
The Magnetic Behaviour of CoTPP Supported on Coinage Metal Surfaces in the Presence of Small Molecules: A Molecular Cluster Study of the Surface trans-Effect

 , , and
, , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Details
3. Results and Discussion
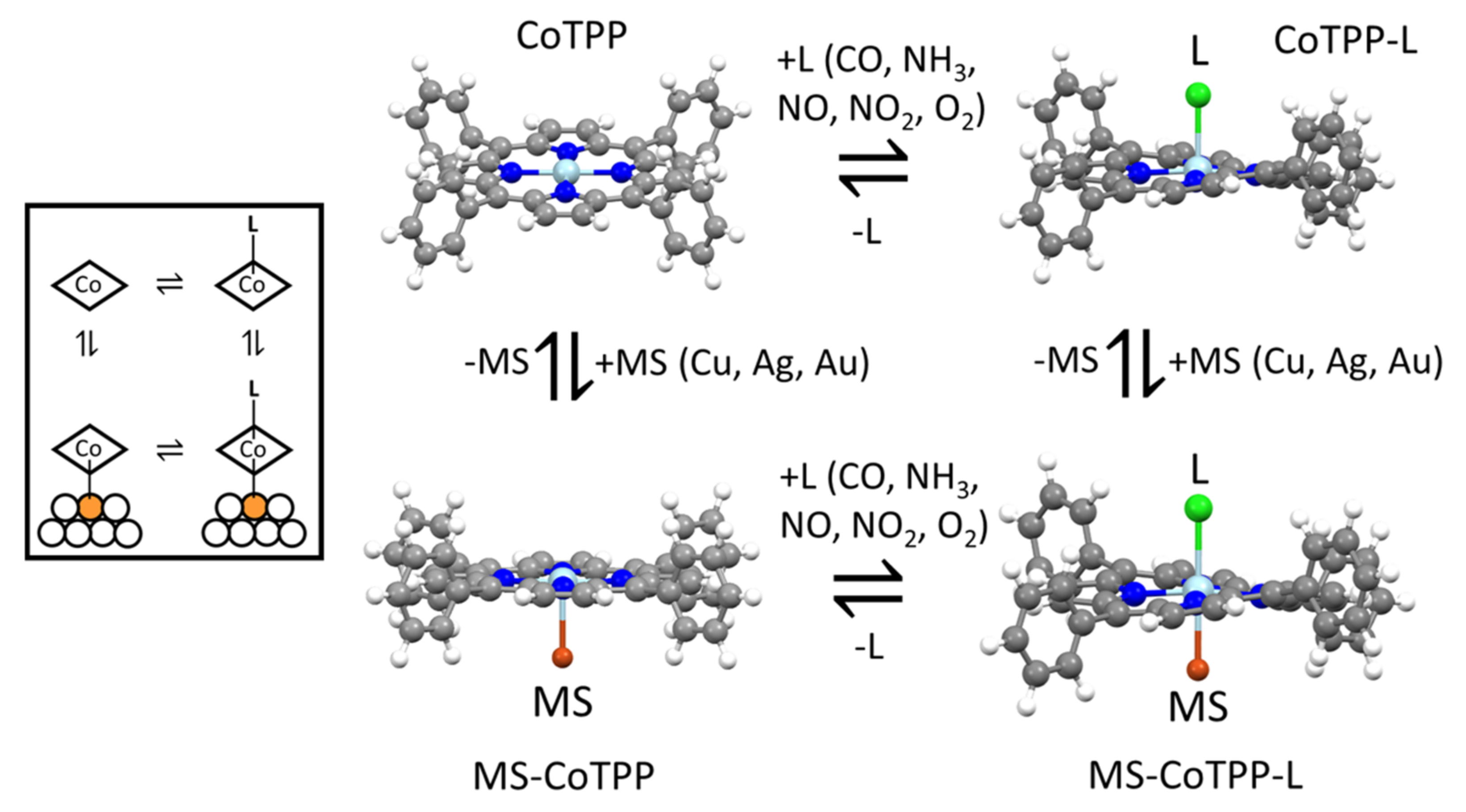
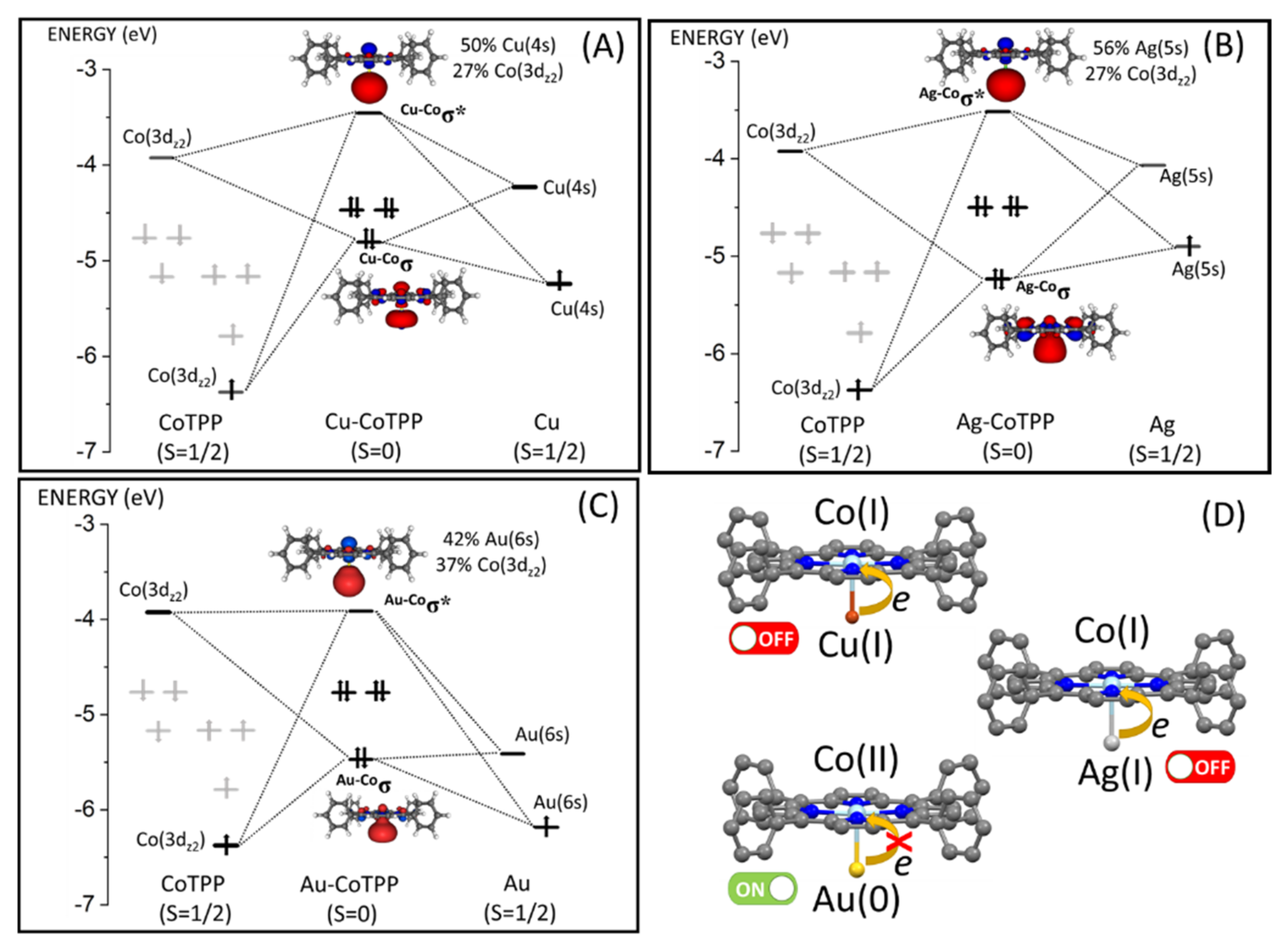
3.1. CoTPP on Cu, Ag and Au Substrates
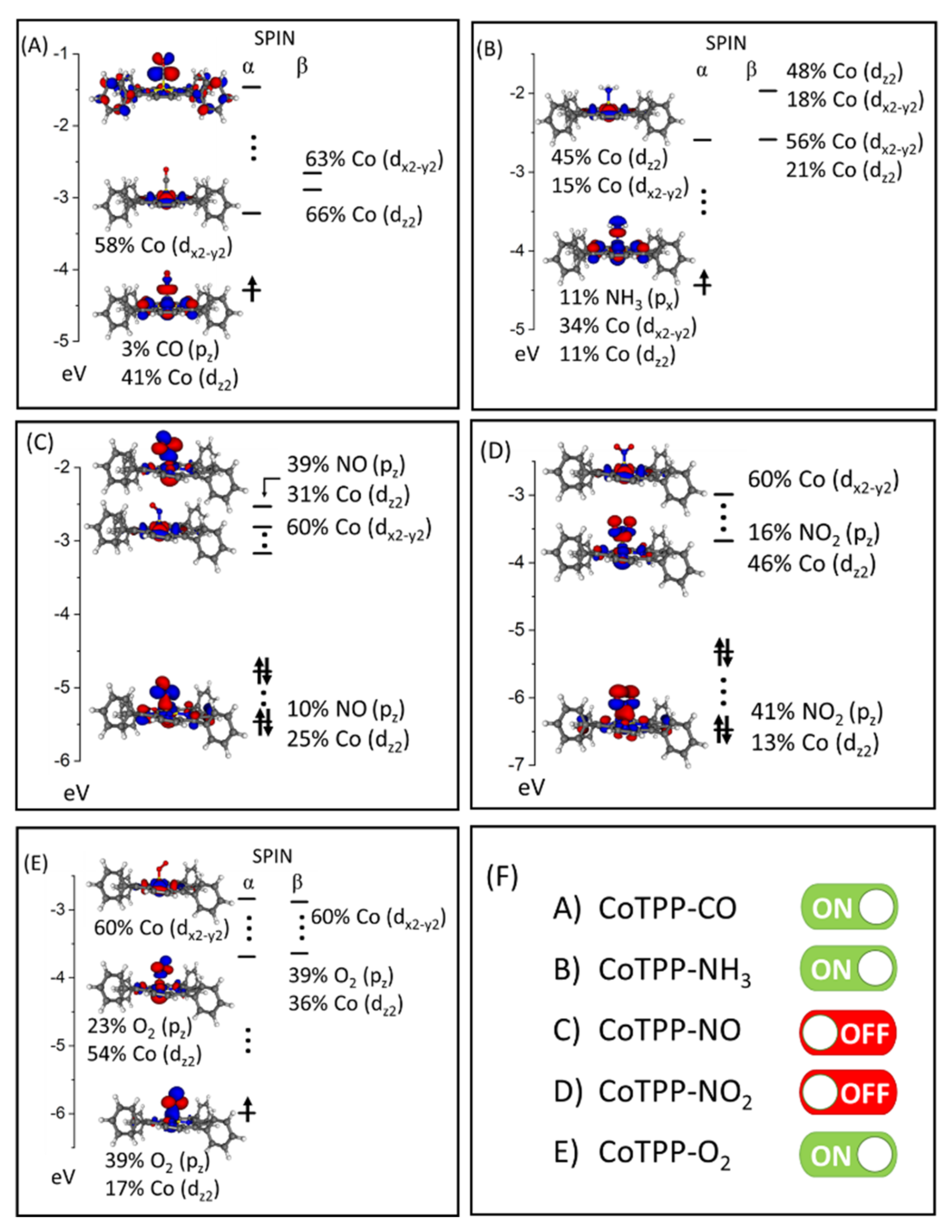
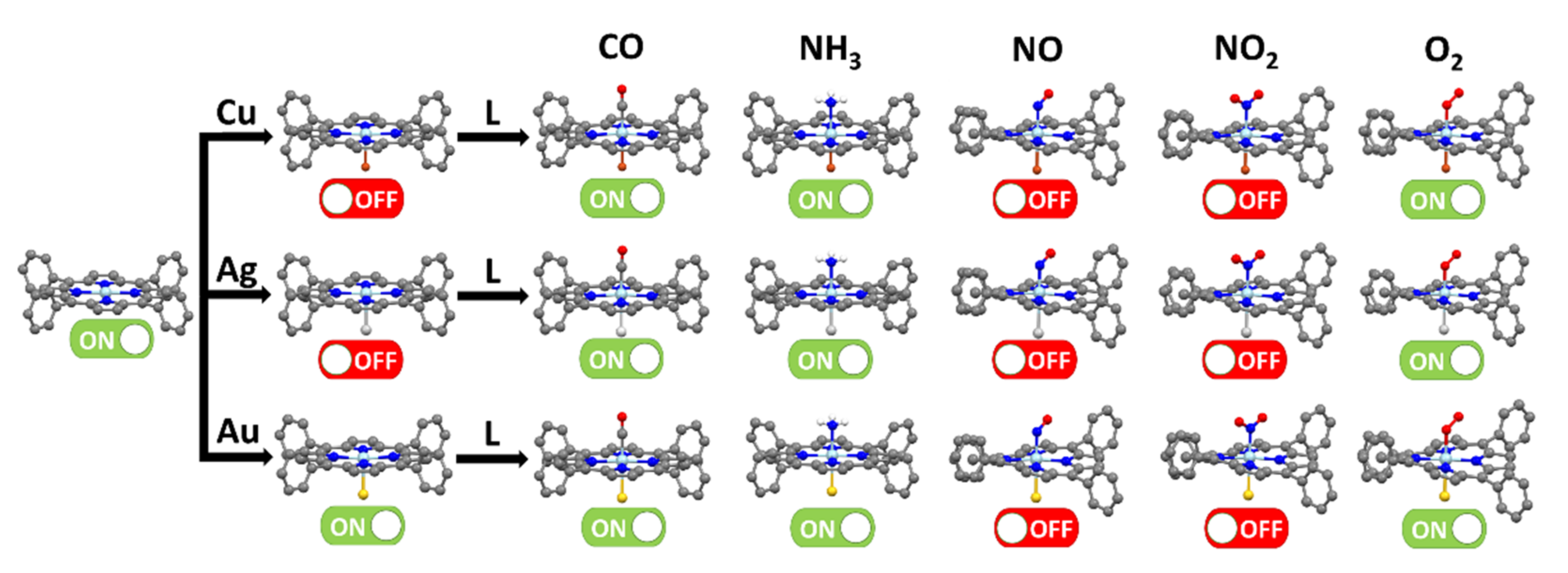
3.2. CoTPP–L Adducts (L = CO, NH3, NO, NO2, O2)
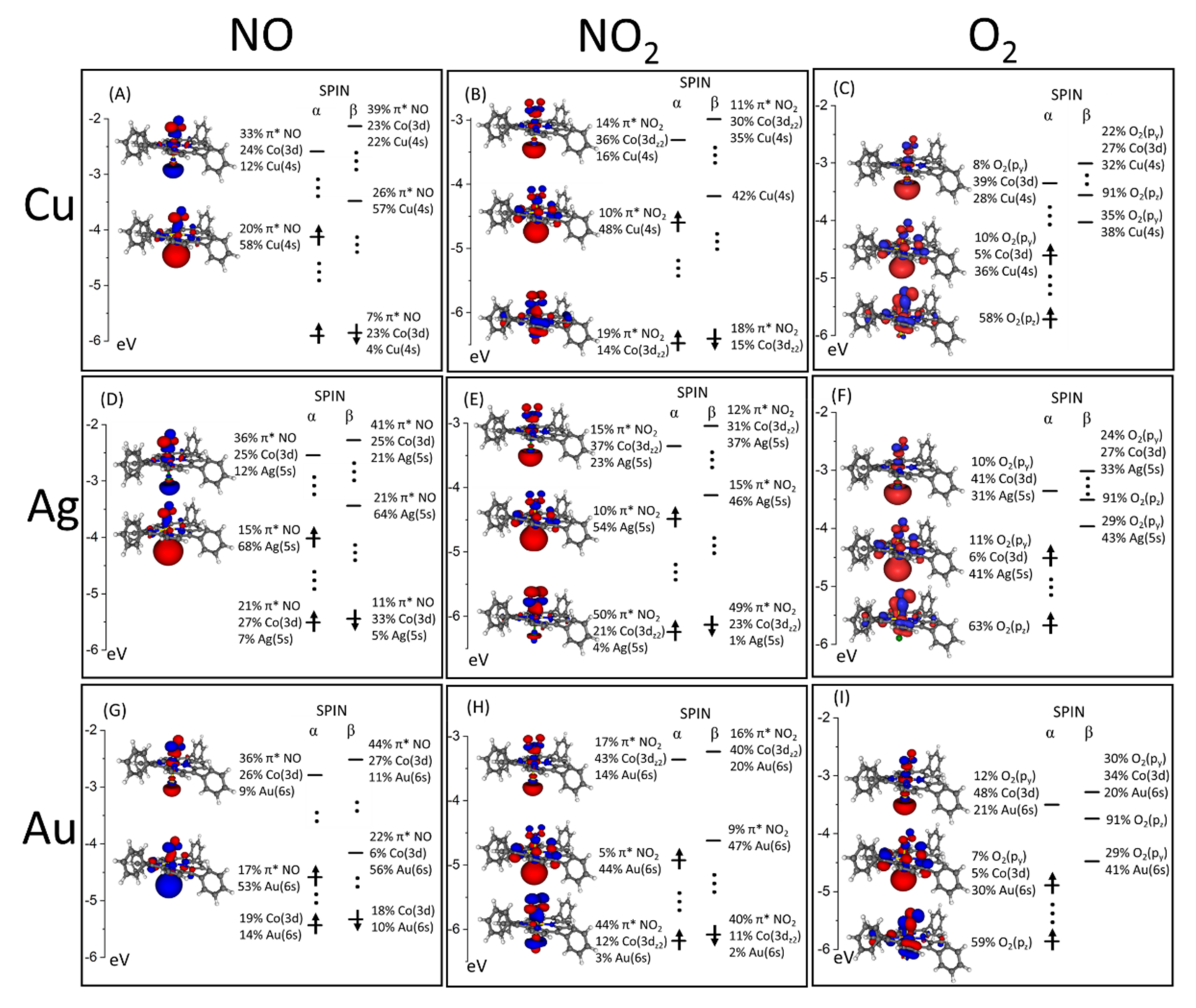
3.3. CoTPP–L Adducts on Cu, Ag and Au Substrates: The Trans-Effect
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brand, H.; Arnold, J. Recent developments in the chemistry of early transition metal porphyrin compounds. Coord. Chem. Rev. 1995, 140, 137–168. [Google Scholar] [CrossRef]
- Gounden, D.; Nombona, N.; van Zyl, W.E. Recent advances in phthalocyanines for chemical sensor, non-linearoptics (NLO) and energy storage applications. Coord. Chem. Rev. 2020, 420, 213359. [Google Scholar] [CrossRef]
- Rakow, N.A.; Suslick, K.S. A colorimetric sensor array for odour visualization. Nature 2000, 406, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Beyene, B.B.; Hung, C.H. Recent progress on metalloporphyrin-based hydrogen evolution catalysis. Coord. Chem. Rev. 2020, 410, 213234. [Google Scholar] [CrossRef]
- Singh, R.; Mukherjee, A. Metalloporphyrin Catalyzed C–H Amination. ACS Catal. 2019, 9, 3604–3617. [Google Scholar] [CrossRef]
