

Characteristics of Cellulose Nanofibrils from Transgenic Trees with Reduced Expression of Cellulose Synthase Interacting 1




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Wood Oxidation and Fibrillation
2.2.2. Viscosity Measurement
2.2.3. Carboxylate Content
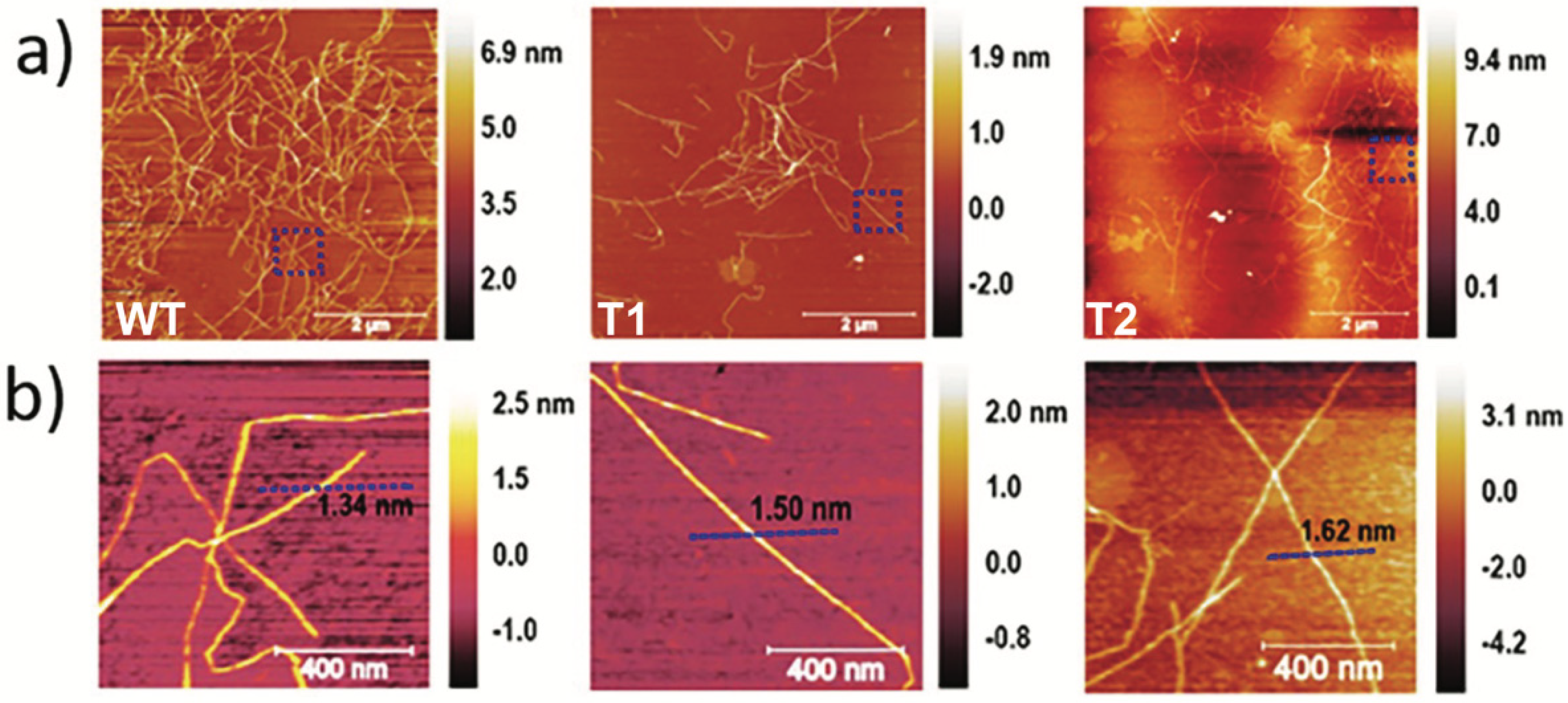
2.2.4. Atomic Force Microscopy
2.2.5. Nanofibril and Process Yield
2.2.6. Nanofibril Surface Area Estimation
2.2.7. Network Manufacturing and Characterization
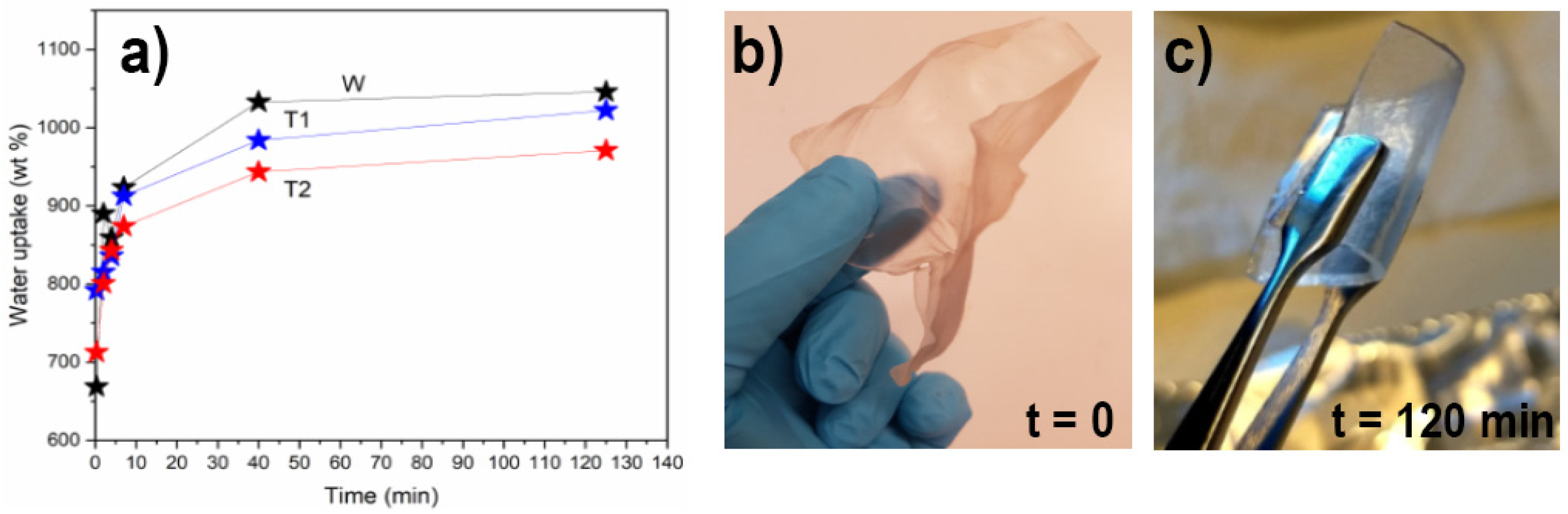
2.2.8. Water Uptake
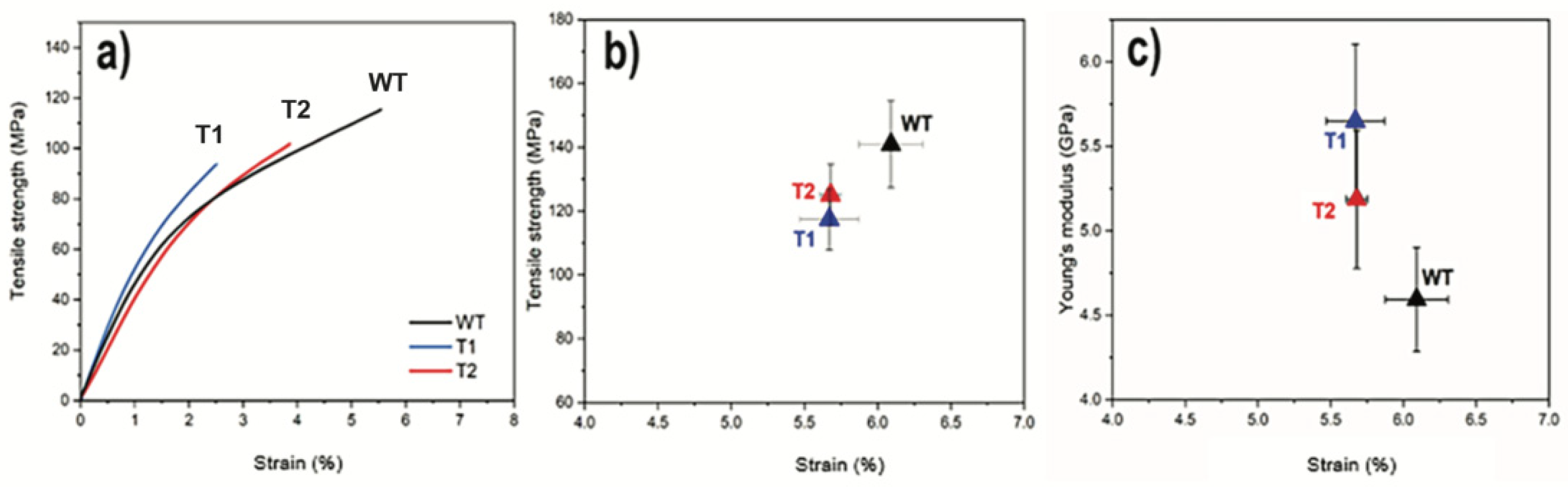
2.2.9. Mechanical Testing
2.2.10. Cellulose, Porosity, and Moisture Analysis
2.2.11. Degree of Polymerization
3. Results
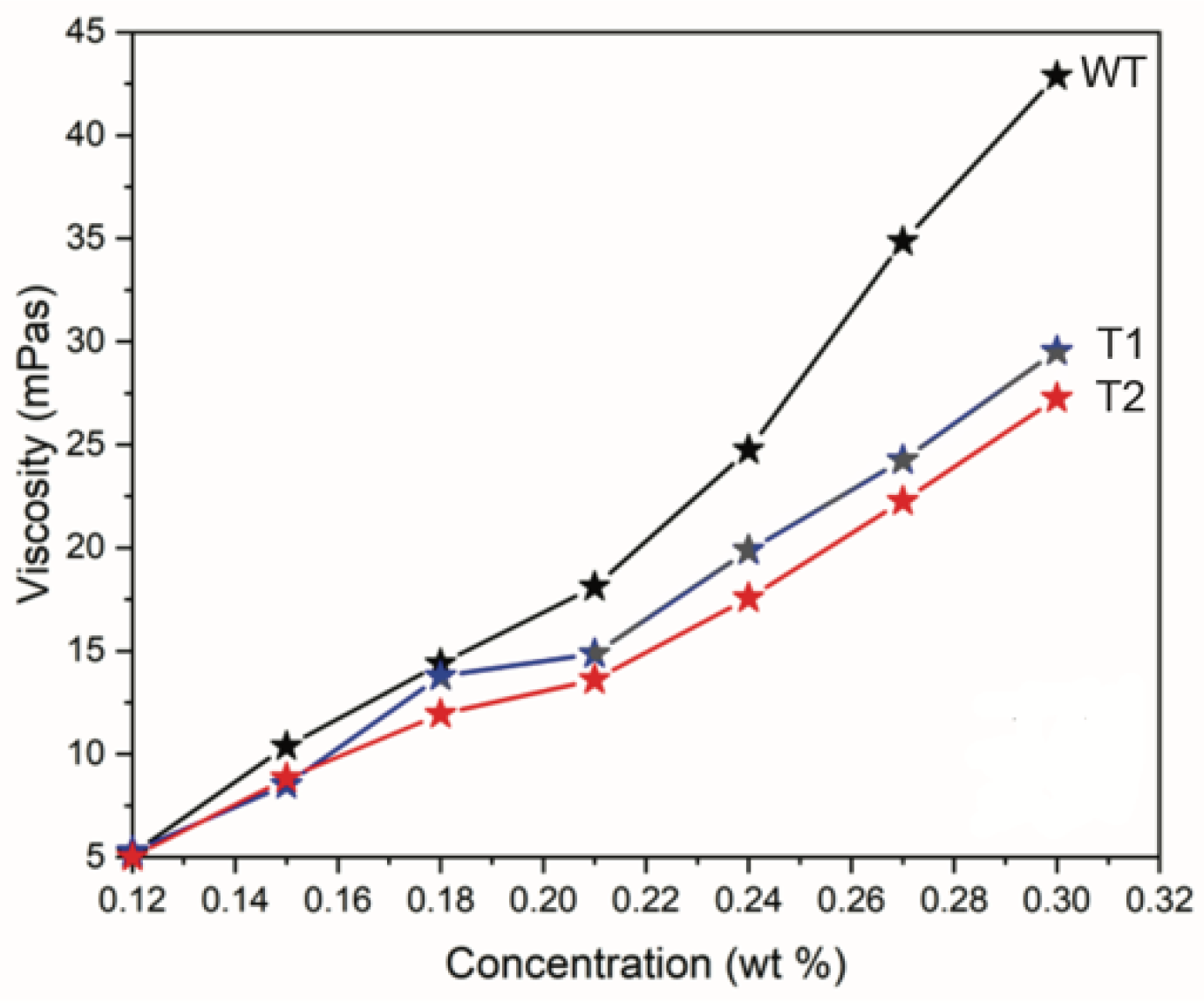
3.1. CNF Properties
3.2. CNF Network Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Groover, A.T.; Nieminen, K.; Helariutta, Y.; Mansfield, S.D. Wood formation in Populus. In Genetics and Genomics of Populus. Plant Genetics and Genomics: Crops and Models; Jansson, S., Bhalerao, R., Groover, A., Eds.; Springer: New York, NY, USA, 2010; Volume 8, pp. 201–224. [Google Scholar] [CrossRef]
- Fernandes, A.N.; Thomas, L.H.; Altaner, C.M.; Callow, P.; Forsyth, V.T.; Apperley, D.C.; Kennedy, C.J.; Jarvis, M.C. Nanostructure of cellulose microfibrils in spruce wood. Proc. Natl. Acad. Sci. USA 2011, 108, E1195–E1203. [Google Scholar] [CrossRef] [PubMed]
