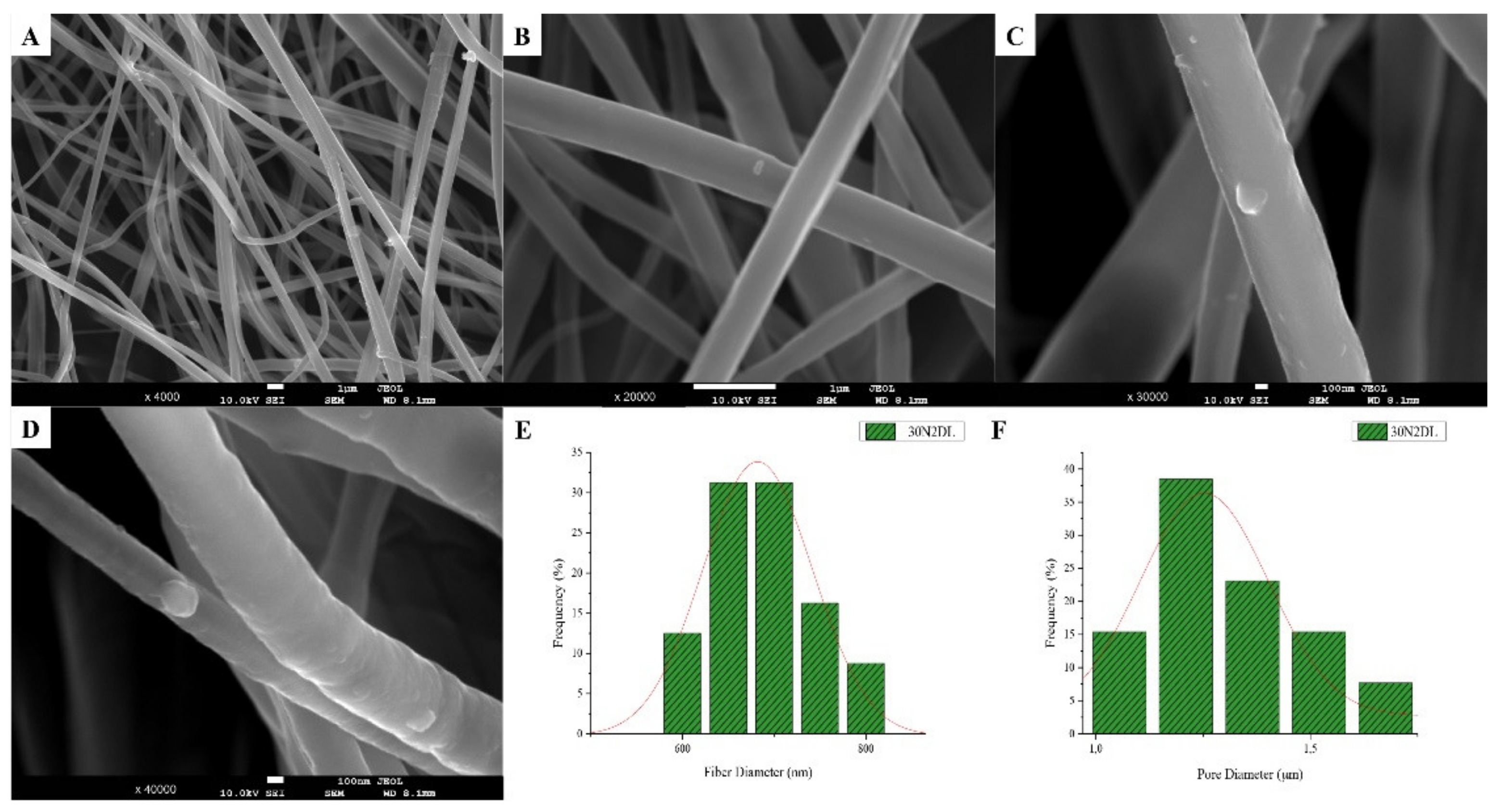
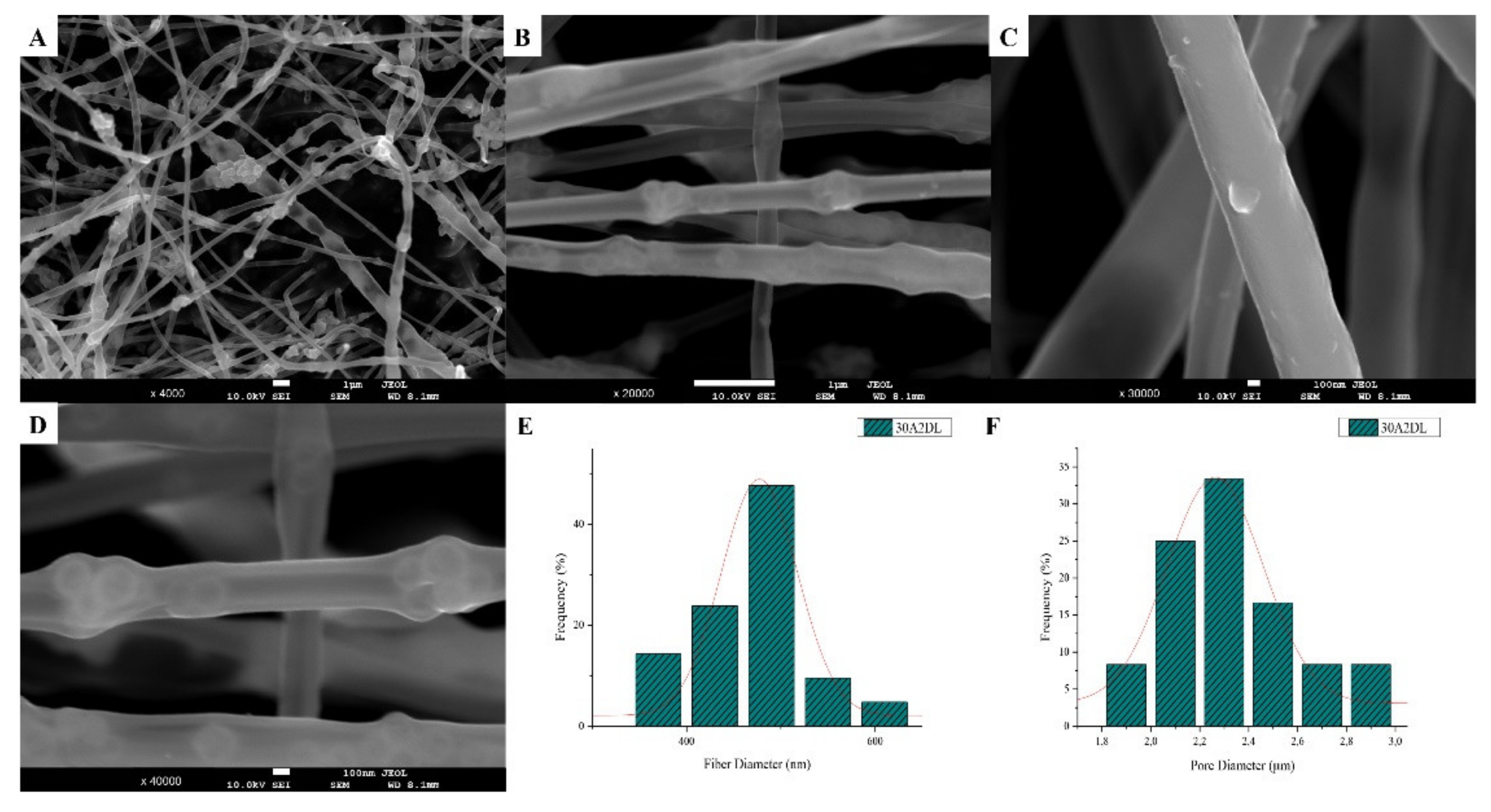
3.1. Scanning Electron Microscopy and Energy Dispersive Spectroscopic Analysis (SEM-EDS)
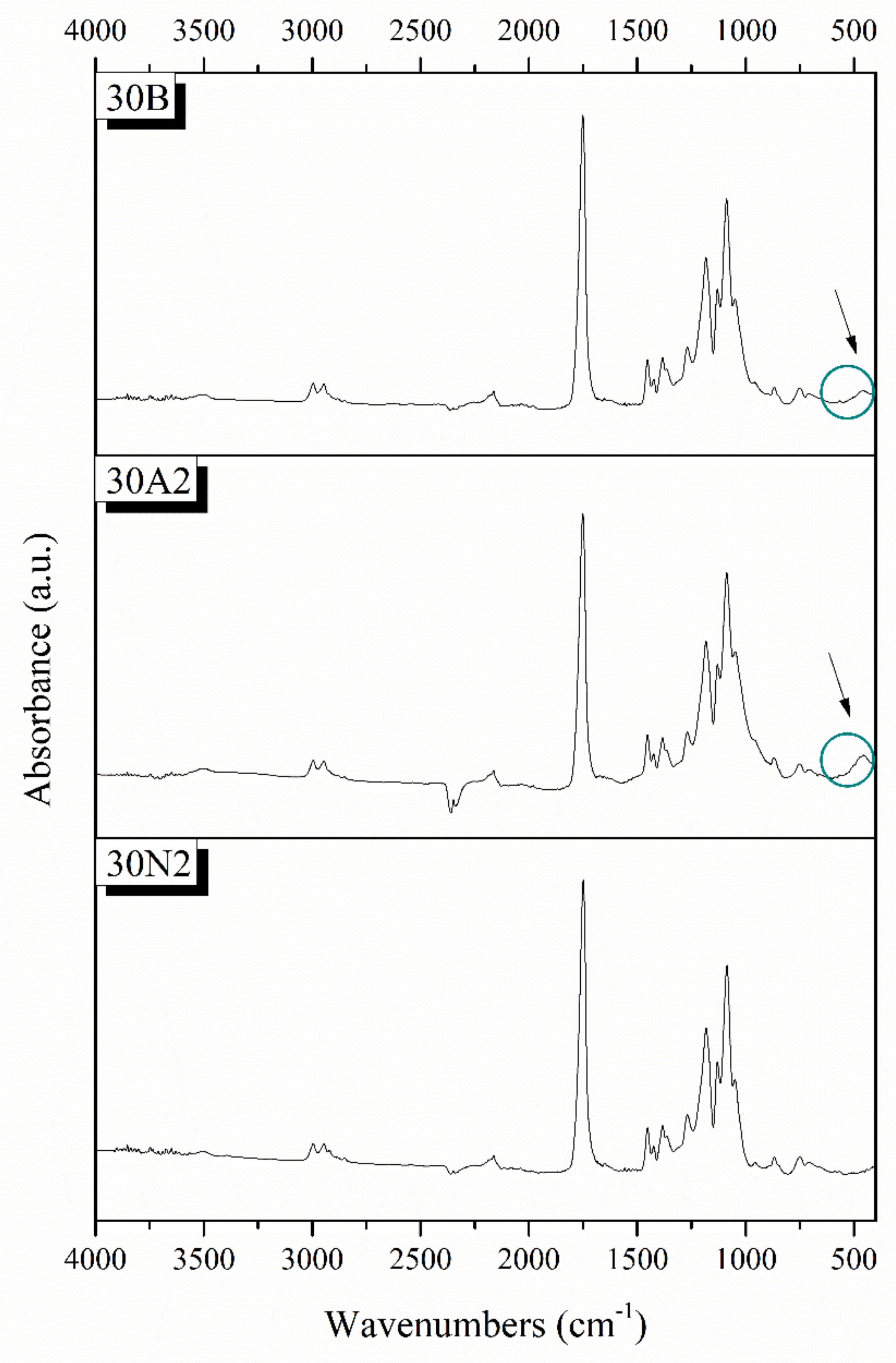
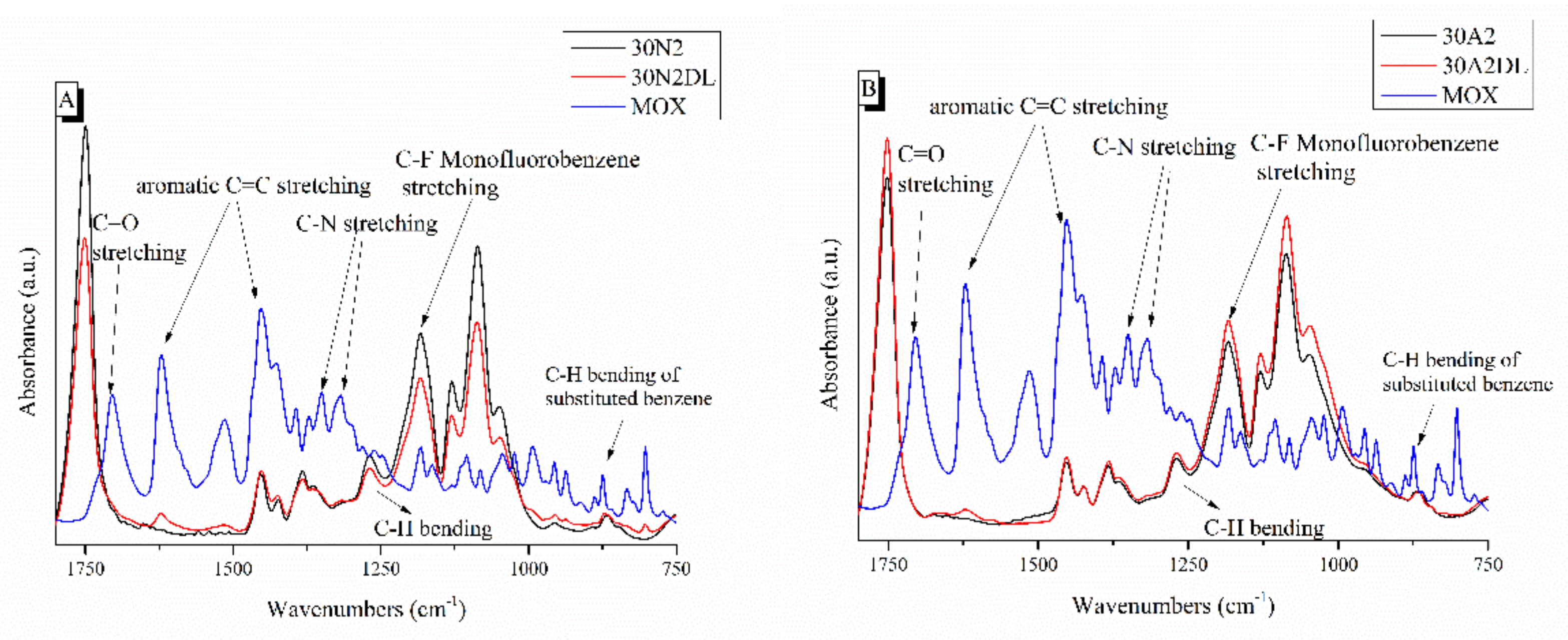
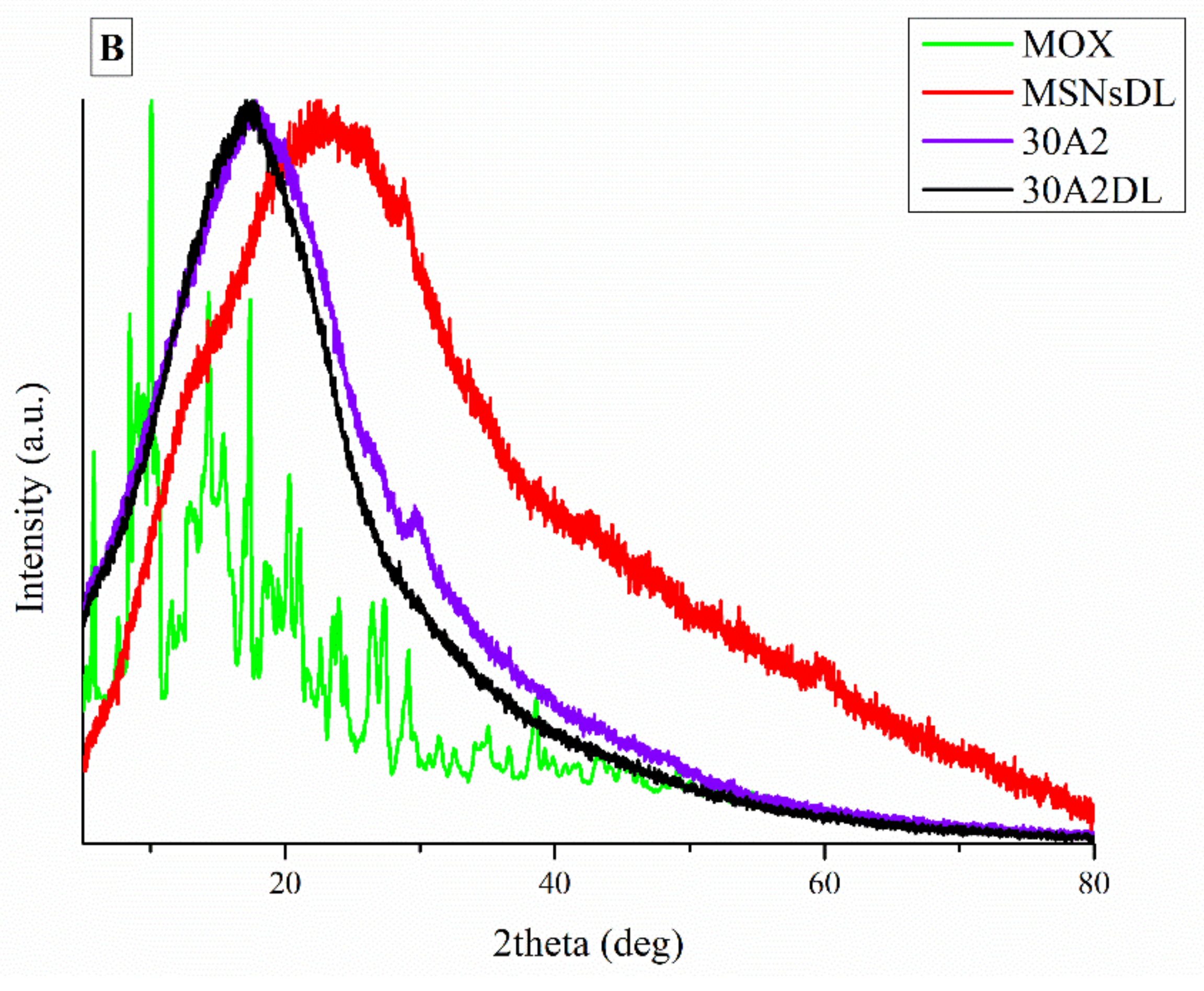
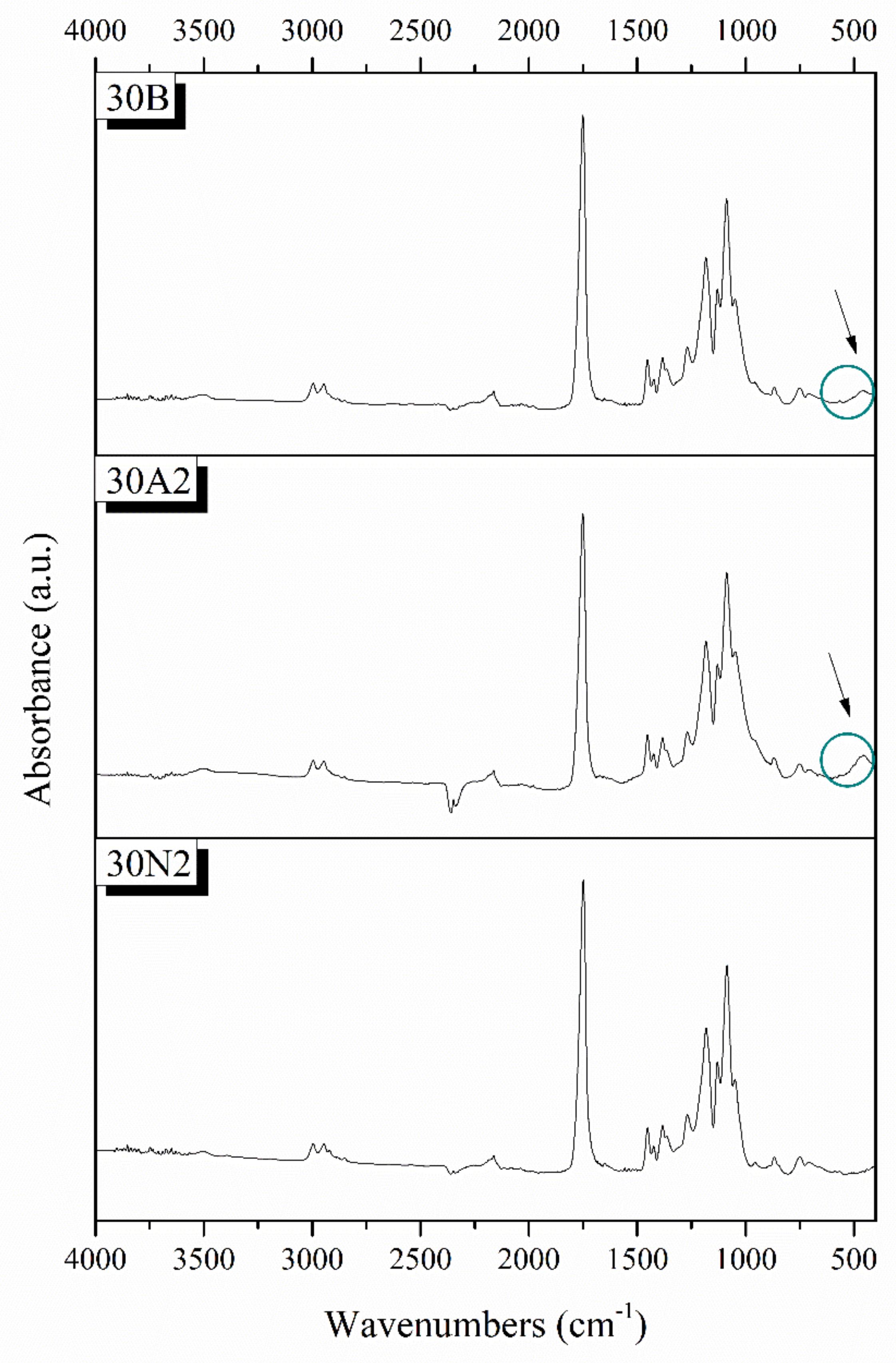
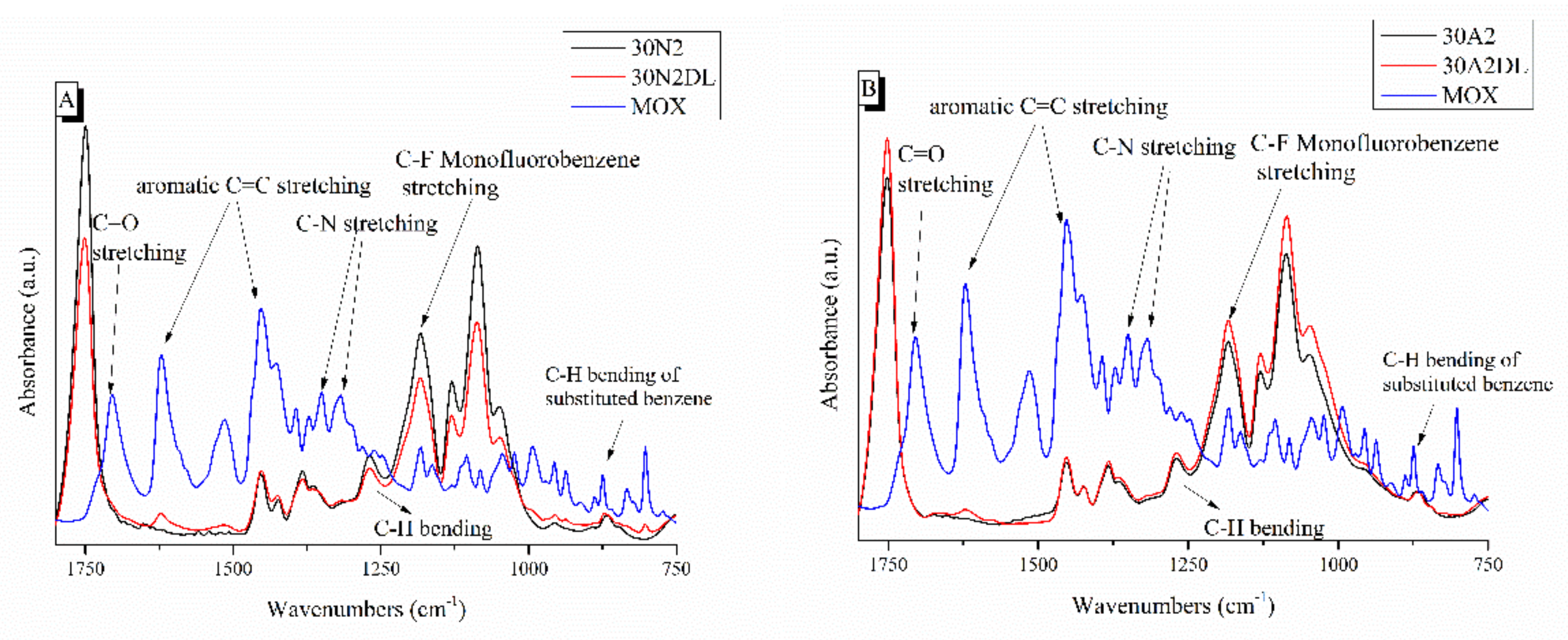
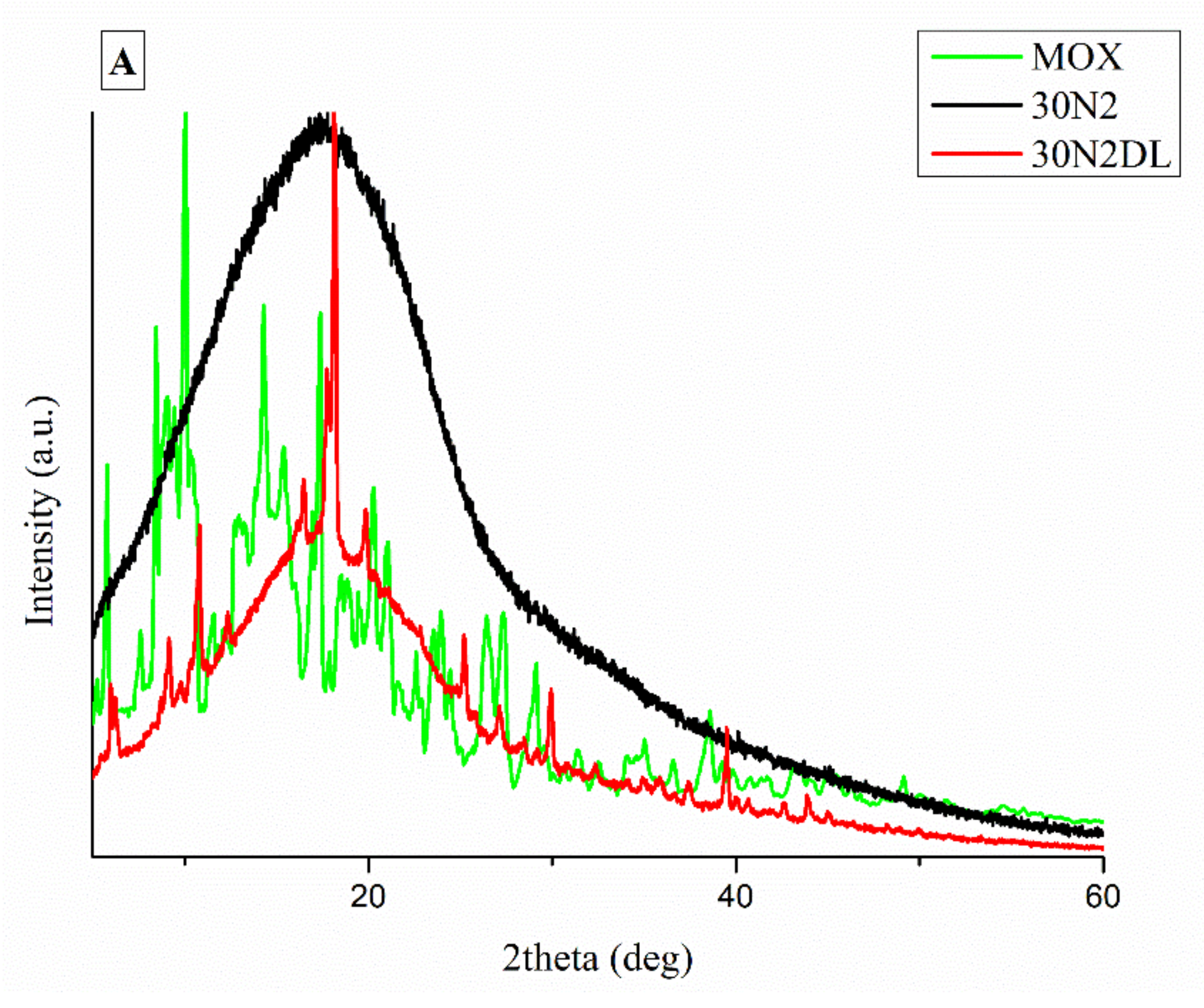
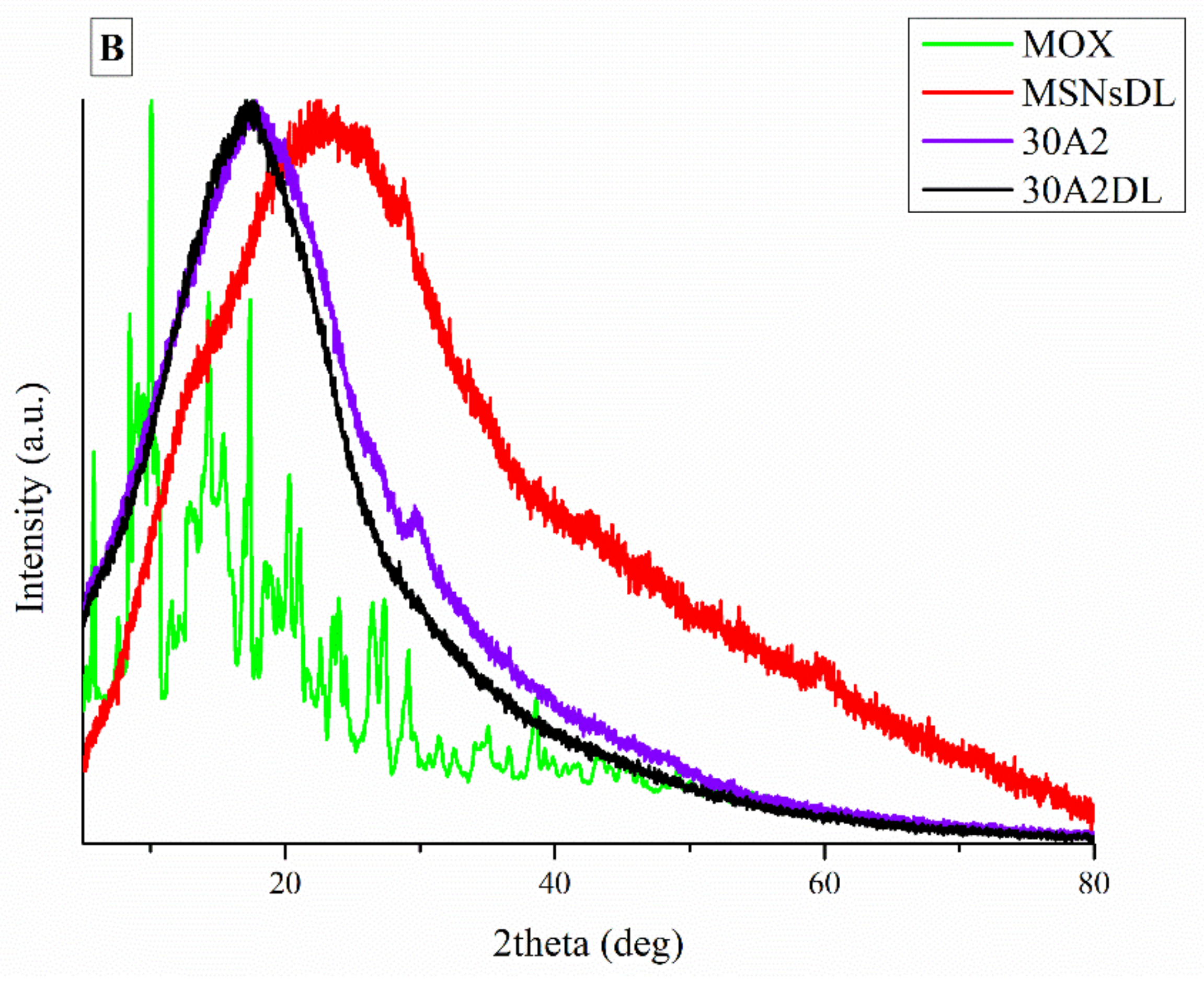
3.2. Attenuated Total Reflectance Spectroscopy (ATR)
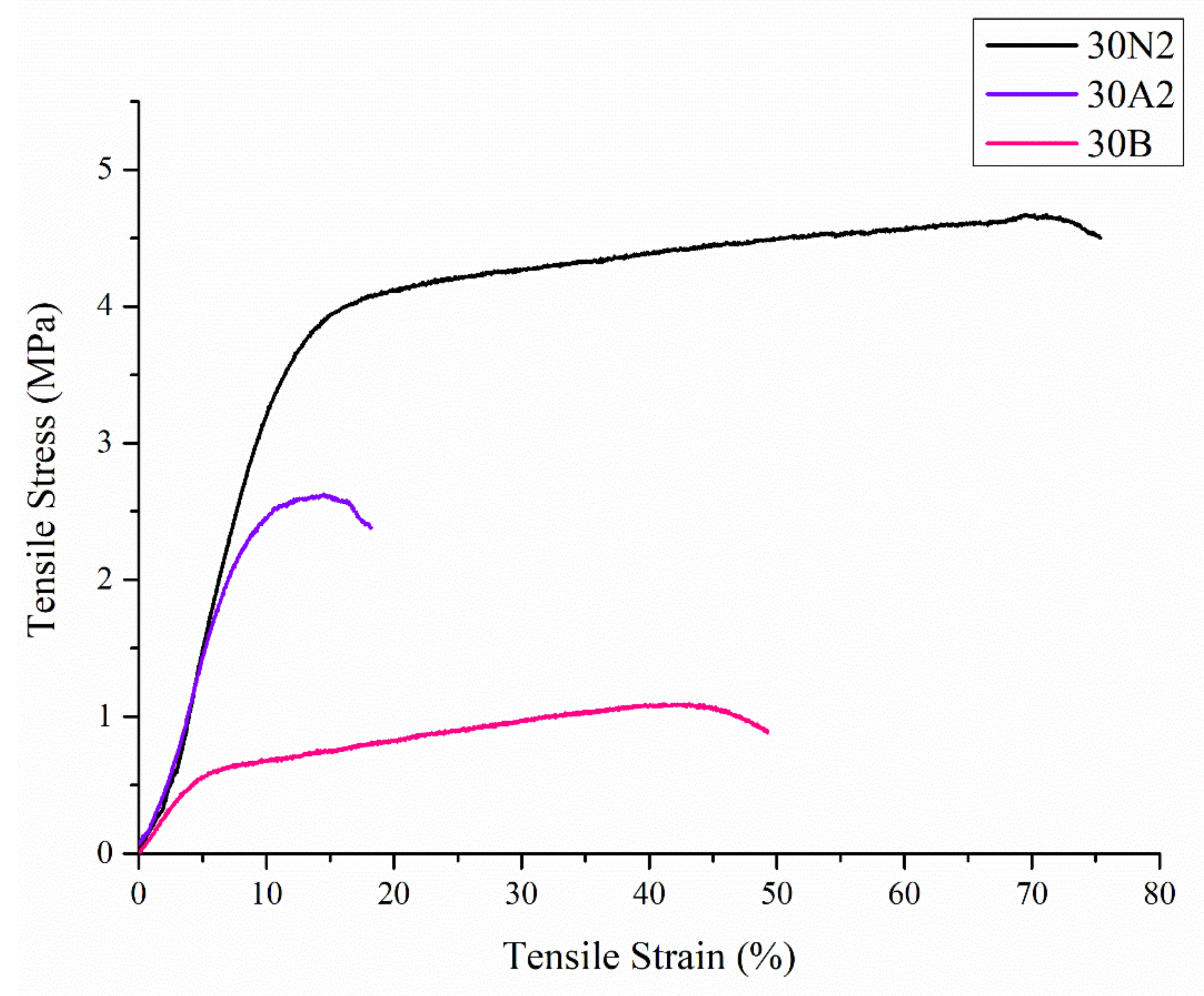
3.3. Mechanical Characterization
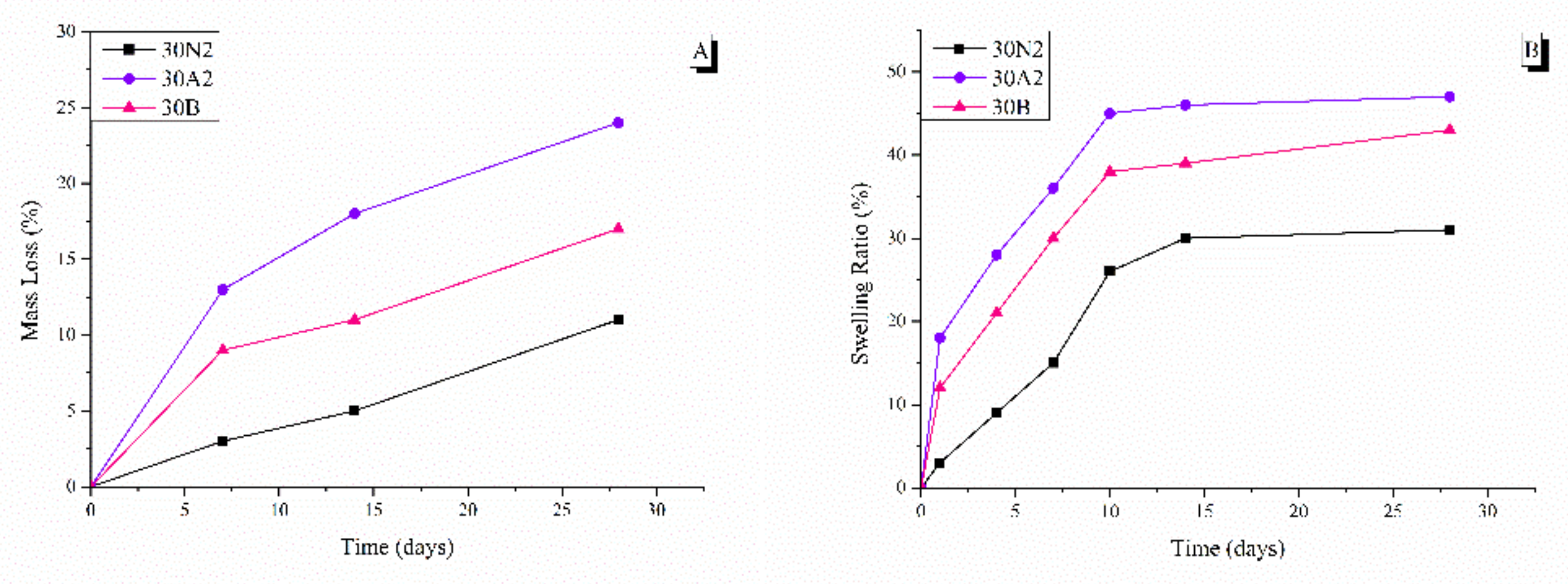
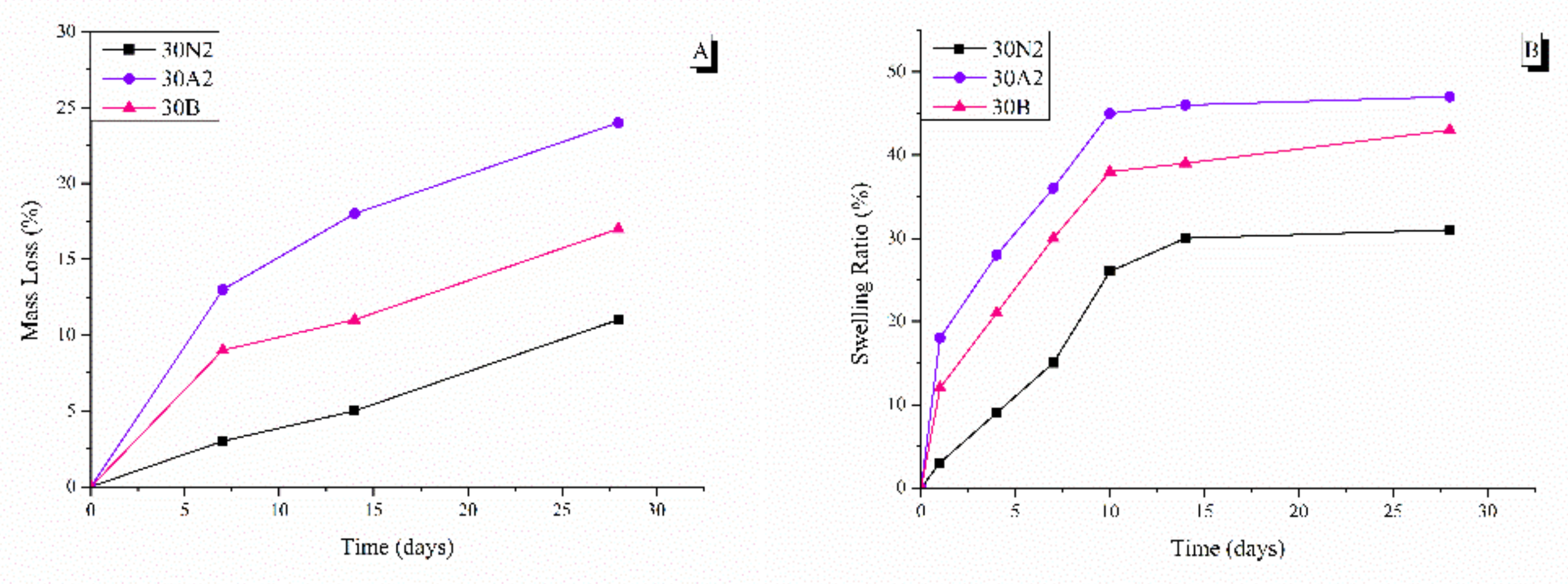
3.4. In Vitro Degradation and Equilibrium Swelling Performance
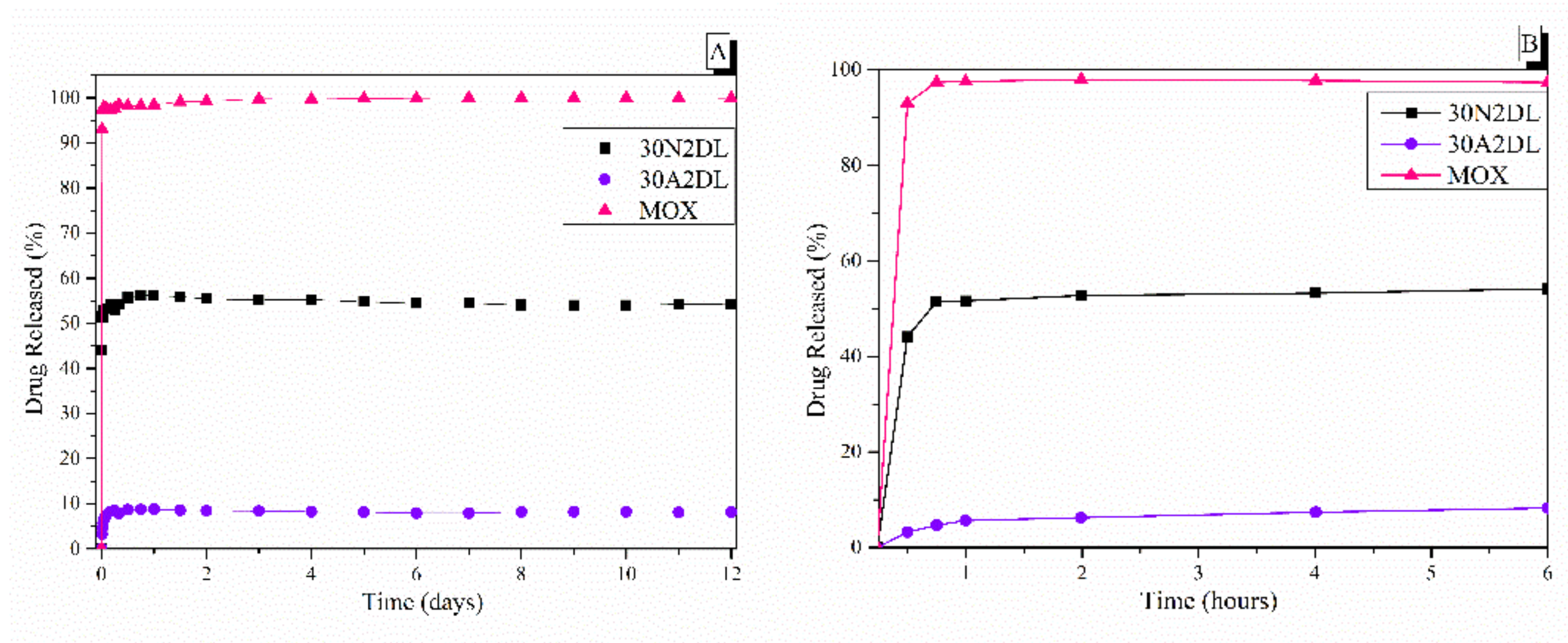
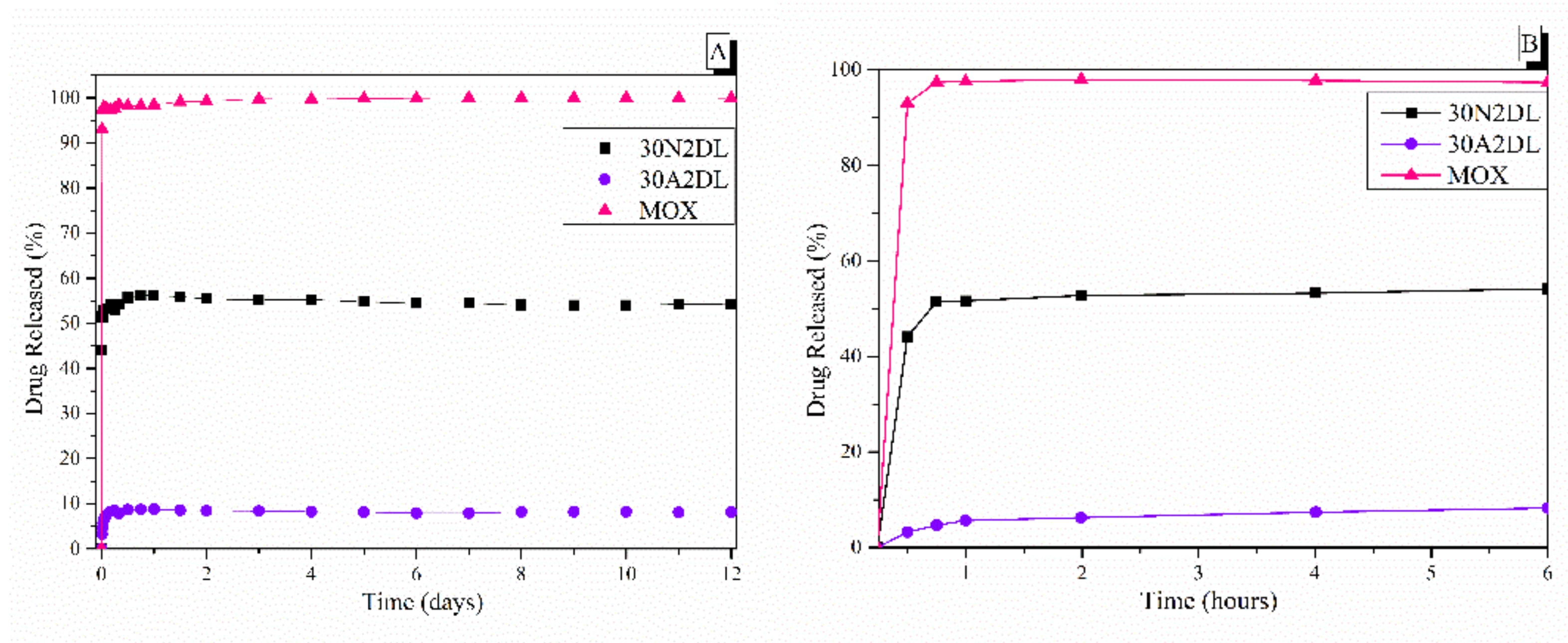
3.5. Drug Loading and Release
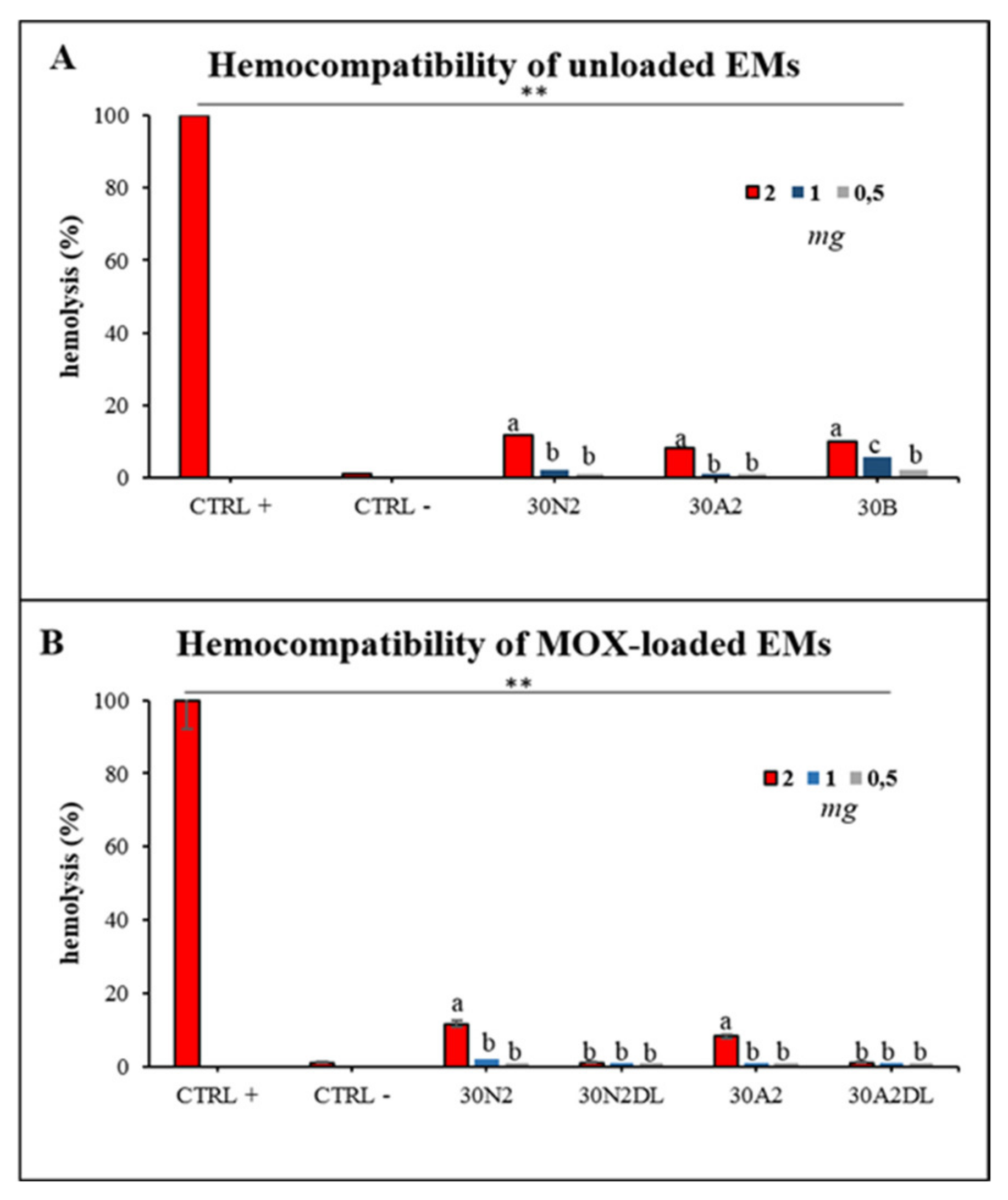
3.6. Hemocompatibility Assay
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martin-Cabezas, R.; Davideau, J.-L.; Tenenbaum, H.; Huck, O. Clinical efficacy of probiotics as an adjunctive therapy to non-surgical periodontal treatment of chronic periodontitis: A systematic review and meta-analysis. J. Clin. Periodontol. 2016, 43, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Feres, M.; Figueiredo, L.C.; Soares, G.M.S.; Faveri, M. Systemic antibiotics in the treatment of periodontitis. Periodontol. 2000 2015, 67, 131–186. [Google Scholar] [CrossRef] [PubMed]
- Karring, T.; Nyman, S.; Gottlow, J.A.N.; Laurell, L. Development of the biological concept of guided tissue regeneration? Animal and human studies. Periodontology 2000 1993, 1, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Bottino, M.C.; Thomas, V.; Schmidt, G.; Vohra, Y.K.; Chu, T.-M.G.; Kowolik, M.J.; Janowski, G. Recent advances in the development of GTR/GBR membranes for periodontal regeneration—A materials perspective. Dent. Mater. 2012, 28, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.N.; Cho, Y.J.; Tarafder, S.; Lee, C.H. The recent advances in scaffolds for integrated periodontal regeneration. Bioact. Mater. 2021, 6, 3328–3342. [Google Scholar] [CrossRef]
- Zhuang, Y.; Lin, K.; Yu, H. Advance of Nano-Composite Electrospun Fibers in Periodontal Regeneration. Front Chem. 2019, 7, 495. [Google Scholar] [CrossRef]
- Agrawal, C.M.; Ray, R.B. Biodegradable polymeric scaffolds for musculoskeletal tissue engineering. J. Biomed. Mater. Res. 2001, 55, 141–150. [Google Scholar] [CrossRef]
- Guo, B.; Ma, P.X. Synthetic biodegradable functional polymers for tissue engineering: A brief review. Sci. China Chem. 2014, 57, 490–500. [Google Scholar] [CrossRef]
- Balla, E.; Daniilidis, V.; Karlioti, G.; Kalamas, T.; Stefanidou, M.; Bikiaris, N.D.; Vlachopoulos, A.; Koumentakou, I.; Bikiaris, D.N. Poly(lactic Acid): A Versatile Biobased Polymer for the Future with Multifunctional Properties—From Monomer Synthesis, Polymerization Techniques and Molecular Weight Increase to PLA Applications. Polymers 2021, 13, 1822. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Turnbull, G.; Clarke, J.; Picard, F.; Riches, P.; Jia, L.; Han, F.; Li, B.; Shu, W. 3D bioactive composite scaffolds for bone tissue engineering. Bioact. Mater. 2018, 3, 278–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachopoulos, A.; Karlioti, G.; Balla, E.; Daniilidis, V.; Kalamas, T.; Stefanidou, M.; Bikiaris, N.D.; Christodoulou, E.; Koumentakou, I.; Karavas, E.; et al. Poly (Lactic Acid)-Based Microparticles for Drug Delivery Applications: An Overview of Recent Advances. Pharmaceutics 2022, 14, 359. [Google Scholar] [CrossRef] [PubMed]