- Sorokin, A.B. Phthalocyanine Metal Complexes in Catalysis. Chem. Rev. 2013, 113, 8152–8192. [Google Scholar] [CrossRef]
- Bartolomé, J.; Monton, C.; Schuller, I.K. Magnetism of Metal Phthalocyanines. In Molecular Magnets; Bartolomé, J., Luis, F., Fernández, J., Eds.; Springer: Berlin, Heidelberg, Germany, 2014; pp. 221–245. [Google Scholar]
- Aykanat, A.; Meng, Z.; Benedetto, G.; Mirica, K.A. Molecular Engineering of Multifunctional Metallophthalocyanine- Containing Framework Materials. Chem. Mater. 2020, 32, 5372–5409. [Google Scholar] [CrossRef]
- Barraud, C.; Seneor, P.; Mattana, R.; Fusil, S.; Bouzehouane, K.; Deranlot, C.; Graziosi, P.; Hueso, L.; Bergenti, I.; Dediu, V.; et al. Unravelling the role of the interface for spin injection into organic semiconductors. Nat. Phys. 2010, 6, 615–620. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Sun, Q. Magnetism of Phthalocyanine-Based Organometallic Single Porous Sheet. J. Am. Chem. Soc. 2011, 133, 15113–15119. [Google Scholar] [CrossRef]
- Paolesse, R.; Nardis, S.; Monti, D.; Stefanelli, M.; Di Natale, C. Porphyrinoids for Chemical Sensor Applications. Chem. Rev. 2017, 117, 2517–2583. [Google Scholar] [CrossRef] [Green Version]
- Drain, C.M.; Varotto, A.; Radivojevic, I. Self-Organized Porphyrinic Materials. Chem. Rev. 2009, 109, 1630–1658. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Yasseri, A.A.; Lindsey, J.S.; Bocian, D.F. Molecular memories that survive silicon device processing and real-world operation. Science 2003, 302, 1543–1545. [Google Scholar] [CrossRef] [Green Version]
