
Spin-Orbit Coupling and Spin-Polarized Electronic Structures of Janus Vanadium-Dichalcogenide Monolayers: First-Principles Calculations



Abstract
:1. Introduction
2. Theoretical Methodology
3. Results and Discussion
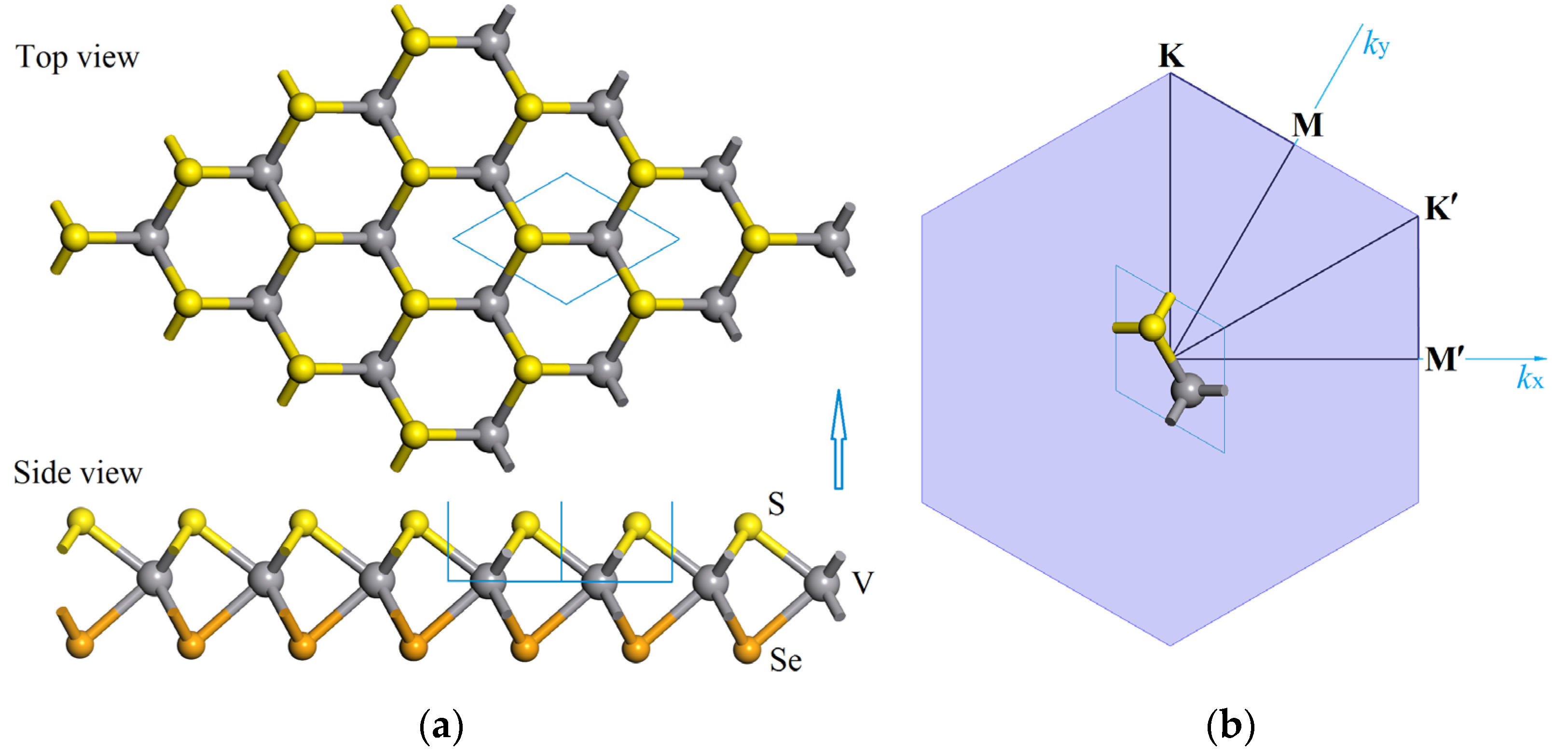
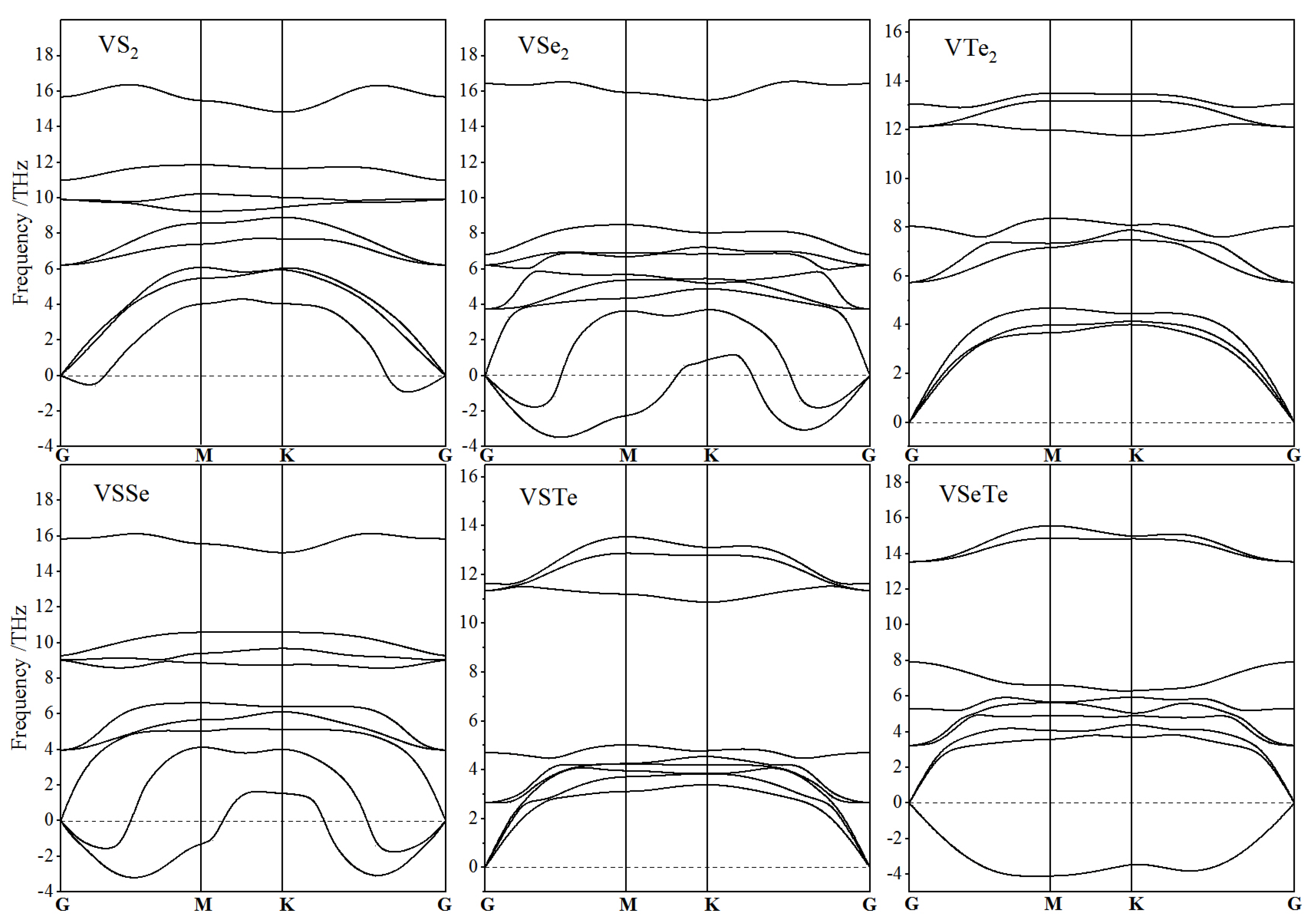
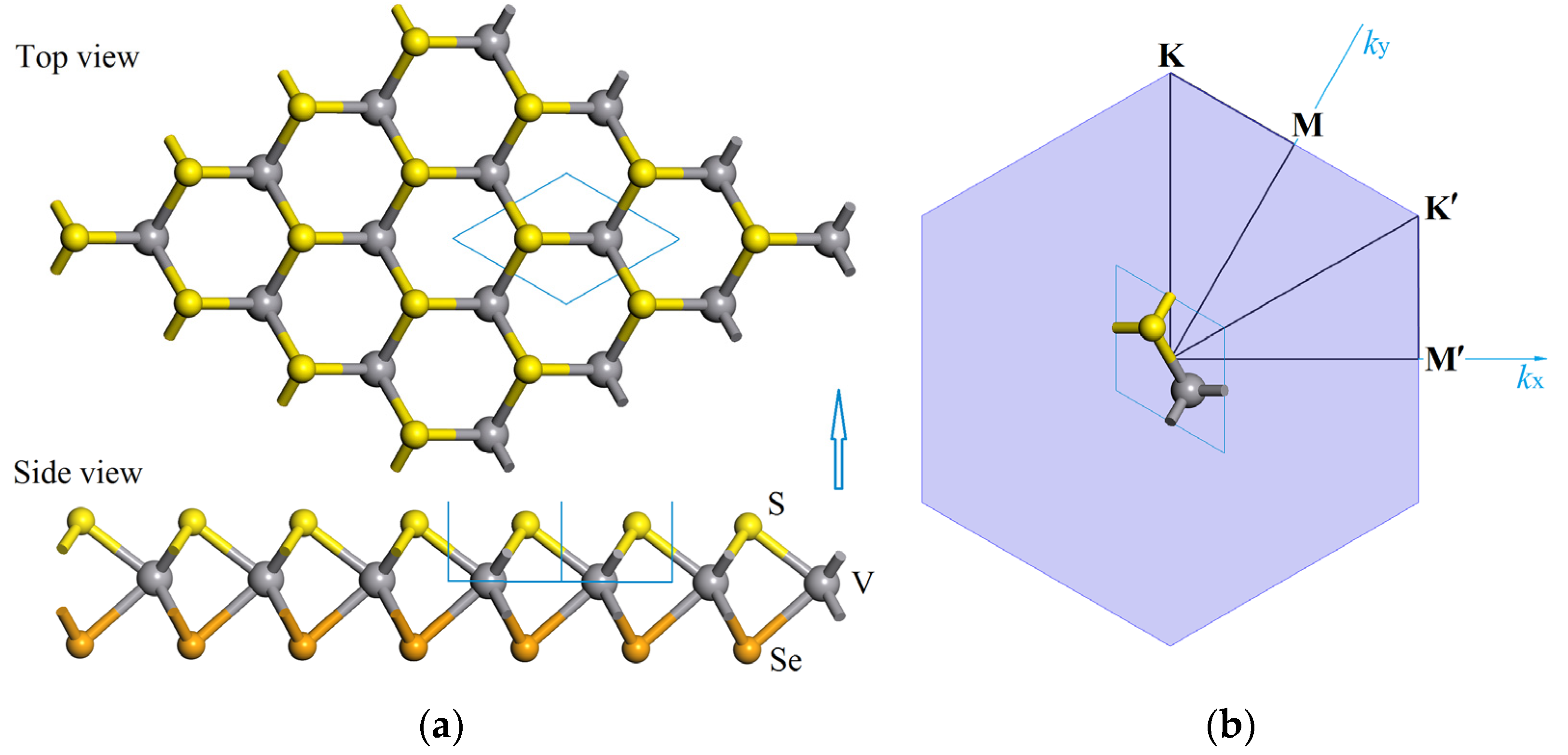
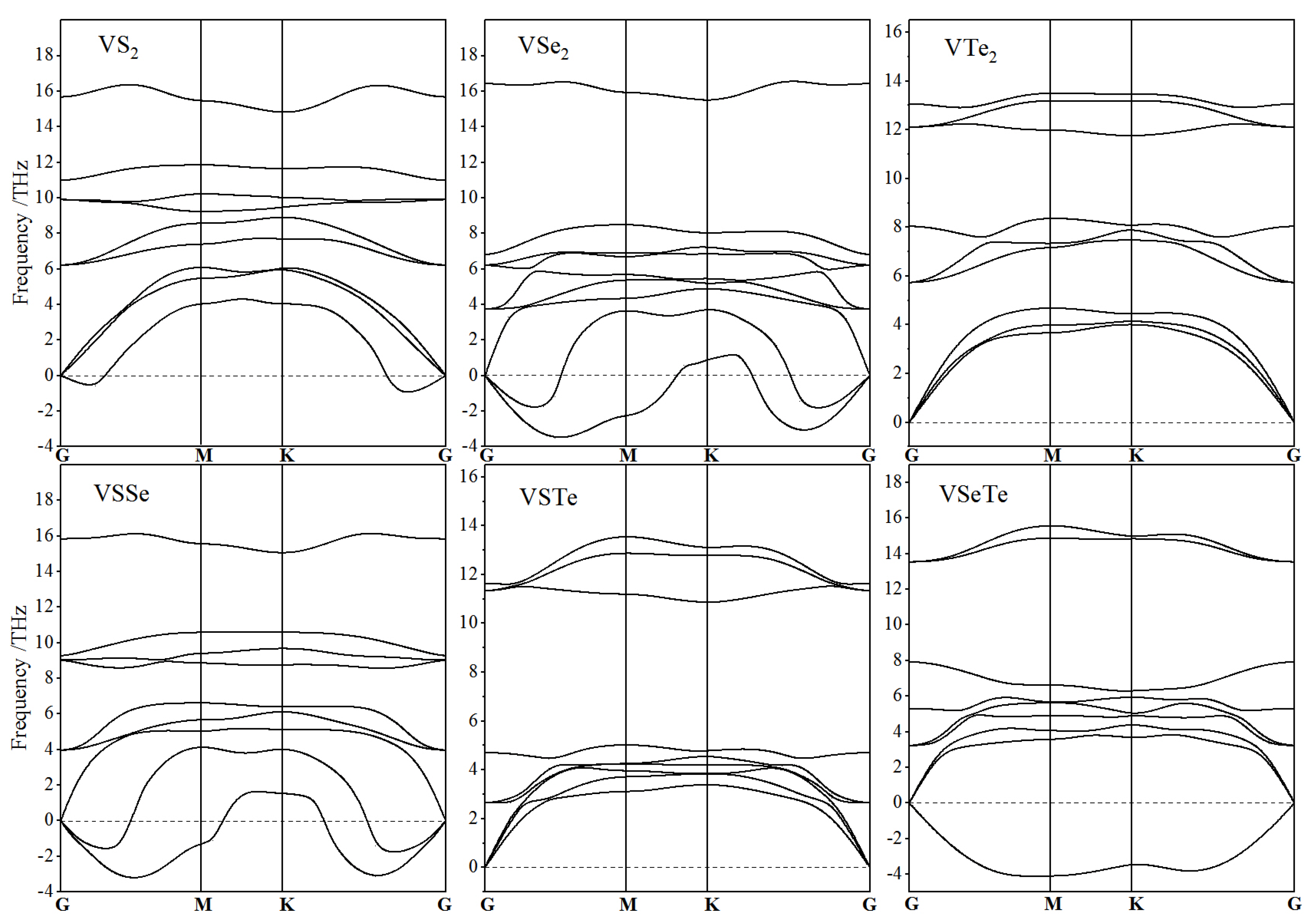
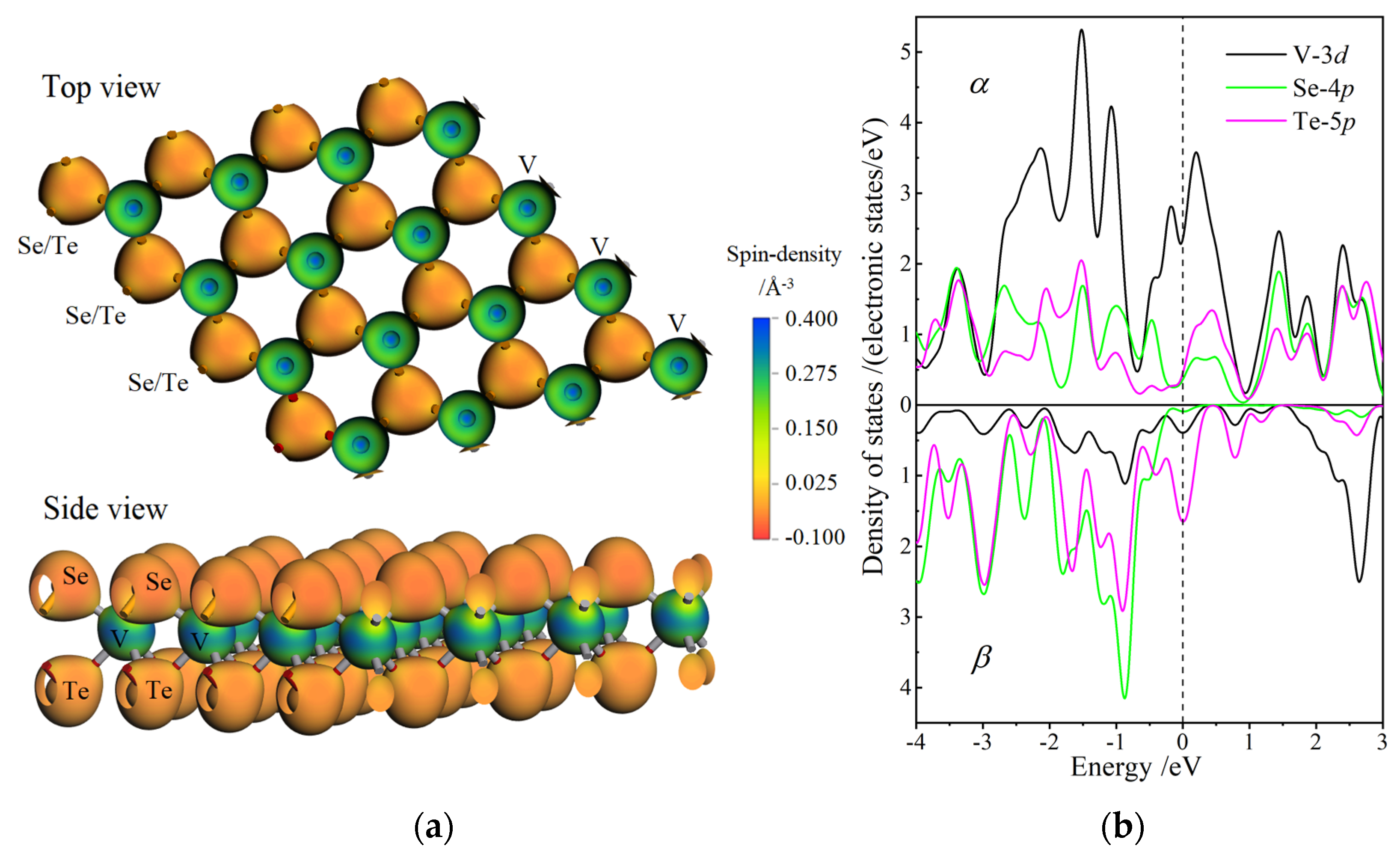
3.1. Atomic Structure and Stability
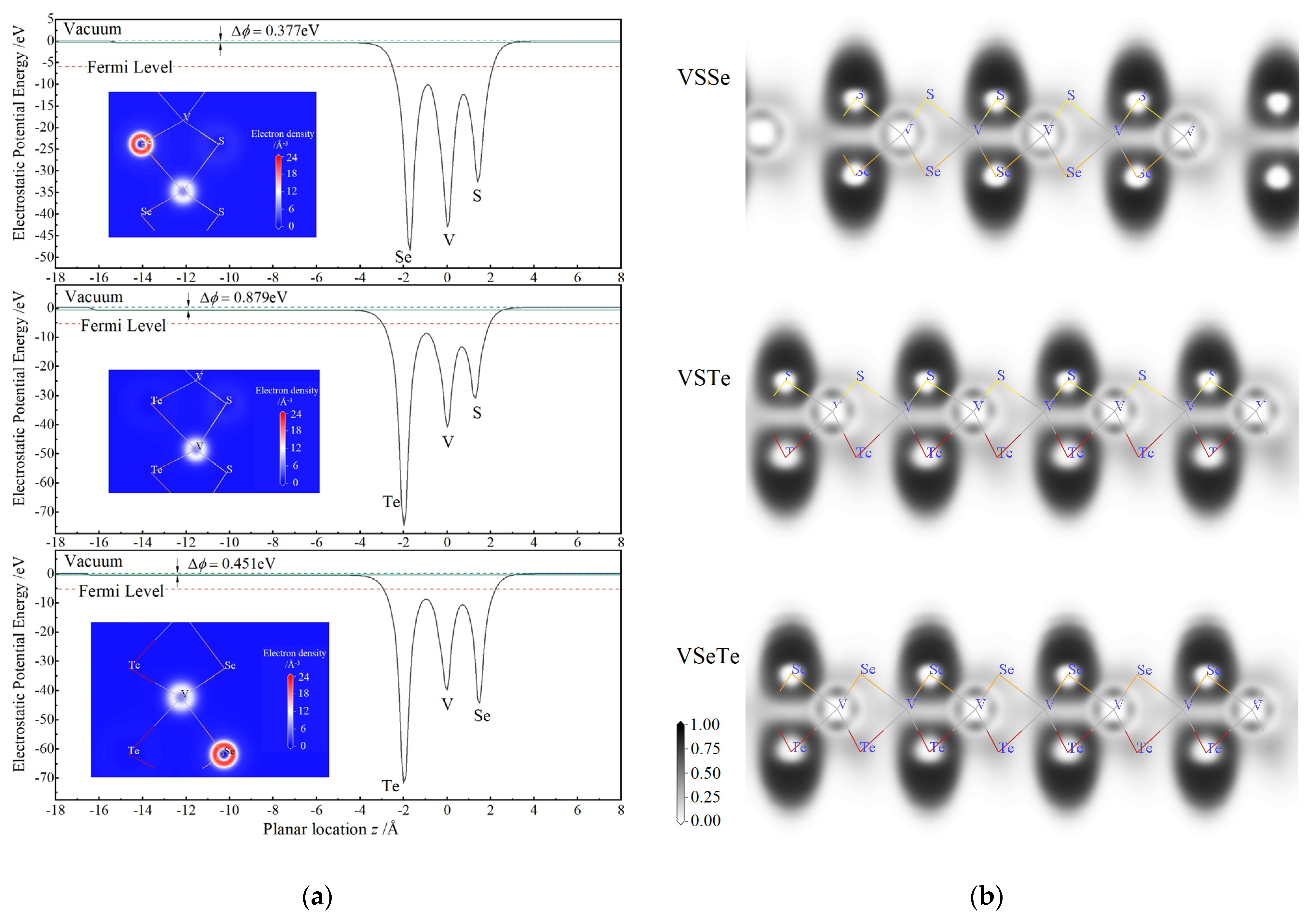
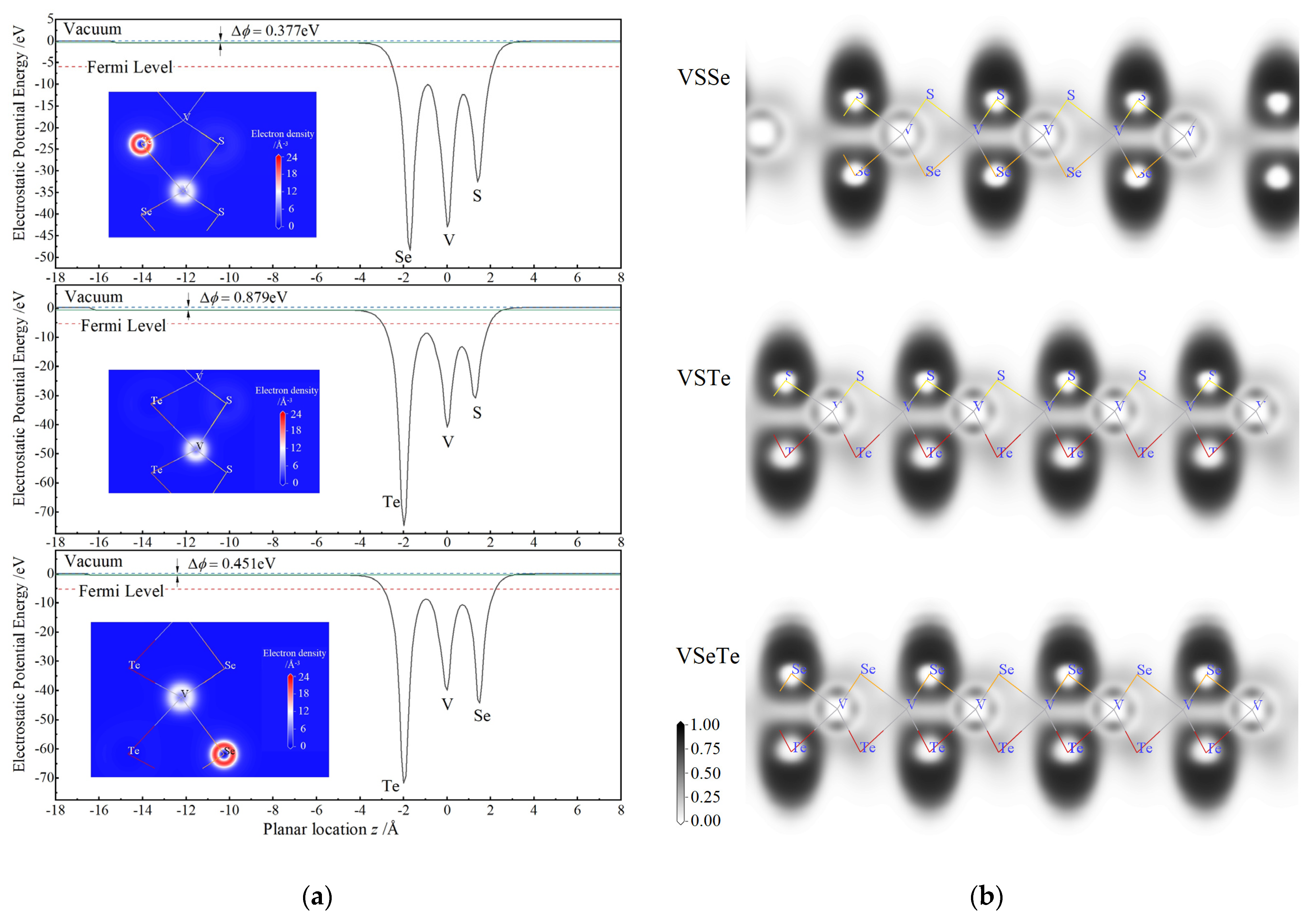
3.2. Internal Gradient of Electrostatic Potential
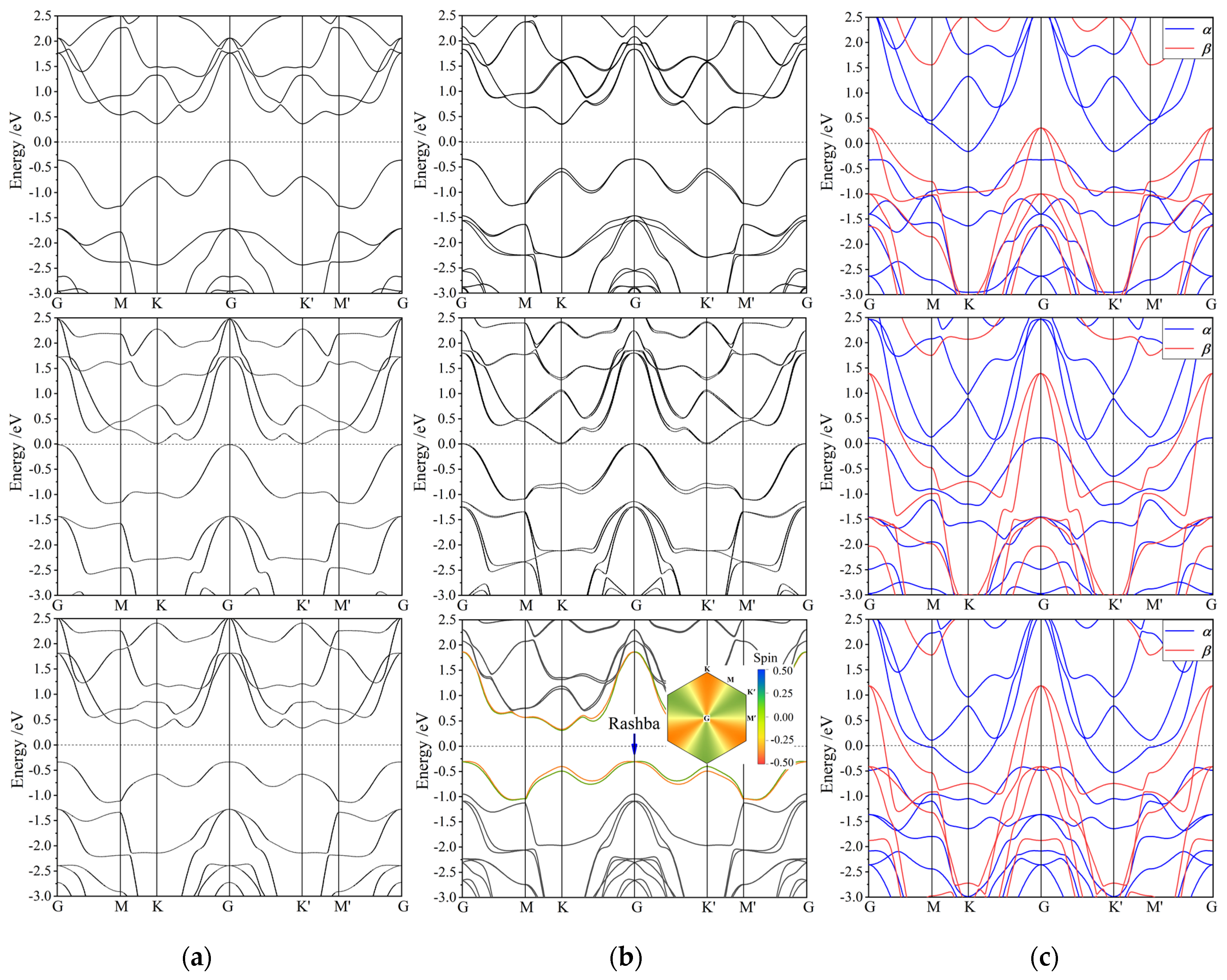
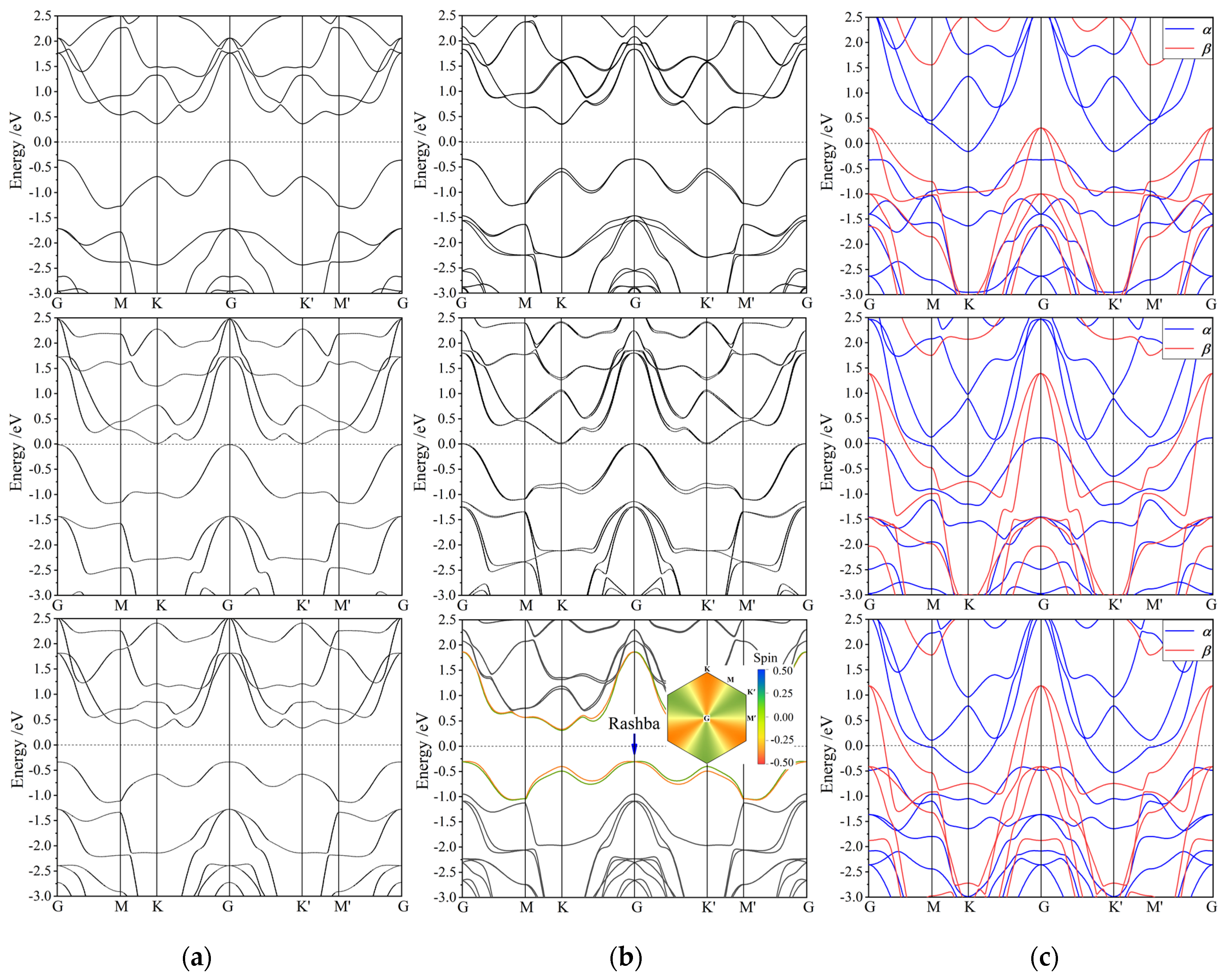
3.3. Band Structure
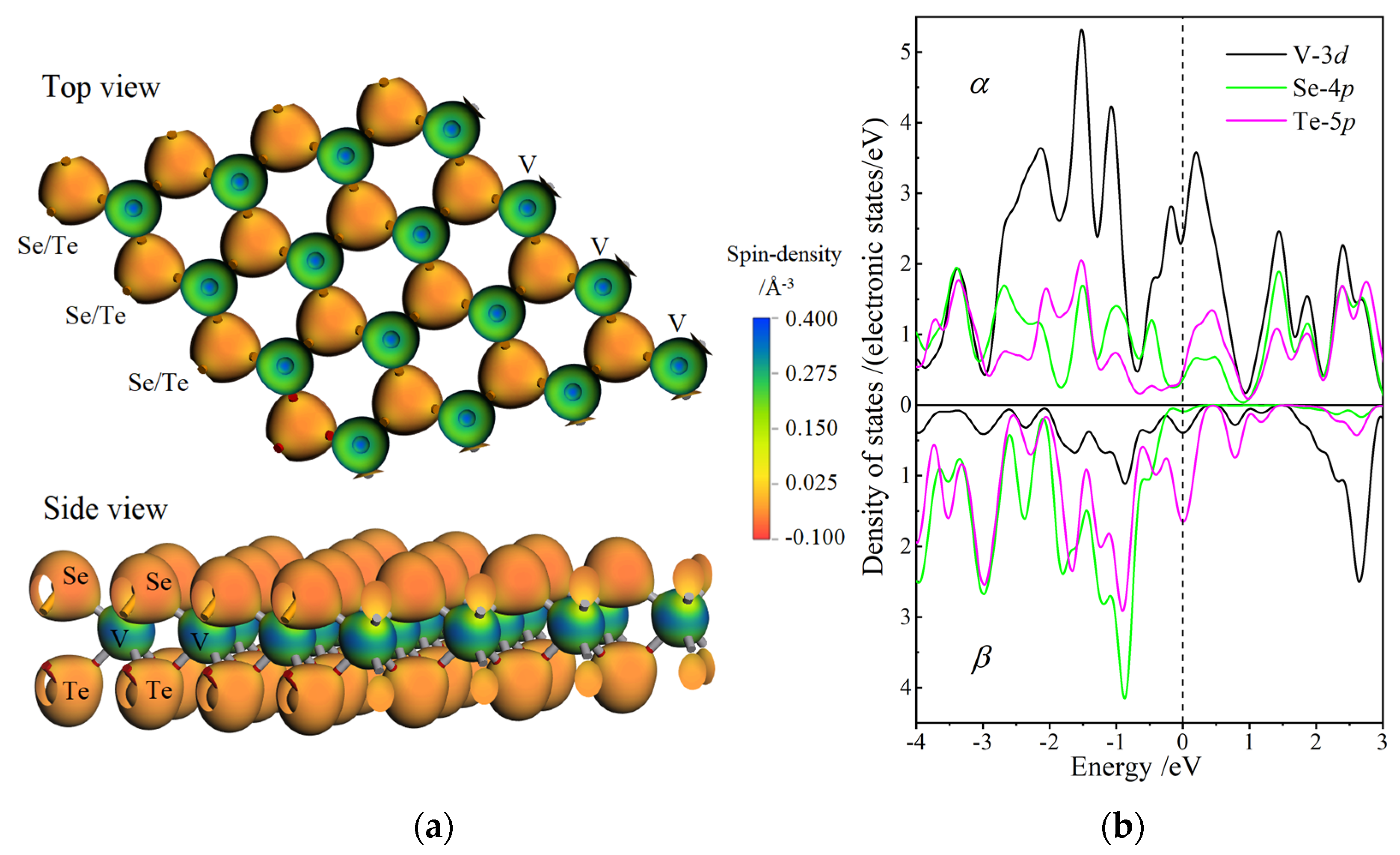
3.4. Magnetic Property
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Dai, Y.; Ma, Y.; Wei, W.; Yu, L.; Huang, B. Prediction of large-gap quantum spin hall insulator and Rashba-Dresselhaus effect in two-dimensional g-TlA (A = N, P, As, and Sb) monolayer films. Nano Res. 2015, 8, 2954–2962. [Google Scholar] [CrossRef]