- Gorth, D.; Webster, T.J. Matrices for tissue engineering and regenerative medicine. In Biomaterials for Artificial Organs; Elsevier: Amsterdam, The Netherlands, 2011; pp. 270–286. [Google Scholar]
- Sun, X.; Xu, C.; Wu, G.; Ye, Q.; Wang, C. Poly(Lactic-co-Glycolic Acid): Applications and Future Prospects for Periodontal Tissue Regeneration. Polymers 2017, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Kyllönen, L.; D’Este, M.; Alini, M.; Eglin, D. Local drug delivery for enhancing fracture healing in osteoporotic bone. Acta Biomater. 2015, 11, 412–434. [Google Scholar] [CrossRef]
- Wei, L.; Ke, J.; Prasadam, I.; Miron, R.J.; Lin, S.; Xiao, Y.; Chang, J.; Wu, C.; Zhang, Y. A comparative study of Sr-incorporated mesoporous bioactive glass scaffolds for regeneration of osteopenic bone defects. Osteoporos. Int. 2014, 25, 2089–2096. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, I.; Dahman, Y. Applications of nanobiomaterials in hard tissue engineering. In Nanobiomaterials in Hard Tissue Engineering; Elsevier: Berlin/Heidelberg, Germany, 2016; pp. 33–62. [Google Scholar]
- Nassar, E.J.; Neri, C.R.; Calefi, P.S.; Serra, O.A. Functionalized silica synthesized by sol–gel process. J. Non-Cryst. Solids 1999, 247, 124–128. [Google Scholar] [CrossRef]
- Singh, S.; Chen, H.; Shahrokhi, S.; Wang, L.P.; Lin, C.-H.; Hu, L.; Guan, X.; Tricoli, A.; Xu, Z.J.; Wu, T. Hybrid Organic–Inorganic Materials and Composites for Photoelectrochemical Water Splitting. ACS Energy Lett. 2020, 5, 1487–1497. [Google Scholar] [CrossRef]
- Vrancken, K.; Possemiers, K.; Van Der Voort, P.; Vansant, E. Surface modification of silica gels with aminoorganosilanes. Colloids Surfaces A Physicochem. Eng. Asp. 1995, 98, 235–241. [Google Scholar] [CrossRef]
- Beari, F.; Brand, M.; Jenkner, P.; Lehnert, R.; Metternich, H.J.; Monkiewicz, J.; Siesler, H.W. Organofunctional alkoxysilanes in dilute aqueous solution: New accounts on the dynamic structural mutability. J. Organomet. Chem. 2001, 625, 208–216. [Google Scholar] [CrossRef]
- Nassar, E.J.; Ciuffi, K.J.; Ribeiro, S.J.L.; Messaddeq, Y. Europium incorporated in silica matrix obtained by sol-gel: Luminescent materials. Mater. Res. 2003, 6, 557–562. [Google Scholar] [CrossRef] [Green Version]
- Jackson, C.L.; Bauer, B.J.; Nakatani, A.I.; Barnes, J.D. Synthesis of Hybrid Organic−Inorganic Materials from Interpenetrating Polymer Network Chemistry. Chem. Mater. 1996, 8, 727–733. [Google Scholar] [CrossRef]
- Shea, K.J.; Loy, D.A.; Webster, O. Arylsilsesquioxane gels and related materials. New hybrids of organic and inorganic networks. J. Am. Chem. Soc. 1992, 114, 6700–6710. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Leclercq, D. Recent Developments of Molecular Chemistry for Sol–Gel Processes. Angew. Chem. Int. Ed. Engl. 1996, 35, 1420–1436. [Google Scholar] [CrossRef]
- Corriu, R. A new trend in metal-alkoxide chemistry: The elaboration of monophasic organic-inorganic hybrid materials. Polyhedron 1998, 17, 925–934. [Google Scholar] [CrossRef]
- Cerveau, G.; Corriu, R.J.P.; Lepeytre, C.; Mutin, P.H. Influence of the nature of the organic precursor on the textural and chemical properties of silsesquioxane materials. J. Mater. Chem. 1998, 8, 2707–2713. [Google Scholar] [CrossRef]
- Pouroutzidou, G.K.; Liverani, L.; Theocharidou, A.; Tsamesidis, I.; Lazaridou, M.; Christodoulou, E.; Beketova, A.; Pappa, C.; Triantafyllidis, K.S.; Anastasiou, A.D.; et al. Synthesis and Characterization of Mesoporous Mg- and Sr-Doped Nanoparticles for Moxifloxacin Drug Delivery in Promising Tissue Engineering Applications. Int. J. Mol. Sci. 2021, 22, 577. [Google Scholar] [CrossRef] [PubMed]