- Wende, H.; Bernien, M.; Luo, J.; Sorg, C.; Ponpandian, N.; Kurde, J.; Miguel, J.; Piantek, M.; Xu, X.; Eckhold, P.; et al. Substrate-induced magnetic ordering and switching of iron porphyrin molecules. Nat. Mater. 2007, 6, 516–520. [Google Scholar] [CrossRef]
- Cojocariu, I.; Carlotto, S.; Sturmeit, H.M.; Zamborlini, G.; Cinchetti, M.; Cossaro, A.; Verdini, A.; Floreano, L.; Jugovac, M.; Puschnig, P.; et al. Ferrous to Ferric Transition in Fe-Phthalocyanine Driven by NO2 Exposure. Chem. A Eur. J. 2021, 27, 3526–3535. [Google Scholar] [CrossRef]
- Carlotto, S.; Sambi, M.; Sedona, F.; Vittadini, A.; Bartolomé, J.; Bartolomé, F.; Casarin, M. L2,3-edges absorption spectra of a 2D complex system: A theoretical modelling. Phys. Chem. Chem. Phys. 2016, 18, 28110–28116. [Google Scholar] [CrossRef]
- Cojocariu, I.; Carlotto, S.; Zamborlini, G.; Jugovac, M.; Schio, L.; Floreano, L.; Casarin, M.; Feyer, V.; Schneider, C. Reversible redox reactions in metal-supported porphyrin: The role of spin and oxidation state. J. Mater. Chem. C 2021, 9, 12559–12565. [Google Scholar] [CrossRef]
- Chang, M.H.; Kim, N.Y.; Chang, Y.H.; Lee, Y.; Jeon, U.S.; Kim, H.; Kim, Y.H.; Kahng, S.J. O2, NO2 and NH3 coordination to Co-porphyrin studied with scanning tunneling microscopy on Au(111). Nanoscale 2019, 11, 8510–8517. [Google Scholar] [CrossRef]