- Knox, J.P. Revealing the structural and functional diversity of plant cell walls. Curr. Opin. Plant Biol. 2008, 11, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E. Wood: A natural fibre reinforced composite. Met. Mater. 1992, 8, 333–336. [Google Scholar]
- Wu, X.; Moon, R.J.; Martini, A. Tensile strength of I β crystalline cellulose predicted by molecular dynamics simulation. Cellulose 2014, 21, 2233–2245. [Google Scholar] [CrossRef]
- Iwamoto, S.; Kai, W.; Isogai, A.; Iwata, T. Elastic modulus of single cellulose microfibrils from tunicate measured by atomic force microscopy. Biomacromolecules 2009, 10, 2571–2576. [Google Scholar] [CrossRef]
- Varanasi, S.; He, R.; Batchelor, W. Estimation of cellulose nanofibre aspect ratio from measurements of fibre suspension gel point. Cellulose 2013, 20, 1885–1896. [Google Scholar] [CrossRef]
- Fukuzumi, H.; Saito, T.; Isogai, A. Influence of TEMPO-oxidized cellulose nanofibril length on film properties. Carbohydr. Polym. 2013, 93, 172–177. [Google Scholar] [CrossRef]
- Saito, T.; Isogai, A. TEMPO-mediated oxidation of native cellulose. The effect of oxidation conditions on chemical and crystal structures of the water-insoluble fractions. Biomacromolecules 2004, 5, 1983–1989. [Google Scholar] [CrossRef]