- Landersdorfer, C.B.; Kinzig, M.; Hennig, F.F.; Bulitta, J.B.; Holzgrabe, U.; Drusano, G.L.; Sörgel, F.; Gusinde, J. Penetration of Moxifloxacin into Bone Evaluated by Monte Carlo Simulation. Antimicrob. Agents. Chemother. 2009, 53, 2074–2081. [Google Scholar] [CrossRef] [Green Version]
- Flemmig, T.F.; Petersilka, G.; Völp, A.; Gravemeier, M.; Zilly, M.; Mross, D.; Prior, K.; Yamamoto, J.; Beikler, T. Efficacy and Safety of Adjunctive Local Moxifloxacin Delivery in the Treatment of Periodontitis. J. Periodontol. 2011, 82, 96–105. [Google Scholar] [CrossRef]
- Milazzo, I.; Blandino, G.; Musumeci, R.; Nicoletti, G.; Lo Bue, A.; Speciale, A. Antibacterial activity of moxifloxacin against periodontal anaerobic pathogens involved in systemic infections. Int. J. Antimicrob. Agents. 2002, 20, 451–456. [Google Scholar] [CrossRef]
- Müller, H.-P.; Holderrieth, S.; Burkhardt, U.; Höffler, U. In vitro antimicrobial susceptibility of oral strains of Actinobacillus actinomycetemcomitans to seven antibiotics. J Clin. Periodontol. 2002, 29, 736–742. [Google Scholar] [CrossRef]
- Eick, S.; Seltmann, T.; Pfister, W. Efficacy of antibiotics to strains of periodontopathogenic bacteria within a single species biofilm—An in vitro study. J. Clin. Periodontol. 2004, 31, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Guentsch, A.; Jentsch, H.; Pfister, W.; Hoffmann, T.; Eick, S. Moxifloxacin as an Adjunctive Antibiotic in the Treatment of Severe Chronic Periodontitis. J. Periodontol. 2008, 79, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Eick, S.; Pfister, W. Efficacy of Antibiotics Against Periodontopathogenic Bacteria Within Epithelial Cells: An In Vitro Study. J. Periodontol. 2004, 75, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, S.; Rakhorst, G.; Blanton, J.; van Oeveren, W. Standardization of incubation conditions for hemolysis testing of biomaterials. Mater. Sci. Eng. C 2009, 29, 1650–1654. [Google Scholar] [CrossRef]
- Albrektsson, T.; Johansson, C. Osteoinduction, osteoconduction and osseointegration. Eur. Spine J. 2001, 10, S96–S101. [Google Scholar]
- Cho, Y.-D.; Kim, K.-H.; Lee, Y.-M.; Ku, Y.; Seol, Y.-J. Periodontal Wound Healing and Tissue Regeneration: A Narrative Review. Pharmaceuticals 2021, 14, 456. [Google Scholar] [CrossRef]
- Alehosseini, M.; Golafshan, N.; Kharaziha, M.; Fathi, M.; Edris, H. Hemocompatible and Bioactive Heparin-Loaded PCL-α-TCP Fibrous Membranes for Bone Tissue Engineering. Macromol. Biosci. 2018, 18, 1800020. [Google Scholar] [CrossRef]
- Cao, C.; Song, Y.; Yao, Q.; Yao, Y.; Wang, T.; Huang, B.; Gong, P. Preparation and preliminary in vitro evaluation of a bFGF-releasing heparin-conjugated poly(ε-caprolactone) membrane for guided bone regeneration. J. Biomater. Sci. Polym. Ed. 2015, 26, 600–616. [Google Scholar] [CrossRef]
- Shoba, E.; Lakra, R.; Kiran, M.S.; Korrapati, P.S. 3D nano bilayered spatially and functionally graded scaffold impregnated bromelain conjugated magnesium doped hydroxyapatite nanoparticle for periodontal regeneration. J. Mech. Behav. Biomed. Mater. 2020, 109, 103822. [Google Scholar] [CrossRef]
- Padalhin, A.R.; Thuy Ba Linh, N.; Ki Min, Y.; Lee, B.-T. Evaluation of the cytocompatibility hemocompatibility in vivo bone tissue regenerating capability of different PCL blends. J. Biomater. Sci. Polym. Ed. 2014, 25, 487–503. [Google Scholar] [CrossRef]