- Flechtner, K.; Kretschmann, A.; Steinrück, H.P.; Gottfried, J.M. NO-Induced Reversible Switching of the Electronic Interaction between a Porphyrin-Coordinated Cobalt Ion and a Silver Surface. J. Am. Chem. Soc. 2007, 129, 12110–12111. [Google Scholar] [CrossRef]
- Gottfried, J.M.; Marbach, H. Surface-Confined Coordination Chemistry with Porphyrins and Phthalocyanines: Aspects of Formation, Electronic Structure, and Reactivity. Zeitschrift fur Phys. Chem. 2009, 223, 53–74. [Google Scholar] [CrossRef]
- Chang, M.H.; Chang, Y.H.; Kim, N.Y.; Kim, H.; Lee, S.H.; Choi, M.S.; Kim, Y.H.; Kahng, S.J. Tuning and sensing spin interactions in Co-porphyrin/Au with NH3 and NO2 binding. Phys. Rev. B 2019, 100, 245406. [Google Scholar] [CrossRef]
- Ballav, N.; Wäckerlin, C.; Siewert, D.; Oppeneer, P.M.; Jung, T.A. Emergence of On-Surface Magnetochemistry. J. Phys. Chem. Lett. 2013, 4, 2303–2311. [Google Scholar] [CrossRef] [Green Version]
- Wäckerlin, C.; Chylarecka, D.; Kleibert, A.; Müller, K.; Iacovita, C.; Nolting, F.; Jung, T.A.; Ballav, N. Controlling spins in adsorbed molecules by a chemical switch. Nat. Commun. 2010, 1, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wäckerlin, C.; Tarafder, K.; Siewert, D.; Girovsky, J.; Hählen, T.; Iacovita, C.; Kleibert, A.; Nolting, F.; Jung, T.A.; Oppeneer, P.M.; et al. On-surface coordination chemistry of planar molecular spin systems: Novel magnetochemical effects induced by axial ligands. Chem. Sci. 2012, 3, 3154–3160. [Google Scholar] [CrossRef]