- Saito, T.; Hirota, M.; Tamura, N.; Kimura, S.; Fukuzumi, H.; Heux, L.; Isogai, A. Individualization of nano-sized plant cellulose fibrils by direct surface carboxylation using TEMPO catalyst under neutral conditions. Biomacromolecules 2009, 10, 1992–1996. [Google Scholar] [CrossRef]
- Wågberg, L.; Decher, G.; Norgren, M.; Lindström, T.; Ankerfors, M.; Axnäs, K. The build-up of polyelectrolyte multilayers of microfibrillated cellulose and cationic polyelectrolytes. Langmuir 2008, 24, 784–795. [Google Scholar] [CrossRef]
- Henriksson, M.; Berglund, L.A.; Isaksson, P.; Lindstrom, T.; Nishino, T. Cellulose nanopaper structures of high toughness. Biomacromolecules 2008, 9, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, R.; Saito, T.; Okita, Y.; Isogai, A. Relationship between length and degree of polymerization of TEMPO-oxidized cellulose nanofibrils. Biomacromolecules 2012, 13, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Saxena, I.M.; Brown Jr, R.M. Cellulose biosynthesis: Current views and evolving concepts. Ann. Bot. 2005, 96, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Polko, J.K.; Kieber, J.J. The regulation of cellulose biosynthesis in plants. Plant Cell 2019, 31, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Kaplinsky, N.; Bringmann, M.; Cobb, A.; Carroll, A.; Sampathkumar, A.; Baskin, T.I.; Persson, S.; Somerville, C.R. Identification of a cellulose synthase-associated protein required for cellulose biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 12866–12871. [Google Scholar] [CrossRef]
