
Light-Activated Protoporphyrin IX-Based Polysilsesquioxane Nanoparticles Induce Ferroptosis in Melanoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
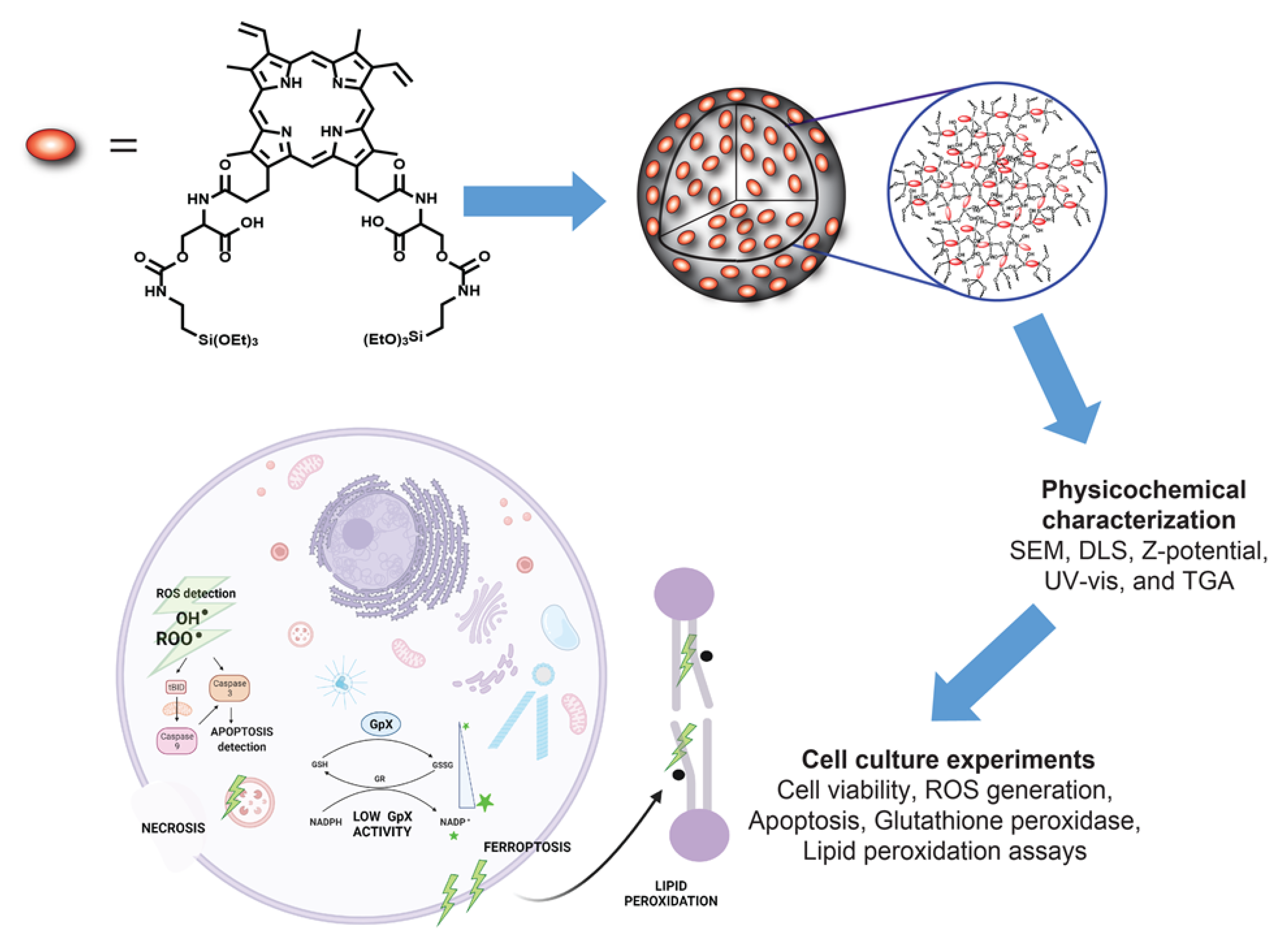{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Stock Solutions for In-Vitro Experiments
2.3. Synthesis of PpIX-PSilQ Nanoparticles