- Hieringer, W.; Flechtner, K.; Kretschmann, A.; Seufert, K.; Auwärter, W.; Barth, J.V.; Görling, A.; Steinrück, H.P.; Gottfried, J.M. The Surface Trans Effect: Influence of Axial Ligands on the Surface Chemical Bonds of Adsorbed Metalloporphyrins. J. Am. Chem. Soc. 2011, 133, 6206–6222. [Google Scholar] [CrossRef] [PubMed]
- Houwaart, T.; Le Bahers, T.; Sautet, P.; Auwärter, W.; Seufert, K.; Barth, J.V.; Bocquet, M.L. Scrutinizing individual CoTPP molecule adsorbed on coinage metal surfaces from the interplay of STM experiment and theory. Surf. Sci. 2015, 635, 108–114. [Google Scholar] [CrossRef]
- Bertoncello, R.; Bettinelli, M.; Casarin, M.; Gulino, A.; Tondello, E.; Vittadini, A. Zn4O(acetate)6, a Well-Tailored Molecular Model of ZnO. An Experimental and Theoretical Investigation of the Electronic Structure of Zn4O(acetate)6 and ZnO by Means of UV and X-ray Photoelectron Spectroscopies and First Principle Local Density Molecular Cluster Calculations. Inorg. Chem. 1992, 31, 1558–1565. [Google Scholar]
- Weber-Bargioni, A.; Auwärter, W.; Klappenberger, F.; Reichert, J.; Lefrançois, S.; Strunskus, T.; Wöll, C.; Schiffrin, A.; Pennec, Y.; Barth, J.V. Visualizing the Frontier Orbitals of a Conformationally Adapted Metalloporphyrin. ChemPhysChem 2008, 9, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Donovan, P.; Robin, A.; Dyer, M.S.; Persson, M.; Raval, R. Unexpected Deformations Induced by Surface Interaction and Chiral Self-Assembly of CoII-Tetraphenylporphyrin (Co-TPP) Adsorbed on Cu(110): A Combined STM and Periodic DFT Study. Chem. A Eur. J. 2010, 16, 11641–11652. [Google Scholar] [CrossRef]