- Chen, Y.L.; Analytis, J.G.; Chu, J.H.; Liu, Z.K.; Mo, S.K.; Qi, X.L.; Zhang, H.J.; Lu, D.H.; Dai, X.; Fang, Z.; et al. Experimental realization of a three-dimensional topological insulator, Bi2Te3. Science 2009, 325, 178–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- König, M.; Wiedmann, S.; Brüne, B.; Roth, A.; Buhmann, H.; Molenhamp, L.W.; Qi, X.L.; Zhang, S.C. Quantum spin Hall insulator state in HgTe quantum wells. Science 2007, 318, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandurin, D.A.; Tyurnina, A.V.; Yu, G.L.; Mishchenko, A.; Zólyomi, V.; Morozov, S.V.; Kumar, R.K.; Gorbachev, R.V.; Kudrynskyi, Z.R.; Pezzini, S. High Electron Mobility, Quantum Hall effect and anomalous optical response in atomically thin InSe. Nat. Nanotechnol. 2017, 12, 223–227. [Google Scholar] [CrossRef]
- Ma, Y.; Kou, L.; Du, A.; Heine, T. Group 14 element-based non-centrosymmetric quantum spin Hall insulators with large bulk gap. Nano Res. 2015, 8, 3412–3420. [Google Scholar] [CrossRef]
- Xu, Y.; Yan, B.; Zhang, H.J.; Wang, J.; Xu, G.; Tang, P.; Duan, W.; Zhang, S.C. Large-gap quantum spin hall insulators in thin films. Phys. Rev. Lett. 2013, 111, 136804. [Google Scholar] [CrossRef]
