1. Introduction
Among all modern energy sources, supercapacitors demonstrate an extraordinary power density and an extremely long cycling life [
1]. Such rapid charging and discharging occur due to fast adsorption-electrostatic processes, making supercapacitor technology environmentally friendly. These advantages open up a wide range of possible applications from small devices [
2] to electric cars [
3]. However, the widespread use of supercapacitor technology is limited by their relatively low energy density [
4]. The implementation of nanoporous electrodes led to a severe increase in energy density through a significant increase in capacitance, first experimentally observed in sub-nanoporous carbon materials [
5]. Moreover, subsequent experiments showed that the pore size giving the highest capacitance corresponds to the diameter of the electrolyte molecules [
6]. The capacitance’s oscillatory behavior as a function of pore size has been successfully described in terms of the classical density functional theory (c-DFT) [
7], accounting for confined properties of the charged hard spheres at an applied electrostatic potential. The state of the art c-DFT approach [
8] allows one to investigate how the supercapacitor parameters, namely electrodes’ pore sizes [
9,
10] and electrolyte composition [
11,
12,
13], affect energy storage performance.
In spite of the long history of [
14,
15,
16] studies of porous electrode charging, the vast majority of existing dynamic models correspond to meso- and macropores (pore size
), where the properties of confined dilute electrolytes are similar to the bulk ones. In this case, the charging time is often estimated using the transmission line model (TLM), first proposed and thoroughly studied in Refs. [
14,
15,
16]. The TLM treats the electrolyte-filled pore as an equivalent circuit of resistors and capacitors. The corresponding total resistance
R and capacitance
C take into account the properties of the electrolyte in the bulk and double electric layer (EDL) states, respectively. More than half a century ago, de Levy proposed a diffusion equation for the electrostatic potential difference between the pore surface and the central plane, which leads to a time scale
[
14,
15]. Significantly, the effective circuit is used nowadays to investigate the various properties of the electrolyte charging [
17,
18,
19]. Biesheuvel and Bazant extended transmission line approach considering pure capacitive process Ref. [
20] and Faradaic reaction Refs. [
21,
22] in porous electrodes. For low applied potentials
model [
20] results in the TLM, which is the linearization of Poisson–Nernst–Planck (PNP) equations [
23] at this limit. Because of the assumption of low applied potential, TLM misses some critical phenomena [
24] at conditions corresponding to the majority of applications
–
. For example, the nonlinear model [
20] predicts the slower charging at higher potentials, which is crucial for the power performance. However, the PNP calculations [
24] showed that this predicted charging slowdown is overestimated due to the unaccounted surface conductivity. Thus, an accurate description of the electrolyte–electrode interface is crucial for adequately modeling nanopore charging at moderate/high applied potentials.
Very recently, the TLM approach has been applied to nanoporous electrodes (
H ∼ 2 nm), and realistic scaling of the charging time has been obtained in terms of physically determined parameters [
19]. The authors of ref. [
19] have also shown that two comparable timescales exist at high applied potentials: the first one corresponds to the equivalent circuit model, and the second timescale is related to adsorption. A more detailed analysis of the charge dynamics in nanopores requires information on the packing properties of finite-sized ions, which goes beyond the [
19] TLM models. The most realistic description of ion transport in nanopores was obtained using the molecular dynamics (MD) method. Molecular dynamics (MD) simulations [
25,
26] demonstrated that the charging of the electrolyte filled pores is described by three consecutive regimes (modes). More precisely, the charging starts with ions diffusion characterized by the slow squared-root dependence of the total charge on time. Further, ion transport corresponds to two exponential regimes at the intermediate and late times. The second exponential regime with a higher relaxation time becomes crucial in ultra-narrow pores, which results in a significant slowdown of the charging. The authors of ref. [
27] demonstrated how to speed up the nanopores charging using time-dependent applied potential. Such control of the applied potential allows the ions to avoid the molecular clogging which causes the sluggish dynamics. Therefore, modeling the ions’ transport inside nanopores is critical to designing the supercapacitors with the optimal relation between storage and power properties.
In addition, an actual application of the ion transport models is capacitive deionization technologies which use an electrostatic field to remove salt ions and provide the water desalination during the filtration through porous electrodes [
28]. Additionally, the recent experiments [
29] demonstrated that the salt ions could be excluded from the confined water because of the steric effects of the nano-scale slit pores. Thus, it is crucial to combine the electrostatic and steric properties to model the ion transport inside the nanopores properly.