- Lukasczyk, T.; Flechtner, K.; Merte, L.R.; Jux, N.; Maier, F.; Gottfried, J.M.; Steinrück, H.P. Interaction of Cobalt(II) Tetraarylporphyrins with a Ag(111) Surface Studied with Photoelectron Spectroscopy. J. Phys. Chem. C 2007, 111, 3090–3098. [Google Scholar] [CrossRef]
- Wechsler, D.; Franke, M.; Tariq, Q.; Zhang, L.; Lee, T.L.; Thakur, P.K.; Tsud, N.; Bercha, S.; Prince, K.C.; Steinrück, H.P.; et al. Adsorption Structure of Cobalt Tetraphenylporphyrin on Ag(100). J. Phys. Chem. C 2017, 121, 5667–5674. [Google Scholar] [CrossRef]
- Barlow, D.E.; Scudiero, L.; Hipps, K.W. Scanning Tunneling Microscopy Study of the Structure and Orbital-Mediated Tunneling Spectra of Cobalt(II) Phthalocyanine and Cobalt(II) Tetraphenylporphyrin on Au(111): Mixed Composition Films. Langmuir 2004, 20, 4413–4421. [Google Scholar] [CrossRef]
- Chang, Y.H.; Kim, H.; Kahng, S.J.; Kim, Y.H. Axial coordination and electronic structure of diatomic NO, CO, and O2 molecules adsorbed onto Co-tetraphenylporphyrin on Au(111), Ag(111), and Cu(111): A density-functional theory study. Dalton Trans. 2016, 45, 16673–16681. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF2014; SCM. Theoretical Chemistry. Available online: http://www.scm.com (accessed on 7 January 2022).