- Yang, B.; Molina, E.; Kim, J.; David, B.; Mark, L.; Liu, Y.; Xu, Y.; Wu, R.; Ludwig, B.; Kenji, W. Effect of distance on photoluminescence quenching and proximity-induced spin–orbit coupling in graphene/WSe2 heterostructures. Nano Lett. 2018, 18, 3580–3585. [Google Scholar] [CrossRef] [Green Version]
- Eda, G.; Maier, S.A. Two-dimensional crystals: Managing light for optoelectronics. ACS Nano 2013, 7, 5660–5665. [Google Scholar] [CrossRef]
- Ma, Y.; Kou, L.; Dai, Y.; Heine, T. Two-dimensional topological insulators in group-11 chalcogenide compounds: M2Te (M = Cu, Ag). Phys. Rev. B 2016, 93, 235451. [Google Scholar] [CrossRef]
- Mak, K.F.; Shan, J. Photonics and optoelectronics of 2D semiconductor transition metal dichalcogenides. Nat. Photon. 2016, 10, 216–226. [Google Scholar] [CrossRef]
- Zibouche, N.; Kuc, A.; Musfeldt, J.; Heine, T. Transition-metal dichalcogenides for spintronic applications. Ann. Der Phys. 2014, 526, 395–401. [Google Scholar] [CrossRef]