- Maji, K.; Pramanik, K. Electrospun scaffold for bone regeneration. Int. J. Polym. Mater. Polym. Biomater. 2021, 1–16. [Google Scholar] [CrossRef]
- Dzenis, Y. Spinning Continuous Fibers for Nanotechnology. Science 2004, 304, 1917–1919. [Google Scholar] [CrossRef]
- Fong, H.; Chun, I.; Reneker, D. Beaded nanofibers formed during electrospinning. Polymer 1999, 40, 4585–4592. [Google Scholar] [CrossRef]
- Venugopal, J.R.; Low, S.; Choon, A.T.; Kumar, A.B.; Ramakrishna, S. Nanobioengineered Electrospun Composite Nanofibers and Osteoblasts for Bone Regeneration. Artif. Organs 2008, 32, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Li, J.; Zhang, X.; Du, L.; Li, Y.; Li, D.; Kong, B.; Ge, S. Super-assembled core/shell fibrous frameworks with dual growth factors for in situ cementum–ligament–bone complex regeneration. Biomater. Sci. 2020, 8, 2459–2471. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yang, F.; Yan, X.; Yang, W.; Yu, N.; Oortgiesen, D.A.W.; Wang, Y.; Jansen, J.A.; Walboomers, X.F. Influence of bone marrow-derived mesenchymal stem cells pre-implantation differentiation approach on periodontal regeneration in vivo. J. Clin. Periodontol. 2015, 42, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Pham, Q.P.; Sharma, U.; Mikos, A.G. Electrospinning of Polymeric Nanofibers for Tissue Engineering Applications: A Review. Tissue Eng. 2006, 12, 1197–1211. [Google Scholar] [CrossRef] [Green Version]
- Teo, W.E.; Ramakrishna, S. A review on electrospinning design and nanofibre assemblies. Nanotechnology 2006, 17, R89–R106. [Google Scholar] [CrossRef]
- Unnithan, A.R.; Arathyram, R.S.; Kim, C.S. Electrospinning of Polymers for Tissue Engineering. In Nanotechnology Applications for Tissue Engineering; Elsevier: Amsterdam, The Netherlands, 2015; pp. 45–55. [Google Scholar]
- Barrientos, I.J.H.; Paladino, E.; Szabó, P.; Brozio, S.; Hall, P.J.; Oseghale, C.I.; Passarelli, M.K.; Moug, S.J.; Black, R.A.; Wilson, C.G.; et al. Electrospun collagen-based nanofibres: A sustainable material for improved antibiotic utilisation in tissue engineering applications. Int. J. Pharm. 2017, 531, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Grafahrend, D.; Heffels, K.-H.; Beer, M.V.; Gasteier, P.; Möller, M.; Boehm, G.; Dalton, P.D.; Groll, J. Degradable polyester scaffolds with controlled surface chemistry combining minimal protein adsorption with specific bioactivation. Nat. Mater. 2011, 10, 67–73. [Google Scholar] [CrossRef]
- Li, D.; Xia, Y. Electrospinning of Nanofibers: Reinventing the Wheel? Adv. Mater. 2004, 16, 1151–1170. [Google Scholar] [CrossRef]
- Yarin, A.L.; Koombhongse, S.; Reneker, D.H. Taylor cone and jetting from liquid droplets in electrospinning of nanofibers. J. Appl. Phys. 2001, 90, 4836–4846. [Google Scholar] [CrossRef] [Green Version]
- Reneker, D.H.; Chun, I. Nanometre diameter fibres of polymer, produced by electrospinning. Nanotechnology 1996, 7, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Doshi, J.; Reneker, D.H. Electrospinning process and applications of electrospun fibers. J. Electrostat. 1995, 35, 151–160. [Google Scholar] [CrossRef]
- Li, H.; Chang, J. pH-compensation effect of bioactive inorganic fillers on the degradation of PLGA. Compos. Sci. Technol. 2005, 65, 2226–2232. [Google Scholar] [CrossRef]
- Hu, C.; Liu, S.; Zhang, Y.; Li, B.; Yang, H.; Fan, C.; Cui, W. Long-term drug release from electrospun fibers for in vivo inflammation prevention in the prevention of peritendinous adhesions. Acta Biomater. 2013, 9, 7381–7388. [Google Scholar] [CrossRef]