This paper describes the ion dynamics inside a nanopore as wide as a molecule. Such sub-nanoporous electrodes are necessary for further advances in supercapacitor-based technologies. However, the sub-nanopore size limitation causes a significant slowing down of ion motion and, consequently, the charge. We develop a theory based on the time-dependent version of the c-DFT approach (see refs. [
30,
31] for one of the first derivations, and ref. [
32] for the comprehensive literature review). More precisely, we derive and numerically solve the transport equations for the charged ions in the three-dimensional narrow slit pores. Such pores exhibit negligibly small values of the width-to-length ratio, which allows us to derive an asymptotic model—a one-dimensional transport equation reflecting the properties of the ion dynamics inside 3D slit nanopores. The proposed theory describes three consecutive charging regimes: an initial root-square process and two exponential regimes with different time scales. The last exponential regime plays a crucial role in the ultra-narrow pores (pore width is around ion diameter) and results in the charging slowdown. Before the current work, the details of the charging inside ultra-narrow pores had been investigated using MD simulations only [
25,
26,
27]. Indeed, since we use an accurate thermodynamic potential, our model accounts for the electric and packaging properties of finite-sized ions within the nanopores. It is an advantage over recently published dynamic models [
18,
19], and allows us to relate charging times to the given characteristics of nanopore supercapacitors.
2. Model
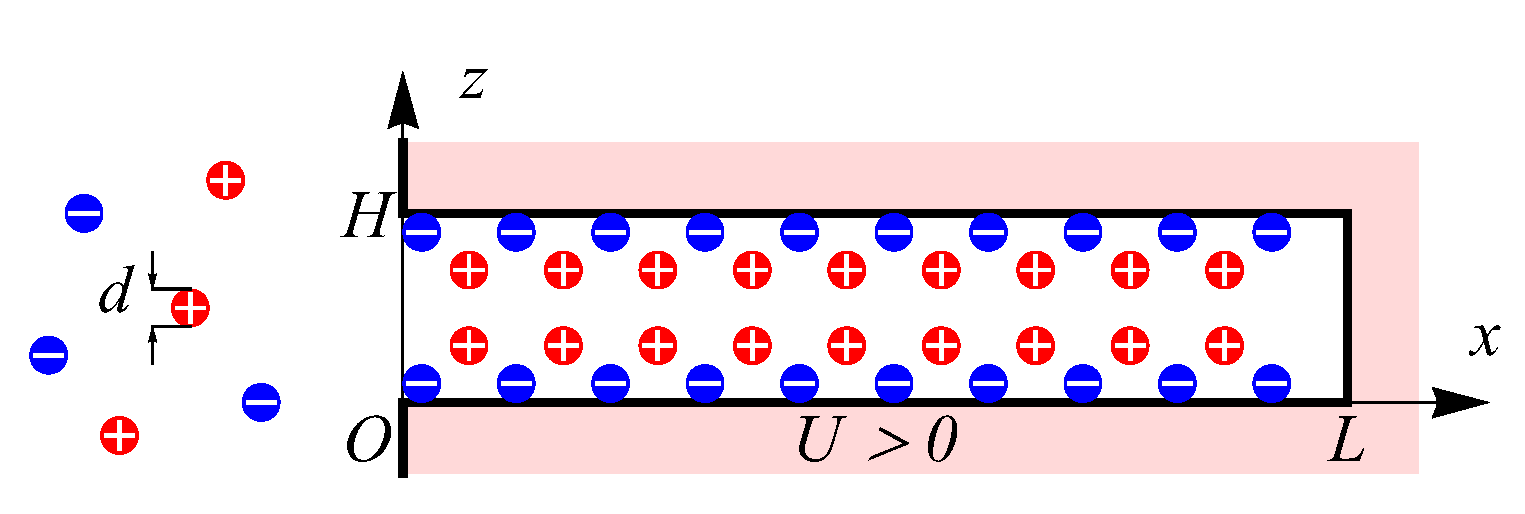
We use the slit pore geometry shown in
Figure 1, which describes the spatial structure of modern porous materials, such as graphene based electrodes [
33,
34] and graphene oxide (GO)/MXene fibers [
35,
36], exhibiting aligned slit-nanopores of width
H comparable to electrolyte diameter
d and length
. The considered materials are used in supercapacitor’s technologies as the porous electrodes which accumulate charge through electric double layer formation without chemical reactions. More precisely, these materials show promising applications for wearable electronics and smart textiles devices due to outstanding energy density and high flexibility [
37]. Moreover, our geometrical representation of the pore closely resembles recent molecular dynamic simulations of the supercapacitors [
26,
27], which allows us to make a comparison of our predictions with the MD-simulated electrolyte’s behavior. The electrodes with inhomogeneous pore-network considered in refs. [
21,
38,
39] are beyond the scope of our work. However, the developed model of individual slit nanopore charging can be implemented as a pore-network building block in the case of a non-trivial pore-size distribution [
39]. A possible implementation follows from ref. [
39], where our transport equations can replace the Navier–Stokes equation for the effective medium approximation—the porous reservoir is a uniform network of equal short pores.