- Kuc, A.; Heine, T. The electronic structure calculations of two-dimensional transition-metal dichalcogenides in the presence of external electric and magnetic fields. Chem. Soc. Rev. 2015, 44, 2603–2614. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Liu, G.B.; Feng, W.X.; Xu, X.D.; Yao, W. Coupled spin and valley physics in monolayers of mos2 and other group-VI dichalcogenides. Phys. Rev. Lett. 2012, 108, 196802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, C.; Bihlmayer, G.; Zhang, H.; Wortmann, D.; Blügel, S.; Mokrousov, Y. Functionalized bismuth films: Giant gap quantum spin Hall and valley-polarizedquantum anomalous Hall states. Phys. Rev. B 2015, 91, 041303. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.C.; Zhu, Z.Y.; Tahir, M.; Schwingenschlögl, U. Spin-orbit–induced spin splittings in polar transition metal dichalcogenide monolayers. Europhys. Lett. 2013, 102, 57001. [Google Scholar] [CrossRef]
- Yao, Q.F.; Cai, J.; Tong, W.Y.; Gong, S.J.; Wang, J.Q.; Wan, X.G.; Duan, C.G.; Chu, J.H. Manipulation of the large Rashba spin splitting in polar two-dimensional transition-metal dichalcogenides. Phys. Rev. B 2017, 95, 165401. [Google Scholar] [CrossRef] [Green Version]