- Li, S.; Lei, L.; Somerville, C.R.; Gu, Y. Cellulose synthase interactive protein 1 (CSI1) links microtubules and cellulose synthase complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 185–190. [Google Scholar] [CrossRef]
- Paredez, A.R.; Somerville, C.R.; Ehrhardt, D.W. Visualization of cellulose synthase demonstrates functional association with microtubules. Science 2006, 312, 1491–1495. [Google Scholar] [CrossRef]
- Bringmann, M.; Li, E.; Sampathkumar, A.; Kocabek, T.; Hauser, M.-T.; Persson, S. POM-POM2/cellulose synthase interacting1 is essential for the functional association of cellulose synthase and microtubules in Arabidopsis. Plant Cell 2012, 24, 163–177. [Google Scholar] [CrossRef]
- Schneider, R.; Tang, L.; Lampugnani, E.R.; Barkwill, S.; Lathe, R.; Zhang, Y.; McFarlane, H.E.; Pesquet, E.; Niittyla, T.; Mansfield, S.D. Two complementary mechanisms underpin cell wall patterning during xylem vessel development. Plant Cell 2017, 29, 2433–2449. [Google Scholar] [CrossRef]
- Bünder, A.; Sundman, O.; Mahboubi, A.; Persson, S.; Mansfield, S.D.; Rüggeberg, M.; Niittylä, T. Cellulose synthase interacting 1 is required for wood mechanics and leaf morphology in aspen. Plant J. 2020, 103, 1858–1868. [Google Scholar] [CrossRef]