- Van Lenthe, E.; Ehlers, A.; Baerends, E.J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two—component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Carlotto, S.; Sambi, M.; Sedona, F.; Vittadini, A.; Casarin, M. A Theoretical Study of the Occupied and Unoccupied Electronic Structure of High- and Intermediate-Spin Transition Metal Phthalocyaninato (Pc) Complexes: VPc, CrPc, MnPc, and FePc. Nanomaterials 2021, 11, 54. [Google Scholar] [CrossRef]
- Carlotto, S.; Sambi, M.; Rancan, M.; Casarin, M. Theoretical Investigation of the Electronic Properties of Three Vanadium Phthalocyaninato (Pc) Based Complexes: PcV, PcVO, and PcVI. Inorg. Chem. 2018, 57, 1859–1869. [Google Scholar] [CrossRef]
- Mangione, G.; Carlotto, S.; Sambi, M.; Ligorio, G.; Timpel, M.; Vittadini, A.; Nardi, M.V.; Casarin, M. Electronic structures of CuTPP and CuTPP(F) complexes. A combined experimental and theoretical study I. Phys. Chem. Chem. Phys. 2016, 18, 18727–18738. [Google Scholar] [CrossRef]
- Mangione, G.; Sambi, M.; Carlotto, S.; Vittadini, A.; Ligorio, G.; Timpel, M.; Pasquali, L.; Giglia, A.; Nardi, M.V.; Casarin, M. Electronic structures of CuTPP and CuTPP(F) complexes. A combined experimental and theoretical study II. Phys. Chem. Chem. Phys. 2016, 18, 24890–24904. [Google Scholar] [CrossRef]
- Nardi, M.V.; Detto, F.; Aversa, L.; Verucchi, R.; Salviati, G.; Iannotta, A.; Casarin, M. Electronic properties of CuPc and H2Pc: An experimental and theoretical study. Phys. Chem. Chem. Phys. 2013, 15, 12864–12881. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Casarin, M.; Forrer, D.; Garau, F.; Pandolfo, L.; Pettinari, C.; Vittadini, A. Density Functional Theory Study of the Binding Capability of Tris(pyrazol-1-yl)methane toward Cu(I) and Ag(I) Cations. J. Phys. Chem. A 2008, 112, 6723–6731. [Google Scholar] [CrossRef]
- Auwärter, W.; Seufert, K.; Klappenberger, F.; Reichert, J.; Weber-Bargioni, A.; Verdini, A.; Cvetko, D.; Dell’Angela, M.; Floreano, L.; Cossaro, A.; et al. Site-specific electronic and geometric interface structure of Co-tetraphenyl-porphyrin layers on Ag(111). Phys. Rev. B 2010, 81, 245403. [Google Scholar] [CrossRef]
- Schwarz, M.; Garnica, M.; Duncan, D.A.; Paz, A.P.; Ducke, J.; Deimel, P.S.; Thakur, P.K.; Lee, T.-L.; Rubio, A.; Barth, J.V.; et al. Adsorption Conformation and Lateral Registry of Cobalt Porphine on Cu(111). J. Phys. Chem. C 2018, 122, 5452–5461. [Google Scholar] [CrossRef]
- Duncan, D.A.; Casado Aguilar, P.; Paszkiewicz, M.; Diller, K.; Bondino, F.; Magnano, E.; Klappenberger, F.; Píš, I.; Rubio, A.; Barth, J.V.; et al. Local adsorption structure and bonding of porphine on Cu(111) before and after self-metalation. J. Chem. Phys. 2019, 150, 094702. [Google Scholar] [CrossRef] [Green Version]
- Buimaga-Iarinca, L.; Morari, C. The effect of translation on the binding energy for transition-metal porphyrines adsorbed on Ag(111) surface. Beilstein J. Nanotechnol. 2019, 10, 706–717. [Google Scholar] [CrossRef]
- Eaton, S.S.; Eaton, G.R. Magnetic susceptibility of porphyrins. Inorg. Chem. 1980, 19, 1095–1096. [Google Scholar] [CrossRef]
- Douglas, B.E.; Hollingsworth, C.A. Symmetry in Bonding and Spectra: An Introduction; Academic Press: Orlando, FL, USA, 1985. [Google Scholar]
- Li, J.; Schreckenbach, G.; Ziegler, T. A Reassessment of the First Metal—Carbonyl Dissociation Energy in M(CO)4 (M = Ni, Pd, Pt), M(CO)5 (M = Fe, Ru, Os), and M(CO)6 (M = Cr, Mo, W) by a Quasirelativistic Density Functional Method. J. Am. Chem. Soc. 1995, 117, 486–494. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J. Modified valence indices from the two-particle density matrix. Int. J. Quantum Chem. 1994, 51, 187–200. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J.; Formosinho, S.J.; Varandas, A.J.C. Quantum mechanical valence study of a bond-breaking−bond forming process in triatomic systems. Int. J. Quantum Chem. 1994, 52, 1153–1176. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J. Hartree-Fock difference approach to chemical valence: Three-electron indices in UHF approximation. Int. J. Quantum Chem. 1996, 57, 377–389. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J.; Mazur, G. Quantum chemical valence indices from the one-determinantal difference approach. Can. J. Chem. 1996, 74, 1121–1130. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J.; Michalak, A. Two-electron valence indices from the Kohn-Sham orbitals. Int. J. Quantum Chem. 1997, 61, 589–601. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J.; Michalak, A. Exploring bonding patterns of molecular systems using quantum mechanical bond multiplicities. Polym. J. Chem. 1998, 72, 1779–1791. [Google Scholar]