- Song, B.; Wu, C.; Chang, J. Dual drug release from electrospun poly(lactic-co-glycolic acid)/mesoporous silica nanoparticles composite mats with distinct release profiles. Acta Biomater. 2012, 8, 1901–1907. [Google Scholar] [CrossRef]
- Shin, H.J.; Lee, C.H.; Cho, I.H.; Kim, Y.-J.; Lee, Y.-J.; Kim, I.A.; Park, K.-D.; Yui, N.; Shin, J.-W. Electrospun PLGA nanofiber scaffolds for articular cartilage reconstruction: Mechanical stability, degradation and cellular responses under mechanical stimulation in vitro. J. Biomater. Sci. Polym. Ed. 2006, 17, 103–119. [Google Scholar] [CrossRef]
- Lu, L.; Peter, S.J.; Lyman, M.D.; Lai, H.-L.; Leite, S.M.; Tamada, J.A.; Uyama, S.; Vacanti, J.P.; Langer, R.; Mikos, A.G. In vitro and in vivo degradation of porous poly(dl-lactic-co-glycolic acid) foams. Biomaterials 2000, 21, 1837–1845. [Google Scholar] [CrossRef]
- Lu, L.; Garcia, C.A.; Mikos, A.G. In vitro degradation of thin poly(DL-lactic-co-glycolic acid) films. J. Biomed. Mater. Res. 1999, 46, 236–244. [Google Scholar] [CrossRef]
- Paragkumar, N.T.; Edith, D.; Six, J.-L. Surface characteristics of PLA and PLGA films. Appl. Surf. Sci. 2006, 253, 2758–2764. [Google Scholar] [CrossRef]
- Akl, M.A.; Kartal-Hodzic, A.; Oksanen, T.; Ismael, H.R.; Afouna, M.M.; Yliperttula, M.; Samy, A.M.; Viitala, T. Factorial design formulation optimization and in vitro characterization of curcumin-loaded PLGA nanoparticles for colon delivery. J. Drug Deliv. Sci. Technol. 2016, 32, 10–20. [Google Scholar] [CrossRef]
- Jia, Y.; Zhang, H.; Yang, S.; Xi, Z.; Tang, T.; Yin, R.; Zhang, W. Electrospun PLGA membrane incorporated with andrographolide-loaded mesoporous silica nanoparticles for sustained antibacterial wound dressing. Nanomedicine 2018, 13, 2881–2899. [Google Scholar] [CrossRef] [PubMed]
- Al Omari, M.M.H.; Jaafari, D.S.; Al-Sou’od, K.A.; Badwan, A.A. Moxifloxacin Hydrochloride. In Profiles of Drug Substances, Excipients and Related Methodology; Academic Press Inc.: Cambridge, MA, USA, 2014; pp. 299–431. [Google Scholar]
- Nagarjuna Reddy, Y.; Deepika, P.; Venkatesh, M.; Rajeshwari, K. Evaluation of moxifloxacin-hydroxyapatite composite graft in the regeneration of intrabony defects: A clinical, radiographic, and microbiological study. Contemp. Clin. Dent. 2016, 7, 357. [Google Scholar] [CrossRef]
- Li, Z.; Clemens, D.L.; Lee, B.-Y.; Dillon, B.J.; Horwitz, M.A.; Zink, J.I. Mesoporous Silica Nanoparticles with pH-Sensitive Nanovalves for Delivery of Moxifloxacin Provide Improved Treatment of Lethal Pneumonic Tularemia. ACS Nano 2015, 9, 10778–10789. [Google Scholar] [CrossRef]
- Lee, B.; Li, Z.; Clemens, D.L.; Dillon, B.J.; Hwang, A.A.; Zink, J.I.; Horwitz, M.A. Redox-Triggered Release of Moxifloxacin from Mesoporous Silica Nanoparticles Functionalized with Disulfide Snap-Tops Enhances Efficacy Against Pneumonic Tularemia in Mice. Small 2016, 12, 3690–3702. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, S.; Li, Q. Mathematical modeling of degradation for bulk-erosive polymers: Applications in tissue engineering scaffolds and drug delivery systems. Acta Biomater. 2011, 7, 1140–1149. [Google Scholar] [CrossRef]
- Júlio, T.A.; Garcia, J.S.; Bonfilio, R.; Araújo, M.B.; Trevisan, M.G. Solid-state stability and solubility determination of crystalline forms of moxifloxacin hydrochloride. Int. J. Pharm. Pharm. Sci. 2015, 7, 173–177. [Google Scholar]
- Filippousi, M.; Siafaka, P.I.; Amanatiadou, E.P.; Nanaki, S.G.; Nerantzaki, M.; Bikiaris, D.N.; Vizirianakis, I.S.; Van Tendeloo, G. Modified chitosan coated mesoporous strontium hydroxyapatite nanorods as drug carriers. J. Mater. Chem. B 2015, 3, 5991–6000. [Google Scholar] [CrossRef]