- Jonasson, S.; Bünder, A.; Niittylä, T.; Oksman, K. Isolation and characterization of cellulose nanofibers from aspen wood using derivatizing and non-derivatizing pretreatments. Cellulose 2020, 27, 185–203. [Google Scholar] [CrossRef]
- Kaffashsaie, E.; Yousef, H.; Nishino, T.; Matsumoto, T.; Mashkour, M.; Madhoushi, M.; Kawaguchi, H. Direct conversion of raw wood to TEMPO-oxidized cellulose nanofibers. Carbohydr. Polym. 2021, 262, 117938. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Fu, S.; Zhai, H.; La, K.; Daneault, C. Influence of TEMPO-mediated oxidation on the lignin of thermomechanical pulp. Bioresour. Technol. 2012, 118, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Puangsin, B.; Soeta, H.; Saito, T.; Isogai, A. Characterization of cellulose nanofibrils prepared by direct TEMPO-mediated oxidation of hemp bast. Cellulose 2017, 24, 3767–3775. [Google Scholar] [CrossRef]
- Spence, K.L.; Venditti, R.A.; Rojas, O.J.; Habibi, Y.; Pawlak, J.J. The effect of chemical composition on microfibrillar cellulose films from wood pulps: Water interactions and physical properties for packaging applications. Cellulose 2010, 17, 835–848. [Google Scholar] [CrossRef]
- TAPPI Alpha-, Beta- and Gamma-Cellulose in Pulp. Test Methods T 203 cm-99; Technical Association of the Pulp and Paper Industry: Atlanta, GA, USA, 1999.