- Eremeev, S.V.; Papagno, M.; Grimaldi, I.; De Luca, O.; Ferrari, L.; Kundu, A.K.; Sheverdyaeva, P.M.; Moras, P.; Avvisati, G.; Crepaldi, A.; et al. Insight into the electronic structure of semiconducting ε−GaSe and ε−InSe. Phys. Rev. Mater. 2020, 4, 084603. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, W.; Huang, B.; Dai, Y. The mirror asymmetry induced nontrivial properties of polar WSSe/MoSSe heterostructures. J. Phys. Condens. Matter 2019, 31, 125003. [Google Scholar] [CrossRef]
- Lu, A.Y.; Zhu, H.; Xiao, J.; Chuu, C.P.; Han, Y.; Chiu, M.H.; Cheng, C.C.; Yang, C.W.; Wei, K.H.; Yang, Y.; et al. Janus monolayers of transition metal dichalcogenides. Nat. Nanotechnol. 2017, 12, 744–749. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jia, S.; Iskandar, K.; Dong, L.; Er, D.; Chen, W.; Guo, H.; Jin, Z.; Shenoy, V.B.; Shi, L.; et al. Janus monolayer transition-metal dichalcogenides. ACS Nano 2017, 11, 8192–8198. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.S.; Qteish, A.; Payne, M.C.; Heine, V. Optimized and transferable nonlocal separable ab initio pseudopotentials. Phys. Rev. B 1993, 47, 4174–4180. [Google Scholar] [CrossRef] [PubMed]