2.4. Determination of Intracellular Reactive Oxygen Species (ROS)
2.5. Evaluation of Intracellular ROS Using Confocal Microscopy
2.6. Cellular Uptake of PpIX-PSilQ Nanoparticles
2.7. In Vitro Evaluation of PDT Triggered Apoptosis
2.8. Inhibition of Ferroptosis
2.9. Evaluation of NADPH/NADP+ Kinetics
2.10. Measurement of Intracellular Lipid Peroxides
2.11. Statistics
3. Results
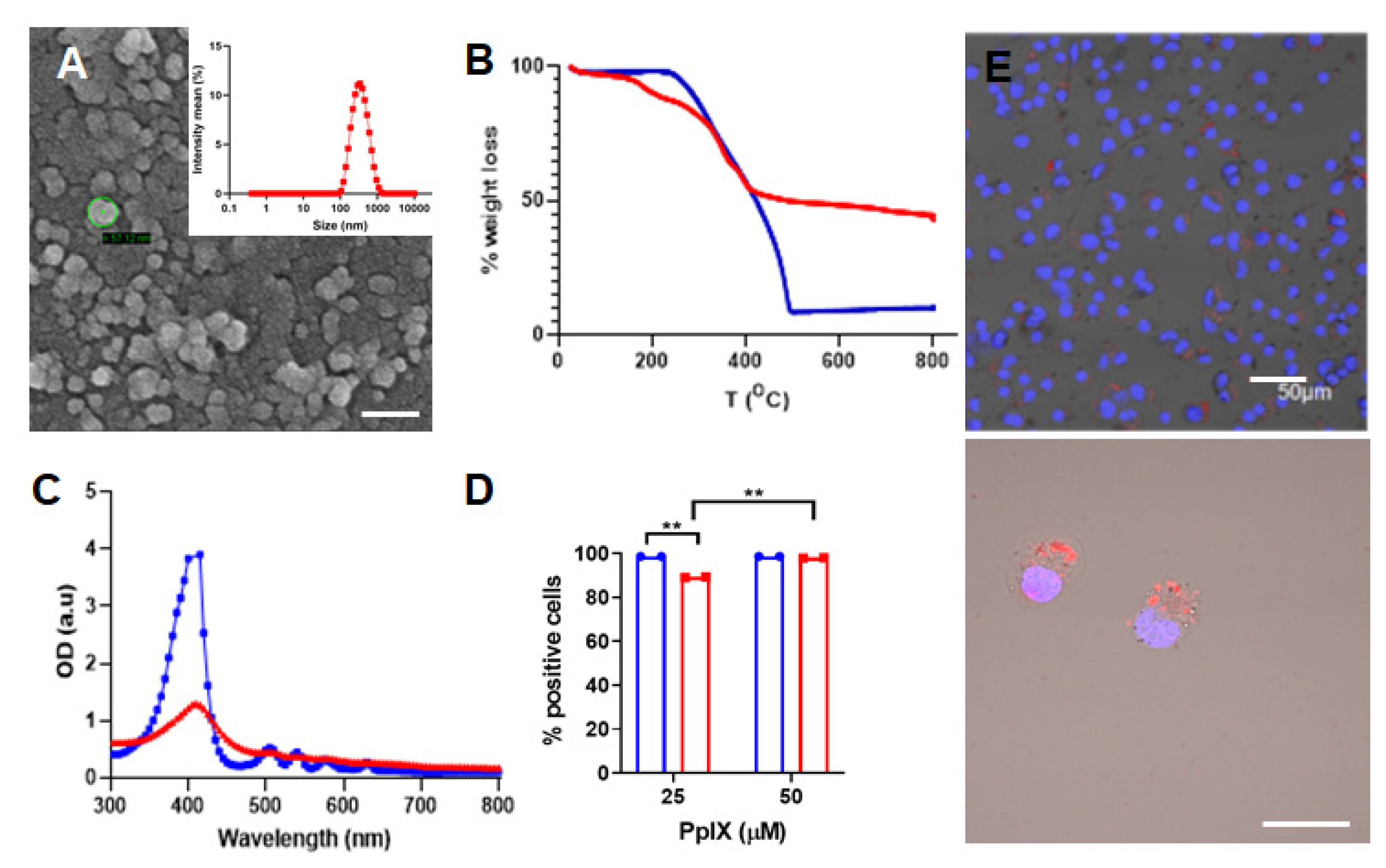
3.1. Synthesis and Characterization of PpIX-PSilQ NPs
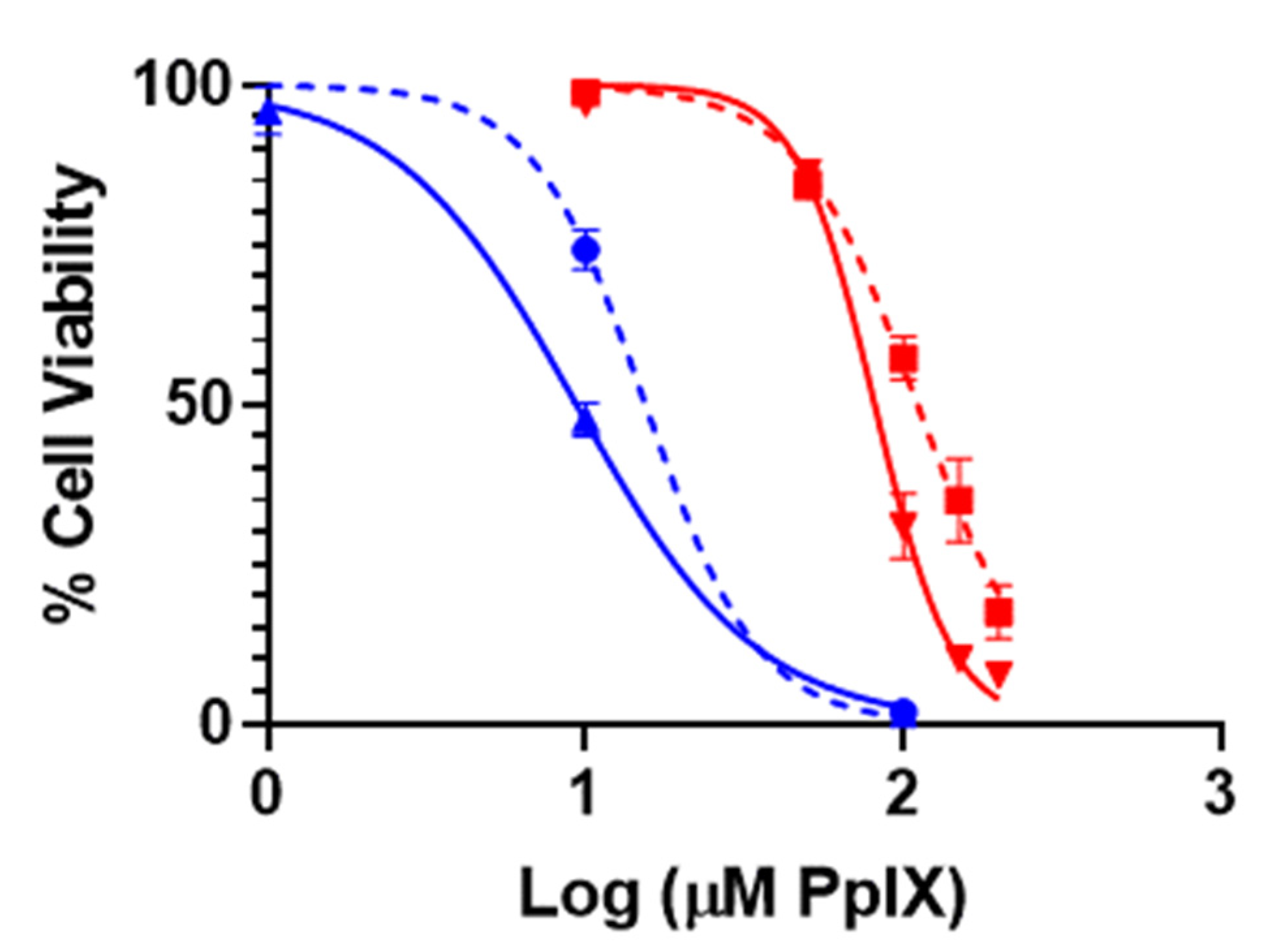
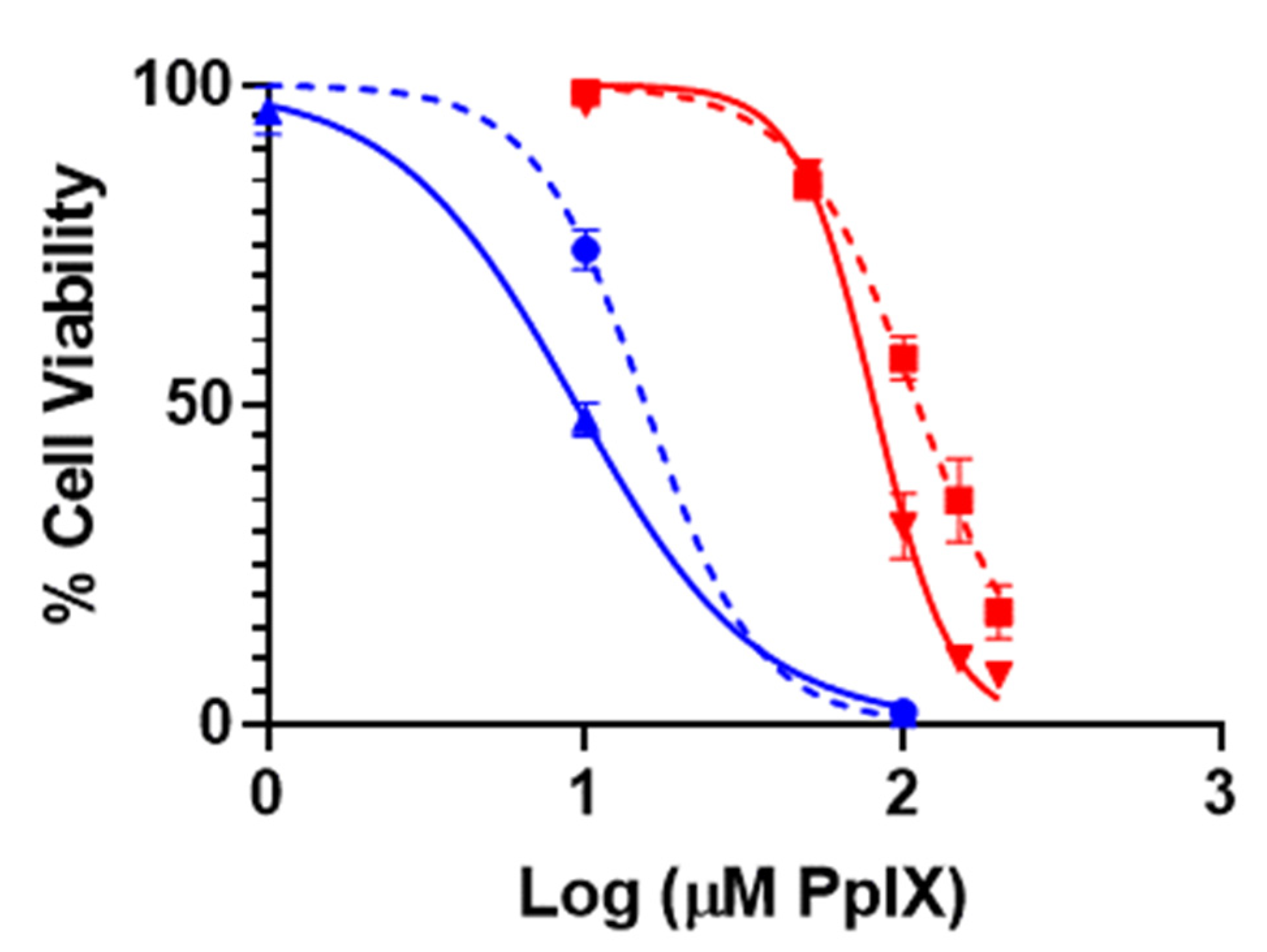
3.2. In Vitro PDT Performance of PpIX-PSilQ NPs
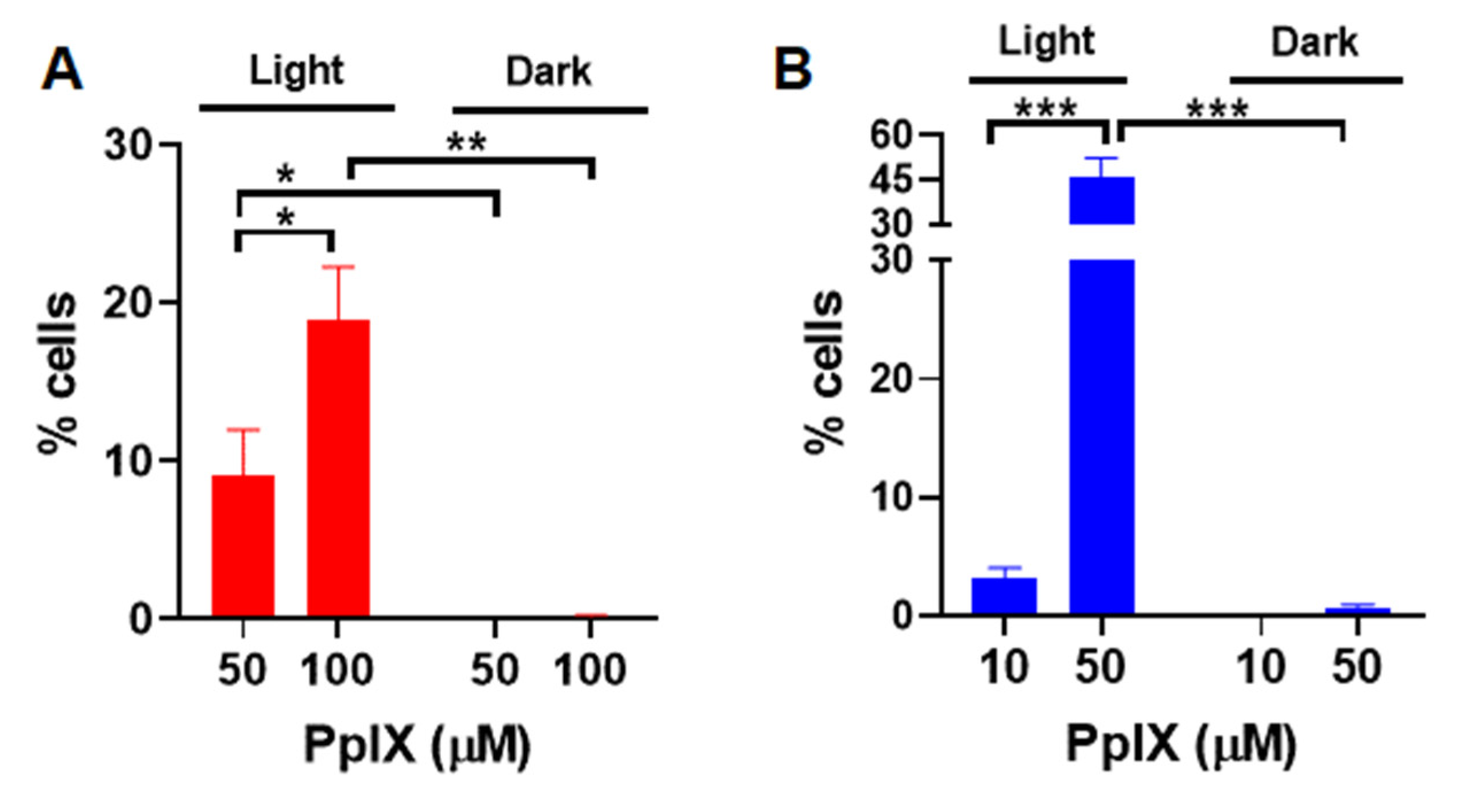
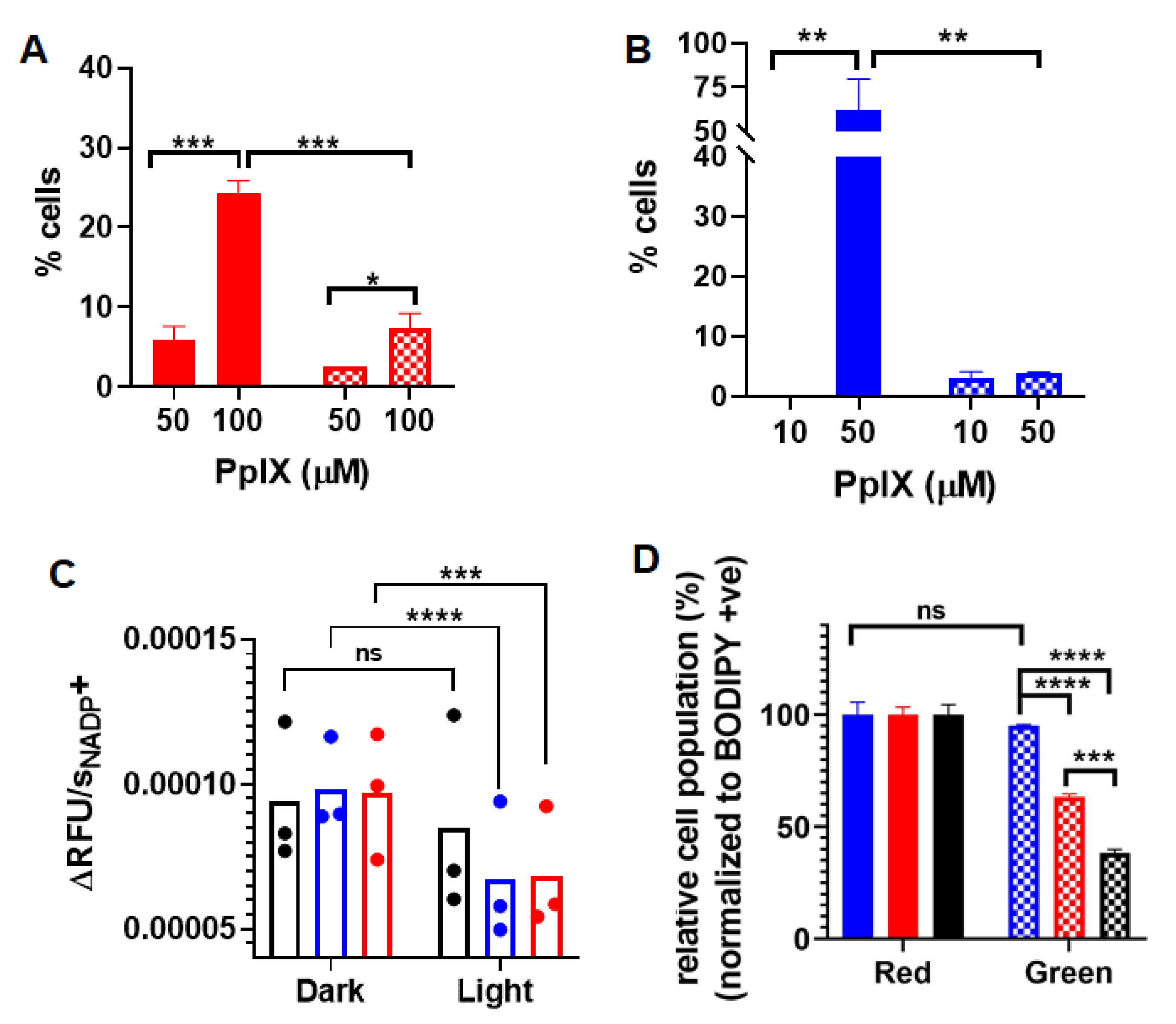
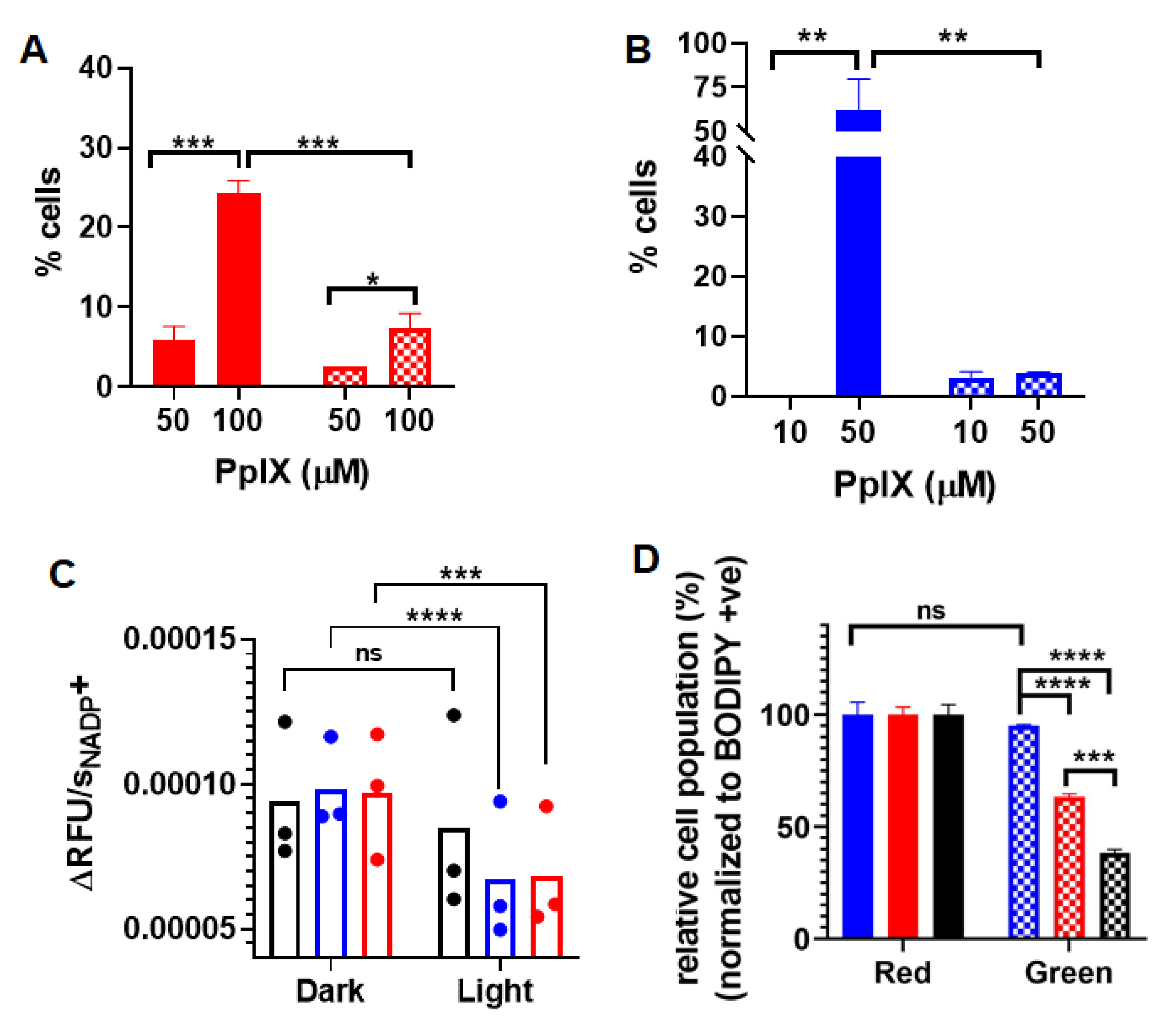