In the case of slit pore geometry (
Figure 1), the dynamic component density
for
(where
n is the number of components) depends on the coordinate
x in lateral direction along the pore’s surface and the normal distance to the surface
z. Notice, that we assume the homogeneous distribution in the direction which is perpendicular to the
x- and
z-coordinates, see
Figure 1. Therefore, to describe dynamics of the charging process, we apply the time-dependent version of c-DFT [
40] in two spatial coordinates
, defined in the general form as
where
is the diffusion coefficient of
i-th component,
,
is the Boltzmann constant,
T is the temperature,
is 2D gradient vector, and
is the local chemical potential of the
i-th component.
To describe the confined electrolyte we use one of the most popular c-DFT approaches based on the confined hard sphere model [
41] and an electrostatic extension [
42] accounting for the contributions from Coulomb interaction and additional finite size residual correlations. The electrode’s pores are considered as an open system connected with a bulk electrolyte. In accordance with Ref. [
40], the chemical potentials
have the following form
where
is the electric potential,
e is the proton charge,
is the ion valency,
is the de Broglie wavelength. The electric potential
satisfies the Poisson equation
where
is the 2D Laplace operator and
is the Bjerrum length. A detailed description of the used c-DFT approach including particular form of the excess term
can be found in
Appendix D. Additionally, see in
Appendix D the static the capacitance’s properties from implemented c-DFT which correspond to model [
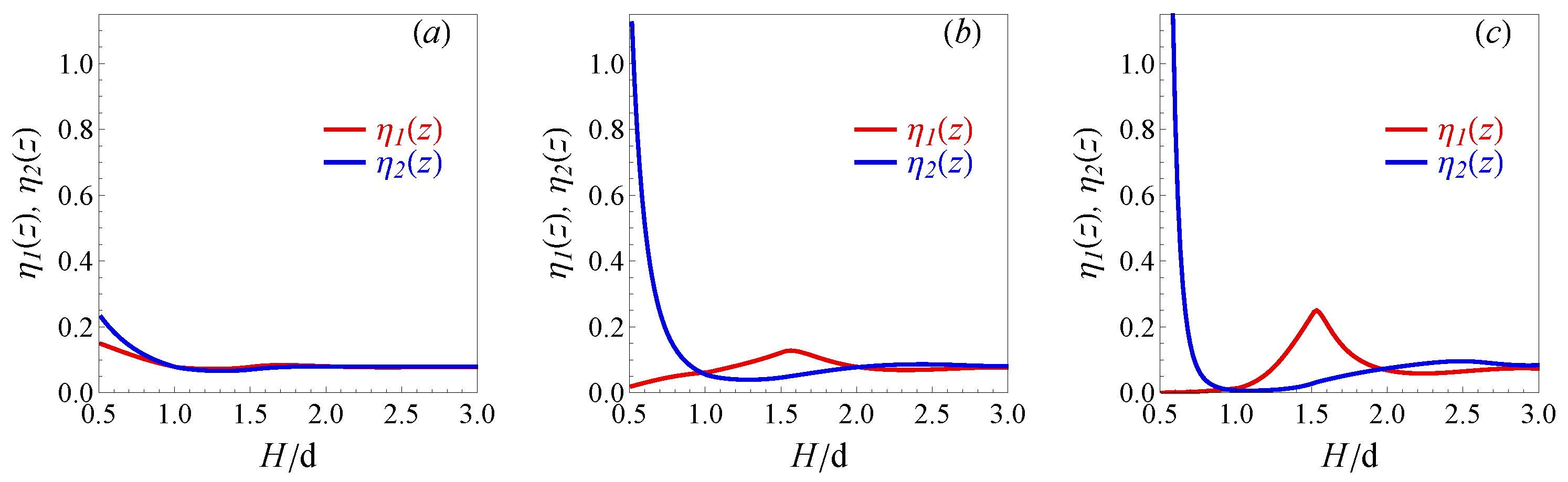
9] and show the realistic behavior.