- Milman, V.; Lee, M.H.; Payne, M.C. Ground-state properties of CoSi2 determined by a total-energy pseudopotential method. Phys. Rev. B 1994, 49, 16300–16308. [Google Scholar] [CrossRef] [PubMed]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative minimization techniques for ab initio total energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations—A reply. Phys. Rev. B 1977, 16, 1748. [Google Scholar]
- Packwood, D.; Kermode, J.; Mones, L.; Bernstein, N.; Woolley, J.; Gould, N.; Ortner, C.; Csanyi, G. A universal preconditioner for simulating condensed phase materials. J. Chem. Phys. 2016, 144, 164109. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Cao, D.; Wang, J.; Liang, P.; Chen, X.; Shu, H. Semiconducting edges and flake-shape evolution of monolayer GaSe: Role of edge reconstructions. Nanoscale 2018, 10, 12133–12140. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, S.; Bai, Y.; Zhao, J. Enhanced piezoelectric effect in Janus group-III chalcogenide monolayers. Appl. Phys. Lett. 2017, 110, 163102. [Google Scholar] [CrossRef]
- Xu, X.D.; Sun, W.F. First-principles investigation of GaInSe2 monolayer as a janus material. J. Chin. Ceram. Soc. 2020, 48, 507–513. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Space Group | VXY Monolayer | a/Å | dVX and dVY/Å | dXY/Å | Ecoh/(eV/Unitcell) | qM/e | k/(eV/Unitcell) | ||
|---|---|---|---|---|---|---|---|---|---|
| V | X | Y | |||||||
| P6m2 | VS2 | 3.136 | 2.338 | 2.959 | 15.5893 | 1.86 | −0.91 | −0.91 | 167.6 |
| VSe2 | 3.295 | 2.480 | 3.183 | 14.1387 | 1.82 | −0.89 | −0.89 | 186.5 | |
| VTe2 | 3.551 | 2.691 | 3.488 | 11.7610 | −0.30 | 0.19 | 0.19 | 277.8 | |
| P3m1 | VSSe | 3.223 | 2.327, 2.490 | 3.052 | 14.9025 | 1.93 | −1.06 | −0.82 | 203.6 |
| VSTe | 3.356 | 2.319, 2.711 | 3.170 | 13.6388 | 1.00 | −1.20 | 0.26 | 254.8 | |
| VSeTe | 3.421 | 2.476, 2.703 | 3.339 | 12.9642 | 0.81 | −1.06 | 0.31 | 173.9 | |
| VXY Monolayer | λv/meV | λc/meV | RG/eV·Å | Eog/eV | Sn/(ћ/2) |
|---|---|---|---|---|---|
| VSSe | 68 | 11 | 0 | 0.93 | 1.16 |
| VSTe | 85 | 19 | 0 | 0.09 | 1.63 |