3.3. Apoptosis and Necrosis Induced by PpIX-PSilQ Nanoparticles
3.4. Inactivation of Glutathione Peroxidase Triggered by PpIX-PSilQ Nanoparticles
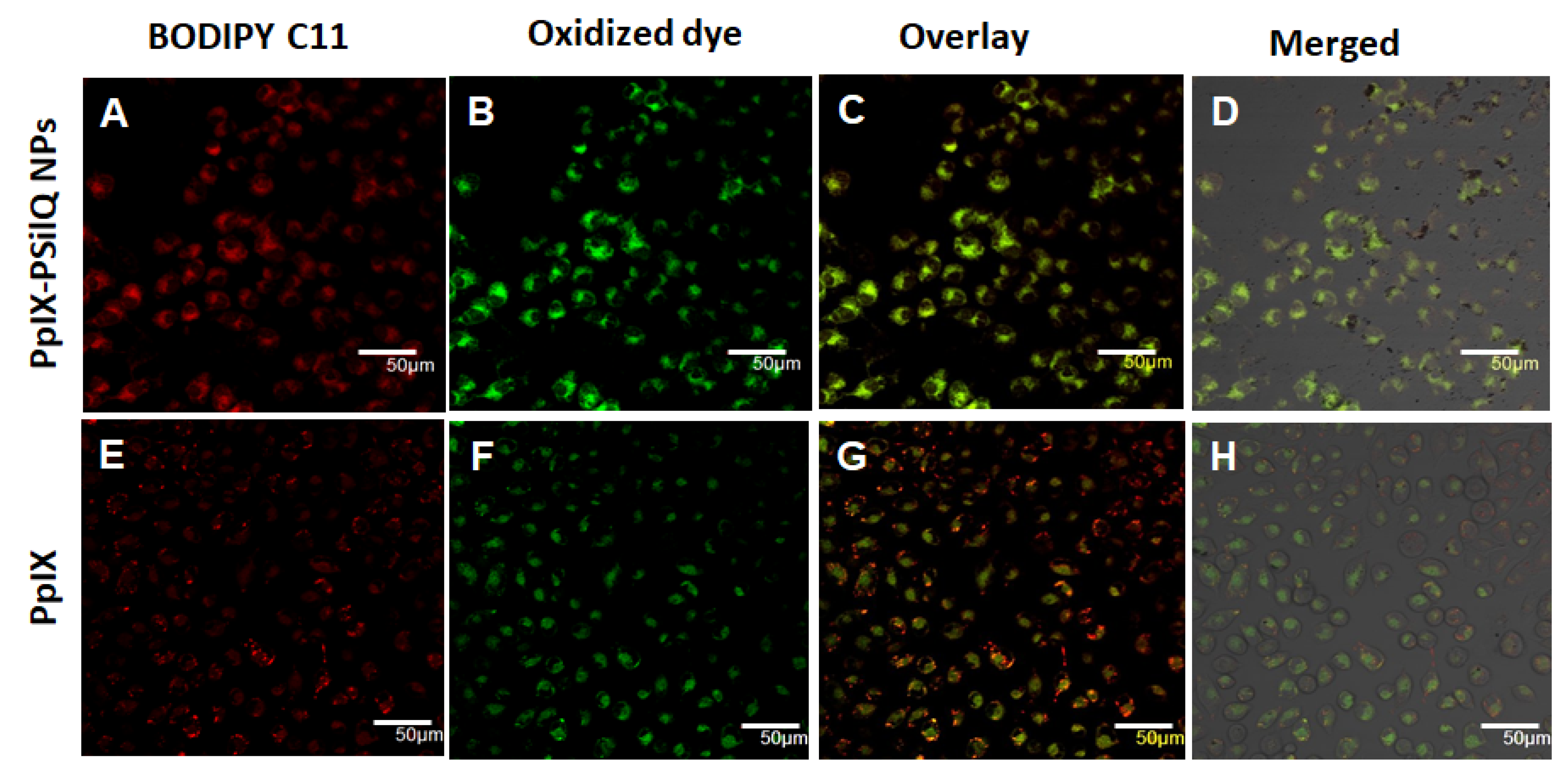
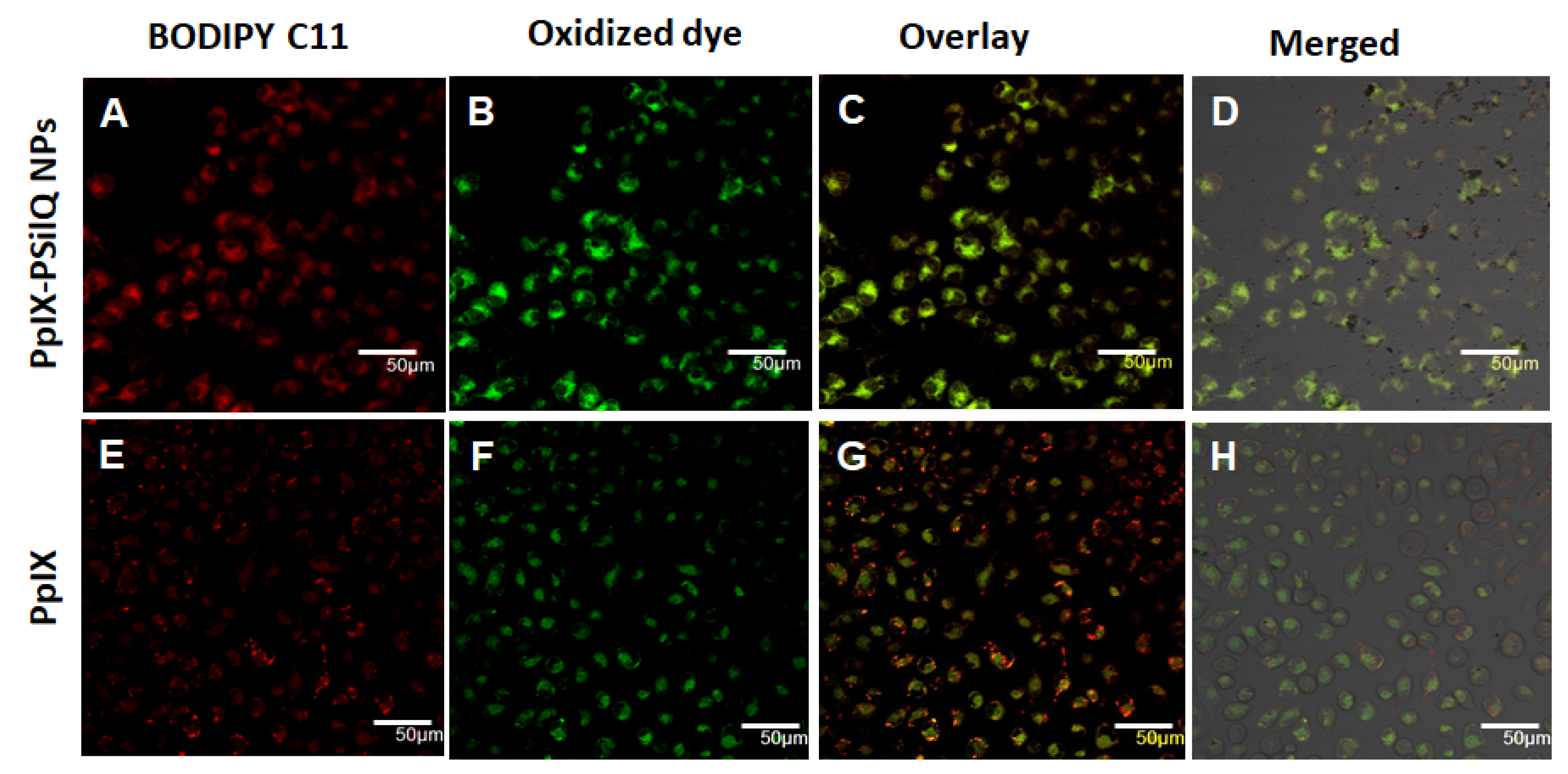
3.5. Lipid ROS Generation Detected by a Lipid Peroxidation Sensor
3.6. Inhibition of Ferroptosis Using