- Michalak, A.; DeKock, R.L.; Ziegler, T. Bond Multiplicity in Transition-Metal Complexes: Applications of Two-Electron Valence Indices. J. Phys. Chem. A 2008, 112, 7256–7263. [Google Scholar] [CrossRef]
- Haller, K.J.; Enemark, J.H. Structural Chemistry of the {CoNO}8 Group. III.* The Structure of N,N’-Ethylenebis(salicylideneiminato) nitrosylcobalt(ll), Co(NO)(Salen). Acta Cryst. 1978, B34, 102–109. [Google Scholar] [CrossRef]
- Kim, H.; Chang, Y.H.; Lee, S.-H.; Lim, S.; Noh, S.-K.; Kim, Y.-H.; Kahng, S.-J. Visualizing tilted binding and precession of diatomic NO adsorbed to Co-porphyrin on Au(111) using scanning tunneling microscopy. Chem. Sci. 2014, 5, 2224–2229. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, R.; Ozarowski, A.; Aroca, R.; de Soares, L.A., II; Trsic, M. The electronic structure of metalated phthalocyanine-NO2 adducts. J. Mol. Struct. 1994, 317, 287–297. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements, 2nd ed.; Butterworth-Heinemann: Cambridge, UK, 1998. [Google Scholar]
- Sharafeldin, I.M.; Allam, N.K. DFT insights into the electronic properties and adsorption of NO2 on metal-doped carbon nanotubes for gas sensing applications. New J. Chem. 2017, 41, 14936–14944. [Google Scholar] [CrossRef]
- Rodley, G.A.; Robinson, W.T. Structure of a Monomeric Oxygen-carrying Complex. Nature 1972, 235, 438–439. [Google Scholar] [CrossRef]
- Melamud, E.; Silver, B.L.; Dori, Z. Electron Paramagnetic Resonance of Mononuclear Cobalt Oxygen Carriers Labeled with Oxygen-17. J. Am. Chem. Soc. 1974, 96, 4690–4692. [Google Scholar] [CrossRef]
- Gall, R.S.; Rogers, J.F.; Schaefer, W.P.; Christoph, G.G. The structure of a monomeric oxygen carrying cobalt complex: Dioxygen-N,N’-(1,1,2,2-tetramethyl)ethylenebis(3-tert-butylsalicylideniminato)(1-benzylimidazole)cobalt(II). J. Am. Chem. Soc. 1976, 98, 5135–5144. [Google Scholar] [CrossRef]
- Gall, R.S.; Schaefer, W.P. Preparation and structural characterization of a monomeric dioxygen adduct of (N,N’-(1,1,2,2-tetramethylethylene)bis(salicylideniminato))-(1-benzylimidazole)cobalt(II). Inorg. Chem. 1976, 15, 2758–2763. [Google Scholar] [CrossRef]
- Avdeef, A.; Schaefer, W.P. Reversible oxygen carriers. The synthesis and low temperature (−171.degree.) structure of an unstable monomeric dioxygen adduct of N,N’-(1,1,2,2-tetramethyl)ethylenebis(3-fluorosalicylideniminato)(1-methylimidazole)cobalt(II), Co(3-F-Saltmen)(1-Me-Imid)(O2).2Me2CO. J. Am. Chem. Soc. 1976, 98, 5153–5159. [Google Scholar]
- Tovrog, B.S.; Kitko, D.J.; Drago, R.S. Nature of the Bound O2 in a Series of Cobalt Dioxygen Adducts. J. Am. Chem. Soc. 1976, 98, 5144–5153. [Google Scholar] [CrossRef]
- Schaefer, W.P.; Huie, B.T.; Kurilla, M.G.; Ealick, S.E. Oxygen-carrying cobalt complexes. 10. Structures of N,N’-ethylenebis(3-tert-butylsalicyliden iminato)cobalt (II) and its monomeric dioxygen adduct. Inorg. Chem. 1980, 19, 340–344. [Google Scholar] [CrossRef]
- Seufert, K.; Auwärter, W.; Barth, J.V. Discriminative Response of Surface-Confined Metalloporphyrin Molecules to Carbon and Nitrogen Monoxide. J. Am. Chem. Soc. 2010, 132, 18141–18146. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlotto, S.; Cojocariu, I.; Feyer, V.; Floreano, L.; Casarin, M. The Magnetic Behaviour of CoTPP Supported on Coinage Metal Surfaces in the Presence of Small Molecules: A Molecular Cluster Study of the Surface trans-Effect. Nanomaterials 2022, 12, 218. https://doi.org/10.3390/nano12020218
Carlotto S, Cojocariu I, Feyer V, Floreano L, Casarin M. The Magnetic Behaviour of CoTPP Supported on Coinage Metal Surfaces in the Presence of Small Molecules: A Molecular Cluster Study of the Surface trans-Effect. Nanomaterials. 2022; 12(2):218. https://doi.org/10.3390/nano12020218
Chicago/Turabian StyleCarlotto, Silvia, Iulia Cojocariu, Vitaliy Feyer, Luca Floreano, and Maurizio Casarin. 2022. "The Magnetic Behaviour of CoTPP Supported on Coinage Metal Surfaces in the Presence of Small Molecules: A Molecular Cluster Study of the Surface trans-Effect" Nanomaterials 12, no. 2: 218. https://doi.org/10.3390/nano12020218
APA StyleCarlotto, S., Cojocariu, I., Feyer, V., Floreano, L., & Casarin, M. (2022). The Magnetic Behaviour of CoTPP Supported on Coinage Metal Surfaces in the Presence of Small Molecules: A Molecular Cluster Study of the Surface trans-Effect. Nanomaterials, 12(2), 218. https://doi.org/10.3390/nano12020218