- Pham, Q.P.; Sharma, U.; Mikos, A.G. Electrospun Poly(ε-caprolactone) Microfiber and Multilayer Nanofiber/Microfiber Scaffolds: Characterization of Scaffolds and Measurement of Cellular Infiltration. Biomacromolecules 2006, 7, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.-M.; Nejad, Z.M.; Hashemi, S.A.; Salari, M.; Gholami, A.; Ramakrishna, S.; Chiang, W.-H.; Lai, C.W. Bioactive Agent-Loaded Electrospun Nanofiber Membranes for Accelerating Healing Process: A Review. Membranes 2021, 11, 702. [Google Scholar] [CrossRef] [PubMed]
- Rnjak-Kovacina, J.; Weiss, A.S. Increasing the Pore Size of Electrospun Scaffolds. Tissue Eng. Part B Rev. 2011, 17, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Lowery, J.L.; Datta, N.; Rutledge, G.C. Effect of fiber diameter, pore size and seeding method on growth of human dermal fibroblasts in electrospun poly(ɛ-caprolactone) fibrous mats. Biomaterials 2010, 31, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.; Huang, Y.; Agarwal, S.; Lannutti, J. Improved Cellular Infiltration in Electrospun Fiber via Engineered Porosity. Tissue Eng. 2007, 13, 2249–2257. [Google Scholar] [CrossRef] [Green Version]
- Soliman, S.; Pagliari, S.; Rinaldi, A.; Forte, G.; Fiaccavento, R.; Pagliari, F.; Franzese, O.; Minieri, M.; Di Nardo, P.; Licoccia, S. Multiscale three-dimensional scaffolds for soft tissue engineering via multimodal electrospinning. Acta Biomater. 2010, 6, 1227–1237. [Google Scholar] [CrossRef] [Green Version]
- Khil, M.-S.; Cha, D.-I.; Kim, H.-Y.; Kim, I.-S.; Bhattarai, N. Electrospun nanofibrous polyurethane membrane as wound dressing. J. Biomed. Mater. Res. 2003, 67B, 675–679. [Google Scholar] [CrossRef]
- Van Tienen, T.G.; Heijkants, R.G.J.; Buma, P.; de Groot, J.H.; Pennings, A.J.; Veth, R.P. Tissue ingrowth and degradation of two biodegradable porous polymers with different porosities and pore sizes. Biomaterials 2002, 23, 1731–1738. [Google Scholar] [CrossRef]
- Sisson, K.; Zhang, C.; Farach-Carson, M.C.; Chase, D.B.; Rabolt, J.F. Fiber diameters control osteoblastic cell migration and differentiation in electrospun gelatin. J. Biomed. Mater. Res. Part A 2010, 94A, 1312–1320. [Google Scholar] [CrossRef]
- Rnjak, J.; Li, Z.; Maitz, P.K.M.; Wise, S.G.; Weiss, A.S. Primary human dermal fibroblast interactions with open weave three-dimensional scaffolds prepared from synthetic human elastin. Biomaterials 2009, 30, 6469–6477. [Google Scholar] [CrossRef]
- Rnjak-Kovacina, J.; Wise, S.G.; Li, Z.; Maitz, P.K.M.; Young, C.J.; Wang, Y.; Weiss, A.S. Tailoring the porosity and pore size of electrospun synthetic human elastin scaffolds for dermal tissue engineering. Biomaterials 2011, 32, 6729–6736. [Google Scholar] [CrossRef]
- Balguid, A.; Mol, A.; van Marion, M.H.; Bank, R.A.; Bouten, C.V.C.; Baaijens, F.P.T. Tailoring Fiber Diameter in Electrospun Poly(ɛ-Caprolactone) Scaffolds for Optimal Cellular Infiltration in Cardiovascular Tissue Engineering. Tissue Eng. Part A 2009, 15, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.W.; Mifflin, R.C.; Valentich, J.D.; Crowe, S.E.; Saada, J.I.; West, A.B. Myofibroblasts. I. Paracrine cells important in health and disease. Am. J. Physiol. Physiol. 1999, 277, C1–C19. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, S.J.; Sampson, W.W. Statistical geometry of pores and statistics of porous nanofibrous assemblies. J. R. Soc. Interface 2005, 2, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Motamedi, A.S.; Mirzadeh, H.; Hajiesmaeilbaigi, F.; Bagheri-Khoulenjani, S.; Shokrgozar, M. Effect of electrospinning parameters on morphological properties of PVDF nanofibrous scaffolds. Prog. Biomater. 2017, 6, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Alhamdani, G.M.; Al-Turaihi, B.A.; Al-Masoody, A.H. Electrospinning approaches for periodontal regeneration: A review. Drug Invent. Today. 2019, 11, 2917–2926. [Google Scholar]