Initially, no electrostatic potentials are applied, and the total charge of the symmetric electrolyte is zero. We consider a step charge when the potential changes abruptly from zero to some positive value
. The applied potential causes the influx into the pores of oppositely charged ions called counter-ions (anions in the case of
), while ions with a charge of the same sign as the applied potential (co-ions, cations for
) are pushed out of the pore volume. Inside sub-nanopores, the local increase in the counter-ion density can strongly influence the co-ion desorption, hindering their release due to counter-ion clogging and thus leading to a notable slowdown of the charging process [
26]. The main variables of interest are the charges associated with individual electrolyte components
and the surface total charge density
. The final charge
corresponds to the steady distributions
, which are in equilibrium with the bulk electrolyte and depend on the applied potential
U.
Considering Equations (
1) and (
3) in the slit-pore geometry (see (
A5) and (
A6)) with very small ratio
, we seek the solutions of Equation (
1) in the following forms:
where upper indices correspond to the
and
problems. As one can see in
Appendix A, we use the
equations to find that
do not depend on the
z-coordinate; and electrostatic potential
can be found from the
-Poisson equation. Therefore, the
-problem allows us to describe the
z-coordinate dependence, while
provides information about dynamics and
x-coordinate distribution. One of our most striking results is the demonstration that a complete solution to the problem
is not required. Indeed, we derive an asymptotic model describing charging dynamics in terms of the pore cross-section averaged quantities for the
-variables
and
, as follows (see details in
Appendix A):
where,
are pore cross-section-averaged densities, and
are functions of averaged densities only, see
Appendix B. We close the 1D transport equation by the following initial and boundary conditions:
where Equations (
7) and (
8) correspond to the open and closed boundaries, respectively; Equation (
9) is the initial condition accounting for the confined electrolyte properties at
. Such partial differential Equation (
6) with (
7)–(
9) can be solved numerically using c-DFT as discussed in
Appendix C.
Notice, that here and below we use
,
and
the dominant terms in the expansions (
4) and (
5) over small parameter
. The chemical potentials
are defined by the popular confined fluid c-DFT model, see
Appendix D. It allows us to account for the realistic behavior of the electrolytes in confinement: alteration of over-screening/crowding interfaces [
43]; the capacity oscillations in narrow nanopores [
6]. Such subnanoscale properties are beyond the scope of the PNP equations [
24], linear TLM models [
23] or their very recent modifications [
17,
19]. Thus, since the derived transport equation correctly describes the ions-packing properties and the electrostatic fluctuations, we can extend the theoretical study to the ultra-narrow pores comparable to molecular size.
3. Results
Similar to some molecular simulations [
25,
26], we assume that electrolyte is a symmetric two component mixture consisting of the molecules with the diameters
and the charge valences
. For the sake of simplicity, we set the diffusion coefficients of the components to be equal
. In the case of this symmetric electrolyte, it is also useful to introduce, following [
9], the dimensionless variables
,
,
,
. It can be shown from dimensional arguments that the characteristic time for the considered problem is
and one can also introduce the scaled time
.
The charges of electrolyte components are conveniently defined in terms of average densities as
. An example of the calculated time-dependent charges
for subnanopore electrodes (
and
) at sufficiently high potential
is shown in
Figure 2a,b. A discussion of the used numerical methods can be found in
Appendix C. As one can see from
Figure 2a, the root-square law
describes the notable part of the charging process at early times well. However, when the charge
Q approaches the saturated value
, the trend changes to the exponential one.
Figure 2b shows that in the case of larger pores with
, the charging up to almost 95% is described by the following equation:
Expression (
10) is the leading term of the analytical solution of the TLM [
24]. Despite that the pores sizes
and potentials
are significantly beyond the ranges of TLM applicability, the published computer simulations [
25,
44] show the adequacy of exponential trend in Equation (
10) for fitting of the charge dependency on time. We observed that at late times the calculated profiles
in
Figure 2b follow another exponential trend succeeding (
10), which is notably slower and describes the charging until almost full saturation. The experimentally observed charging time for supercapacitors is around 10
3 s, while the published dynamic DFT models result in enormously underestimated values ∼10
−9 s [
19]. The proposed model provides both the realistic behavior of the capacitance’s properties (
Appendix D) and correct charging time scales. To provide numerical estimations we have used experimental parameters from refs. [
19,
45]: the pore length is
L = 0.5 mm and confined diffusion coefficient
D = 2 × 10
−10 m
2s
−1. The full charging times estimations in the physical units for the pores
and
shown in
Figure 2b correspond to around 700 s and 400 s, respectively, which agrees with the experimental characteristics.