| VSeTe | 92 | 22 | 0.658 | 0.15 | 1.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, M.-H.; Li, C.-M.; Sun, W.-F. Spin-Orbit Coupling and Spin-Polarized Electronic Structures of Janus Vanadium-Dichalcogenide Monolayers: First-Principles Calculations. Nanomaterials 2022, 12, 382. https://doi.org/10.3390/nano12030382
Lv M-H, Li C-M, Sun W-F. Spin-Orbit Coupling and Spin-Polarized Electronic Structures of Janus Vanadium-Dichalcogenide Monolayers: First-Principles Calculations. Nanomaterials. 2022; 12(3):382. https://doi.org/10.3390/nano12030382
Chicago/Turabian StyleLv, Ming-Hao, Chang-Ming Li, and Wei-Feng Sun. 2022. "Spin-Orbit Coupling and Spin-Polarized Electronic Structures of Janus Vanadium-Dichalcogenide Monolayers: First-Principles Calculations" Nanomaterials 12, no. 3: 382. https://doi.org/10.3390/nano12030382
APA StyleLv, M.-H., Li, C.-M., & Sun, W.-F. (2022). Spin-Orbit Coupling and Spin-Polarized Electronic Structures of Janus Vanadium-Dichalcogenide Monolayers: First-Principles Calculations. Nanomaterials, 12(3), 382. https://doi.org/10.3390/nano12030382