- Zargham, S.; Bazgir, S.; Tavakoli, A.; Rashidi, A.S.; Damerchely, R. The Effect of Flow Rate on Morphology and Deposition Area of Electrospun Nylon 6 Nanofiber. J. Eng. Fiber Fabr. 2012, 7, 155892501200700. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Zhang, Y.; Dong, C.; Sheng, J. Morphology of ultrafine polysulfone fibers prepared by electrospinning. Polym. Int. 2004, 53, 1704–1710. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, L.; Fan, J.; Wang, R.; Yu, J.-Y. Effect of applied voltage on diameter and morphology of ultrafine fibers in bubble electrospinning. J. Appl. Polym. Sci. 2011, 120, 592–598. [Google Scholar] [CrossRef]
- Demir, M.; Yilgor, I.; Yilgor, E.; Erman, B. Electrospinning of polyurethane fibers. Polymer 2002, 43, 3303–3309. [Google Scholar] [CrossRef]
- Zare, Y. Study of nanoparticles aggregation/agglomeration in polymer particulate nanocomposites by mechanical properties. Compos. Part A Appl. Sci. Manuf. 2016, 84, 158–164. [Google Scholar] [CrossRef]
- Heggannavar, G.B.; Vijeth, S.; Kariduraganavar, M.Y. Development of dual drug loaded PLGA based mesoporous silica nanoparticles and their conjugation with Angiopep-2 to treat glioma. J. Drug Deliv. Sci. Technol. 2019, 53, 101157. [Google Scholar] [CrossRef]
- Kikuchi, M.; Koyama, Y.; Yamada, T.; Imamura, Y.; Okada, T.; Shirahama, N.; Akita, K.; Takakuda, K.; Tanaka, J. Development of guided bone regeneration membrane composed of β-tricalcium phosphate and poly (l-lactide-co-glycolide-co-ε-caprolactone) composites. Biomaterials 2004, 25, 5979–5986. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, L.; Zhou, Z.; Lai, H.; Xu, P.; Liao, L.; Wei, J. Biodegradable Polymer Membranes Applied in Guided Bone/Tissue Regeneration: A Review. Polymers 2016, 8, 115. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; He, M.; Liang, Y.; Crawford, A.; Coates, P.; Chen, D.; Shi, R.; Zhang, L. Fabrication and evaluation of electrospun PCL–gelatin micro-/nanofiber membranes for anti-infective GTR implants. J. Mater. Chem. B 2014, 2, 6867–6877. [Google Scholar] [CrossRef] [PubMed]
- Hameed, M.; Rasul, A.; Nazir, A.; Yousaf, A.M.; Hussain, T.; Khan, I.U.; Abbas, G.; Abid, S.; Yousafi, Q.U.A.; Ghori, M.U.; et al. Moxifloxacin-loaded electrospun polymeric composite nanofibers-based wound dressing for enhanced antibacterial activity and healing efficacy. Int. J. Polym. Mater. Polym. Biomater. 2021, 70, 1271–1279. [Google Scholar] [CrossRef]
- Luraghi, A.; Peri, F.; Moroni, L. Electrospinning for drug delivery applications: A review. J. Control. Release 2021, 334, 463–484. [Google Scholar] [CrossRef]
- Seif, S.; Franzen, L.; Windbergs, M. Overcoming drug crystallization in electrospun fibers—Elucidating key parameters and developing strategies for drug delivery. Int. J. Pharm. 2015, 478, 390–397. [Google Scholar] [CrossRef]
- Ammann, K.R.; Hossainy, S.F.A.; Hossainy, S.; Slepian, M.J. Hemocompatibility of polymers for use in vascular endoluminal implants. J. Appl. Polym. Sci. 2021, 138, 51277. [Google Scholar] [CrossRef]