Calculations for a wide range of system parameters show that the full charging (say 95% for the sake of concreteness) in pores with
is described by the first exponential trend in Equation (
10). For this reason, we use Equation (
10) to fit the charging time
from the calculated profiles
.
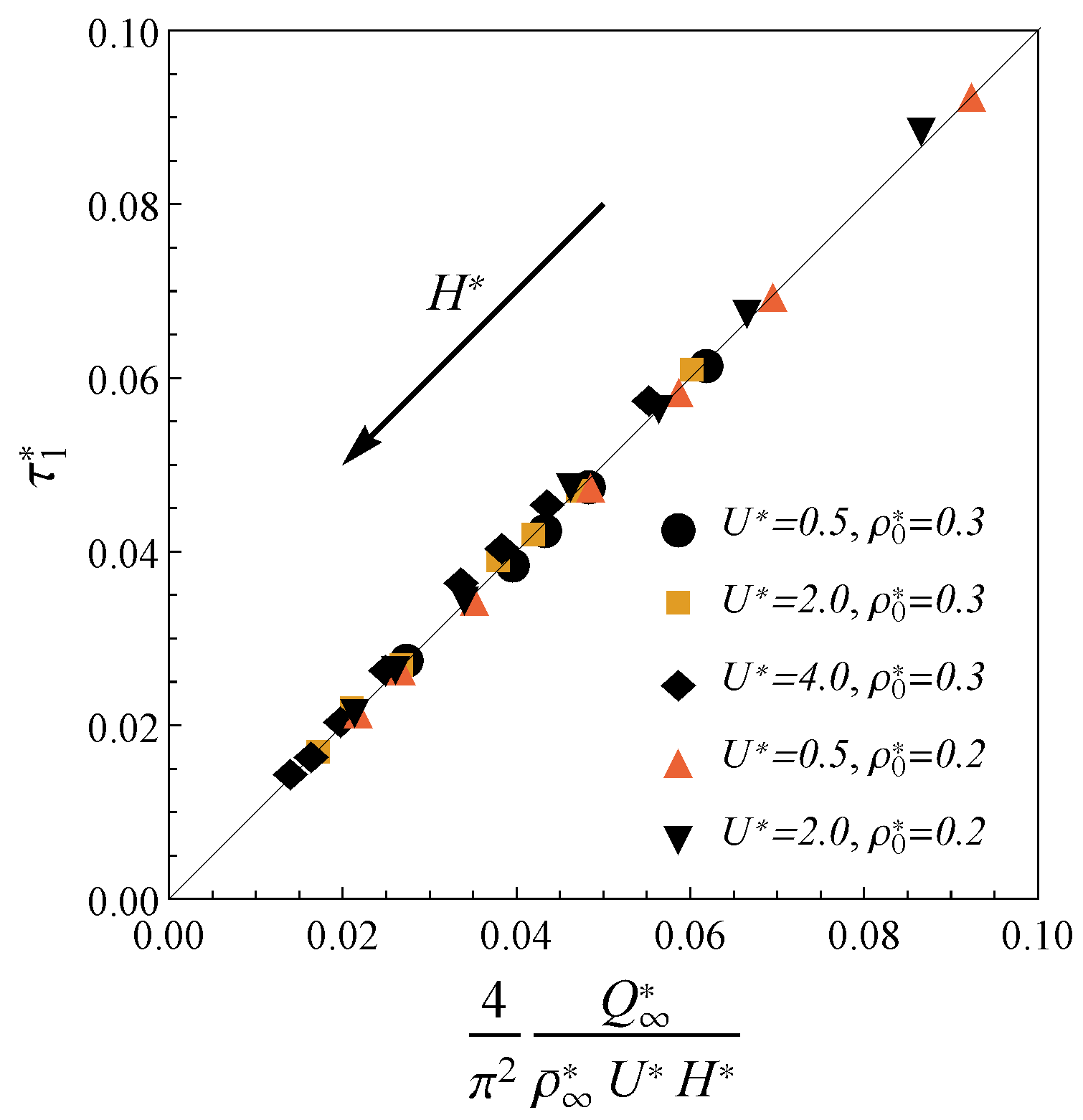
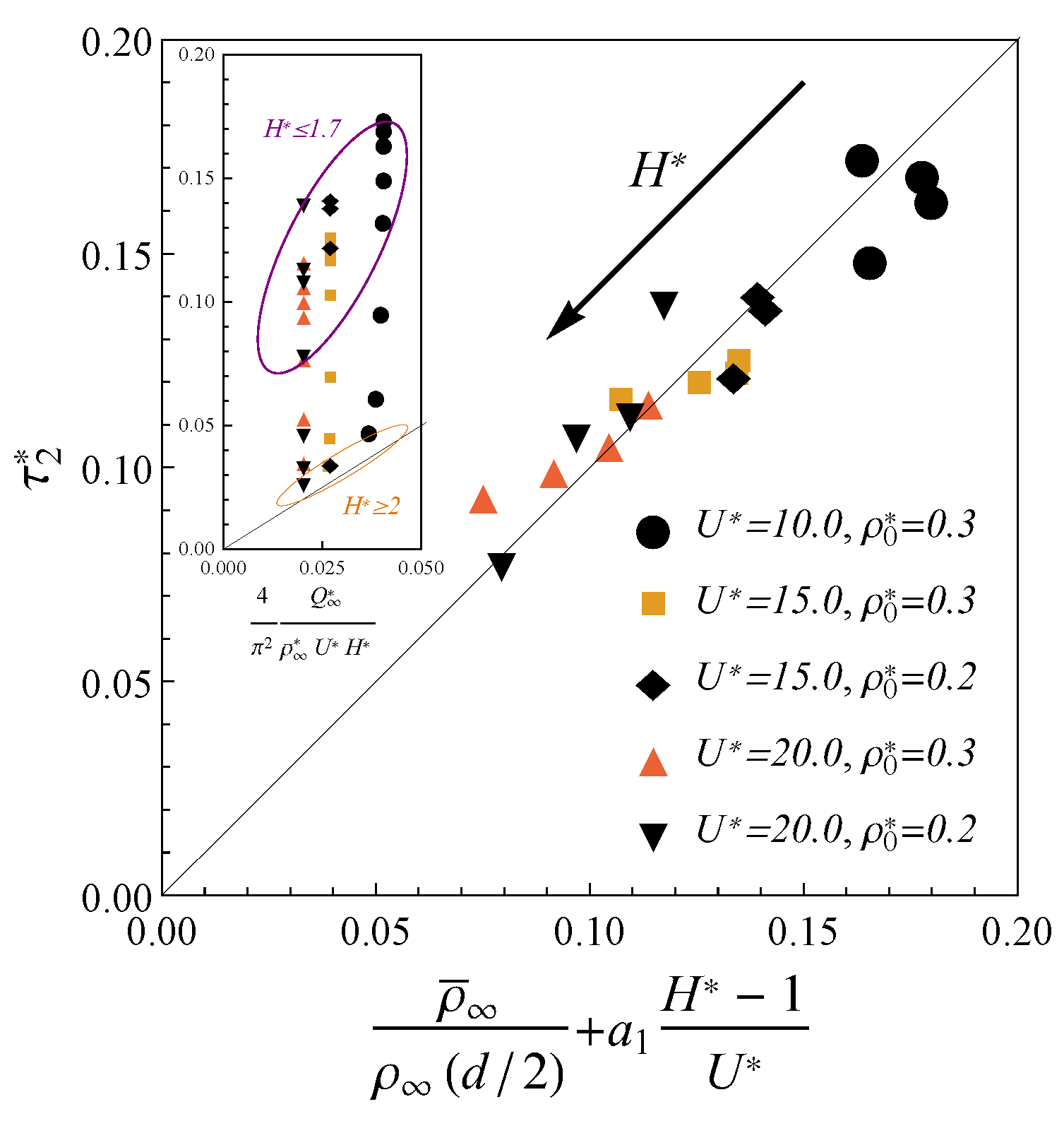
Figure 3 demonstrates that the charging time for the electrodes of width
and various electrolyte bulk densities
and applied potentials
can be explicitly expressed in terms of macroscopic parameters as
Here,
is the total final density; the coefficient
is obtained from the comparison with the TLM. The potentials considered here (up to 0.1 V in dimensional terms) correspond to the lower range of the values used in the modern experiments [
45,
46,
47], but are significantly beyond the formal applicability range of the TLM [
24]. To compare result Equation (
11) with TLM relaxation time we consider the limit of large pores (
) at extremely low potentials (
). Unlike nano-confinement, in this limit, the total density is almost constant
, and the charge can be calculated from the linearization of the Gouy–Chapman theory
, where
is the Debye length. Inserting these approximations in Equation (
11), we obtain
, where
is the ratio of pore cross-sectional area to perimeter. Expression
fits the TLM relaxation time from ref. [
24]. Significantly, such assumptions are not relevant to nanopores charging due to the properties of the confined electrolyte. For example, the total charge
Q becomes an oscillating function of
H, average densities strongly depend on the ion sizes, and electrostatic correlations play a crucial role. Thus, our result Equation (
11) generalizes the TLM predictions for the case of the charging in nanopores at moderate potentials. Thus, our approach is more general than standard TLM since it describes the charging at realistic nanopores conditions.