- TAPPI Viscosity of Pulp (Capillary Viscometer Method). TAPPI T230 om-99; Technical Association of the Pulp and Paper Industry: Atlanta, GA, USA, 1999.
- Iwamoto, S.; Lee, S.-H.; Endo, T. Relationship between aspect ratio and suspension viscosity of wood cellulose nanofibers. Polym. J. 2014, 46, 73–76. [Google Scholar] [CrossRef]
- Lasseuguette, E.; Roux, D.; Nishiyama, Y. Rheological properties of microfibrillar suspension of TEMPO-oxidized pulp. Cellulose 2008, 15, 425–433. [Google Scholar] [CrossRef]
- Arcari, M.; Axelrod, R.; Adamcik, J.; Handschin, S.; Sánchez-Ferrer, A.; Mezzenga, R.; Nyström, G. Structure–property relationships of cellulose nanofibril hydro-and aerogels and their building blocks. Nanoscale 2020, 12, 11638–11646. [Google Scholar] [CrossRef]
- Moberg, T.; Sahlin, K.; Yao, K.; Geng, S.; Westman, G.; Zhou, Q.; Oksman, K.; Rigdahl, M. Rheological properties of nanocellulose suspensions: Effects of fibril/particle dimensions and surface characteristics. Cellulose 2017, 24, 2499–2510. [Google Scholar] [CrossRef]
- Isogai, A.; Saito, T.; Fukuzumi, H. TEMPO-oxidized cellulose nanofibers. Nanoscale 2011, 3, 71–85. [Google Scholar] [CrossRef]
- Nge, T.T.; Lee, S.-H.; Endo, T. Preparation of nanoscale cellulose materials with different morphologies by mechanical treatments and their characterization. Cellulose 2013, 20, 1841–1852. [Google Scholar] [CrossRef]
- Sjöstedt, A.; Wohlert, J.; Larsson, P.T.; Wågberg, L. Structural changes during swelling of highly charged cellulose fibres. Cellulose 2015, 22, 2943–2953. [Google Scholar] [CrossRef]
- Ferrer, A.; Filpponen, I.; Rodríguez, A.; Laine, J.; Rojas, O.J. Valorization of residual Empty Palm Fruit Bunch Fibers (EPFBF) by microfluidization: Production of nanofibrillated cellulose and EPFBF nanopaper. Bioresour. Technol. 2012, 125, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Spence, K.L.; Venditti, R.A.; Rojas, O.J.; Habibi, Y.; Pawlak, J.J. A comparative study of energy consumption and physical properties of microfibrillated cellulose produced by different processing methods. Cellulose 2011, 18, 1097–1111. [Google Scholar] [CrossRef]
- Li, Q.; Renneckar, S. Molecularly thin nanoparticles from cellulose: Isolation of sub-microfibrillar structures. Cellulose 2009, 16, 1025–1032. [Google Scholar] [CrossRef]
- Usov, I.; Nyström, G.; Adamcik, J.; Handschin, S.; Schütz, C.; Fall, A.; Bergström, L.; Mezzenga, R. Understanding nanocellulose chirality and structure–properties relationship at the single fibril level. Nat. Commun. 2015, 6, 7564. [Google Scholar] [CrossRef]
- Lindström, T. Aspects on nanofibrillated cellulose (NFC) processing, rheology and NFC-film properties. Curr. Opin. Colloid Interface Sci. 2017, 29, 68–75. [Google Scholar] [CrossRef]