- Tsamesidis, I.; Gkiliopoulos, D.; Pouroutzidou, G.K.; Lymperaki, E.; Papoulia, C.; Reybier, K.; Perio, P.; Paraskevopoulos, K.M.; Kontonasaki, E.; Theocharidou, A. Effect of Artemisinin-Loaded Mesoporous Cerium-Doped Calcium Silicate Nanopowder on Cell Proliferation of Human Periodontal Ligament Fibroblasts. Nanomaterials 2021, 11, 2189. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | PLGA Concentration (% w/v) | MSNs Concentration (% w/v) | Voltage (kV) | Rotation Speed (rpm) | Average Fiber Diameter (nm) | Average Pore Diameter (μm) |
|---|---|---|---|---|---|---|
| 20Ν1 | 20 | - | 22 | 1100 | 299 | 0.82 |
| 20Ν2 | 20 | - | 22 | 500 | 297 | 0.77 |
| 20Ν3 | 20 | - | 22 | 400 | 283 | 0.62 |
| 20Ν4 | 20 | - | 18 | 1100 | 278 | 0.62 |
| 20Ν5 | 20 | - | 18 | 500 | 239 | 0.54 |
| 20Ν6 | 20 | - | 18 | 400 | 239 | 0.54 |
| 30Ν1 | 30 | - | 22 | 1100 | 728 | 2.15 |
| 30Ν2 | 30 | - | 22 | 800 | 718 | 1.49 |
| 30Ν3 | 30 | - | 22 | 500 | 716 | 1.48 |
| 30Ν4 | 30 | - | 22 | 400 | 707 | 1.47 |
| 30Ν5 | 30 | - | 18 | 1100 | 767 | 1.55 |
| 30Ν6 | 30 | - | 18 | 500 | 759 | 1.50 |
| 30Ν7 | 30 | - | 18 | 400 | 759 | 1.47 |
| 20A1 | 20 | 10 | 22 | 1100 | 421 | 1.72 |
| 20A2 | 20 | 10 | 22 | 500 | 383 | 1.45 |
| 20A3 | 20 | 10 | 22 | 400 | 340 | 1.34 |
| 20A4 | 20 | 10 | 18 | 1100 | 335 | 1.31 |
| 20A5 | 20 | 10 | 18 | 500 | 315 | 1.14 |
| 20A6 | 20 | 10 | 18 | 400 | 312 | 1.11 |
| 30A1 | 30 | 10 | 22 | 1100 | 436 | 2.92 |
| 30A2 | 30 | 10 | 22 | 800 | 434 | 2.45 |
| 30A3 | 30 | 10 | 22 | 500 | 374 | 2.05 |
| 30A4 | 30 | 10 | 22 | 400 | 332 | 1.59 |
| 30A5 | 30 | 10 | 18 | 1100 | 274 | 1.77 |
| 30A6 | 30 | 10 | 18 | 500 | 214 | 1.28 |
| 30A7 | 30 | 10 | 18 | 400 | 211 | 1.20 |
| 30B | 30 | 3 | 22 | 800 | 445 | 1.46 |
| 30N2 | 30A2 | 30B | |
|---|---|---|---|
| UTS Stress (MPa) | 3.8 ± 0.5 | 2.5 ± 0.2 | 1.2 ± 0.2 |
| Tensile strain (%) | 77.3 ± 12.7 | 18.8 ± 3.9 | 55.2 ± 13.7 |
| Young’s modulus (MPa) | 39.0 ± 0.2 | 33.9 ± 0.2 | 13.1 ± 0.1 |
| Sample | Drug Loading (%) |
|---|---|
| 30N2DL | 37.2 |
| 30A2DL | 27.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pouroutzidou, G.K.; Lazaridou, M.; Papoulia, C.; Tsamesidis, I.; Chrissafis, K.; Vourlias, G.; Paraskevopoulos, K.M.; Bikiaris, D.; Kontonasaki, E. Electrospun PLGA Membranes with Incorporated Moxifloxacin-Loaded Silica-Based Mesoporous Nanocarriers for Periodontal Regeneration. Nanomaterials 2022, 12, 850. https://doi.org/10.3390/nano12050850
Pouroutzidou GK, Lazaridou M, Papoulia C, Tsamesidis I, Chrissafis K, Vourlias G, Paraskevopoulos KM, Bikiaris D, Kontonasaki E. Electrospun PLGA Membranes with Incorporated Moxifloxacin-Loaded Silica-Based Mesoporous Nanocarriers for Periodontal Regeneration. Nanomaterials. 2022; 12(5):850. https://doi.org/10.3390/nano12050850
Chicago/Turabian StylePouroutzidou, Georgia K., Maria Lazaridou, Chrysanthi Papoulia, Ioannis Tsamesidis, Konstantinos Chrissafis, George Vourlias, Konstantinos M. Paraskevopoulos, Dimitrios Bikiaris, and Eleana Kontonasaki. 2022. "Electrospun PLGA Membranes with Incorporated Moxifloxacin-Loaded Silica-Based Mesoporous Nanocarriers for Periodontal Regeneration" Nanomaterials 12, no. 5: 850. https://doi.org/10.3390/nano12050850
APA StylePouroutzidou, G. K., Lazaridou, M., Papoulia, C., Tsamesidis, I., Chrissafis, K., Vourlias, G., Paraskevopoulos, K. M., Bikiaris, D., & Kontonasaki, E. (2022). Electrospun PLGA Membranes with Incorporated Moxifloxacin-Loaded Silica-Based Mesoporous Nanocarriers for Periodontal Regeneration. Nanomaterials, 12(5), 850. https://doi.org/10.3390/nano12050850