In addition, we have not met the derived expression (
11) in the literature. However, we observed an interesting connection between expression (
11) and the very recent result for the charging time published in ref. [
44]. The authors of ref. [
44] used the effective circuit to obtain RC-time of the charging inside nanoporous Metal Organic Frameworks electrodes. The application of this adopted TLM approach from ref. [
44] to slit pores gives the following expression for relaxation time
where using the notation adopted in ref. [
44] the areal capacitance can be expressed as
; the ionic conductivity inside the pore can be expressed in terms of the electrical mobility of ions
and ions density
as
. Inserting these electrolyte characteristics into Equation (
A51) one can obtain that
, see details in
Appendix E. Therefore, Equation (
11) is equivalent to one from the nanopores RC-circuit developed in ref. [
44]. The advantage of our approach over the adopted TLM is self contained form provided by c-DFT’s calculations without highly cost simulations.
The three consecutive dynamic regimes predicted in our work describe the results of molecular dynamics simulations published in refs. [
25,
26,
27]. Similarly with ref. [
25,
26] we observed a stark contrast between the charging dynamics inside ultra-narrow
and wider
pores shown in
Figure 2, for comparison, see
Figure 2 and
Figure 3 in ref. [
26] and ref. [
25], respectively. In
Figure 2b the first exponential regime (
10) for
covers the charging up to only
. Therefore, the significant part of the full charging inside ultra-narrow pores (
) is defined by the slower second exponential trend: the charge to
is ∼2 times slower than in the case of wider pores
.
To investigate this effect numerically, we described the charge profiles near the saturation
in terms of another exponential trend
, where
and
are the fitting parameters. The results for the corresponding relaxation time
are shown in the inset of
Figure 4. As one can see from this inset, expression (
11) fits the charging time in larger pores (
). However, we observed that in the case of the ultra-narrow pores
, charging slow down becomes notable. We observed that the charging slowdown starts as the pore width becomes less than two molecular diameters (
). In such confinement, the electrolyte behavior near the wall crucially influence on the ions distribution inside whole pore. Accordingly, it is reasonable to correlate the charging inside narrow pores not only with the average inner density
but the wall-contact density as well. As one can see from
Figure 4, the charging time in ultra-narrow pores (
) at high potential can be fitted to the following expression
where
is the wall-contact density at the final state of complete charging and
is the fitting parameter. The inverse dependence of charging time (
13) on the potential
U contributes as the pore’s width increases. This behavior is consistent with the high potential limit (
) of expression (
11), which fits the data well around
.
To discuss the slowdown dynamics inside ultra-narrow pores we considered the dynamics of the co-ions/counter-ions separately. We observed that the contributions to total charge from the co-ions
and counter-ions
shown in
Figure 2c exhibit significantly different relaxation times. More precisely, the counter-ions adsorption proceed more rapidly than slow outflux of the co-ions. Therefore, at the late times the counter-ions contribution is almost equal to the final charge
, while the co-ions’ slow-release decrease the total charge
. The possible explanation of this phenomenon is that the co-ions are trapped in the crowded counter-ion phase. Such behavior of the ions in nanopores was observed in MD simulations of symmetric electrolyte [
26,
27]. To demonstrate that expression (
13) is in agreement with this explanation, we consider the ultra-narrow pores
. Then, the second term in Equation (
13) is negligible and the counter-ion density dominates
. Therefore, the relaxation time (
13) can be written as
. At the late times, such dynamics is equivalent to a diffusion process of co-ions with the effective diffusion coefficient
, while the counter ions component is already in equilibrium. This coefficient
is defined by the density and structure of the counter-ion component distribution, that confirms the idea that the the abrupt counter-ions adsorption induces slow diffusion of the co-ions.
This paper proved the concept of
-expansion in application to the nanoporous charging compared with the experiment and simulations results. This approach could be helpful in applications beyond the charging time calculations. For example, we recently became aware of paper [
48], where the authors use the same asymptotic approach to calculate the impedance response in slit pores. In the future, the developed model could be perspective in the following research directions.