- Jonasson, S.; Bünder, A.; Das, O.; Niittylä, T.; Oksman, K. Comparison of tension wood and normal wood for oxidative nanofibrillation and network characteristics. Cellulose 2021, 28, 1085–1104. [Google Scholar] [CrossRef]
- Patiño-Masó, J.; Serra-Parareda, F.; Tarrés, Q.; Mutjé, P.; Espinach, F.X.; Delgado-Aguilar, M. TEMPO-oxidized cellulose nanofibers: A potential bio-based superabsorbent for diaper production. Nanomaterials 2019, 9, 1271. [Google Scholar] [CrossRef]
- Meng, Q.; Wang, T.J. Mechanics of strong and tough cellulose nanopaper. Appl. Mech. Rev. 2019, 71, 040801. [Google Scholar] [CrossRef]
- Meng, Q.; Li, B.; Li, T.; Feng, X.-Q. A multiscale crack-bridging model of cellulose nanopaper. J. Mech. Phys. Solids 2017, 103, 22–39. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tree ID | Process Yield (wt%) | Nanofibril Yield (%) | Carboxylate Content (mmol/g) | Viscosity (mPa·s) | Suspension Conductivity (µS/cm) | Surface Area (m2·g−1) |
|---|---|---|---|---|---|---|
| WT | 45.2 (1.3)a | 50.8 (0.3)a | 0.69 (0.10)a | 51.8 | 37 (5)a | 100 |
| T1 | 44.0 (2.0)a | 49.1 (1.0)b | 0.55 (0.08)b | 36.7 | 41 (8)a | 83 |
| T2 | 43.9 (2.1)a | 48.2 (0.8)b | 0.50 (0.06)b | 33.3 | 42 (10)a | 79 |
| Tree ID | Initial Cellulose DPn | CNF DPv | Moisture Content (%) | Porosity (%) | α-Cellulose (%) |
|---|---|---|---|---|---|
| WT | 1802 (214)a | 1185 (64)a | 10.3 (2.1)a | 22 (2)a | 80 |
| T1 | 1644 (137)b | 995 (62)b | 11.3 (2.8)a | 21 (1)a | 82 |
| T2 | 1637 (129)b | 1061 (37)b | 9.7 (1.9)a | 23 (2)a | 80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jonasson, S.; Bünder, A.; Berglund, L.; Niittylä, T.; Oksman, K. Characteristics of Cellulose Nanofibrils from Transgenic Trees with Reduced Expression of Cellulose Synthase Interacting 1. Nanomaterials 2022, 12, 3448. https://doi.org/10.3390/nano12193448
Jonasson S, Bünder A, Berglund L, Niittylä T, Oksman K. Characteristics of Cellulose Nanofibrils from Transgenic Trees with Reduced Expression of Cellulose Synthase Interacting 1. Nanomaterials. 2022; 12(19):3448. https://doi.org/10.3390/nano12193448
Chicago/Turabian StyleJonasson, Simon, Anne Bünder, Linn Berglund, Totte Niittylä, and Kristiina Oksman. 2022. "Characteristics of Cellulose Nanofibrils from Transgenic Trees with Reduced Expression of Cellulose Synthase Interacting 1" Nanomaterials 12, no. 19: 3448. https://doi.org/10.3390/nano12193448
APA StyleJonasson, S., Bünder, A., Berglund, L., Niittylä, T., & Oksman, K. (2022). Characteristics of Cellulose Nanofibrils from Transgenic Trees with Reduced Expression of Cellulose Synthase Interacting 1. Nanomaterials, 12(19), 3448. https://doi.org/10.3390/nano12193448