(I): We considered only the popular version of the Constant Potential Method (CPM)—the charging starts when the surface potential suddenly steps from zero to some value. However, the CPM mode does not cover charging at a constant electric current widely used in Galvanostatic Charge–Discharge (GCD) measurements. Additionally, recent MD simulations in ref. [
27] show that the charging in nanopores can be accelerated applying time-dependent potential
, which allows one to avoid ion clogging near the orifice. First of all, to describe the GCD- and
-modes for the realistic supercapacitor’s setup, one needs to account for the dynamics of the neutral electrolyte stored in the finite bulk volume between two oppositely charged porous electrodes. Therefore, the proposed model should be improved by accounting for the bulk electrolyte, as recently done for the TLM in ref. [
17], and for PNP equations in ref. [
49]. Then, the time-dependent potential can be accounted for in boundary condition (8), assuming that
varies slowly then the diffusion time scale in
z-direction
. Possible realization of GCD mode follows from very recent ref. [
50]—one can use the boundary condition at
which indicates an applied current with the given period and amplitude (see Equation (7c) in [
50]). (II): Our model is applied to the porous electrodes with flat smooth inner surface. Recently we developed model for the electrolytes structure near the electrodes with rough surfaces ref. [
51]. Therefore, the proposed approach could be extended to the porous materials with rough surfaces accounting for the modified Poisson equation from ref. [
51]. (III): We considered purely electrostatic supercapacitors, also known as electric double-layer capacitors. However, our approach can be extended to the pseudocapacitors’ charging mechanism based on the Faradaic processes (redox reactions). For this case, the possible modifications are discussed in ref. [
21], where the authors extended the porous electrode theory from [
20], accounting for the Faradaic reaction rate. Notice that these fast reactions differ from the charging mechanism of Li-ion batteries, where the chemical reactions induce the changes in the electrode molecules making/breaking chemical bonds.




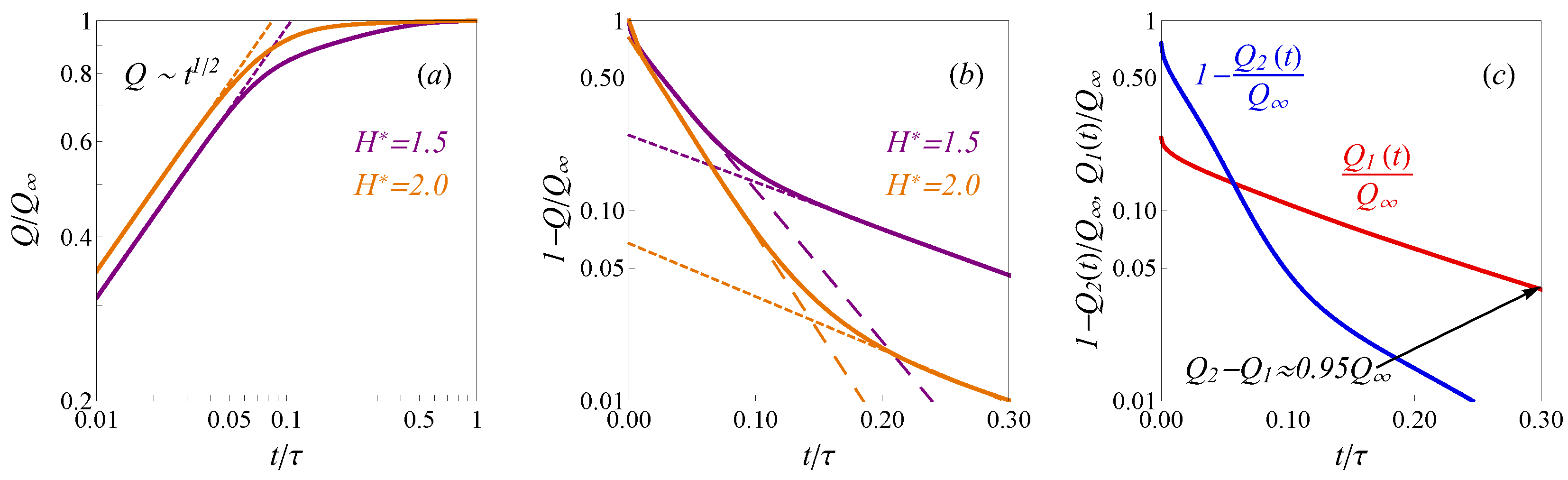
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}