Ferrostatin-1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, X.; Zhang, C.Y.; Gao, J.; Wang, Z. Recent advances in photodynamic therapy for cancer and infectious diseases. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1560. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—Mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Obaid, G.; Broekgaarden, M.; Bulin, A.L.; Huang, H.C.; Kuriakose, J.; Liu, J.; Hasan, T. Photonanomedicine: A convergence of photodynamic therapy and nanotechnology. Nanoscale 2016, 8, 12471–12503. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Alghamdi, M.S.; Alotaibi, A.S.; Alzhrani, R.; Alwuthaynani, F.; Althobaiti, Y.S.; Almalki, A.H.; Sau, S.; Iyer, A.K. Progress in Clinical Trials of Photodynamic Therapy for Solid Tumors and the Role of Nanomedicine. Cancers 2020, 12, 2793. [Google Scholar] [CrossRef] [PubMed]
- Vega, D.L.; Lodge, P.; Vivero-Escoto, J.L. Redox-Responsive Porphyrin-Based Polysilsesquioxane Nanoparticles for Photodynamic Therapy of Cancer Cells. Int. J. Mol. Sci. 2015, 17, 56. [Google Scholar] [CrossRef] [Green Version]
- Vivero-Escoto, J.L.; Vega, D.L. Stimuli-responsive protoporphyrin IX silica-based nanoparticles for photodynamic therapy in vitro. RSC Adv. 2014, 4, 14400–14407. [Google Scholar] [CrossRef]
- Vivero-Escoto, J.; DeCillis, D.; Fritts, L.; Vega, D. Porphyrin-Based Polysilsesquioxane Nanoparticles to Improve Photodynamic Therapy for Cancer Treatment; SPIE BiOS: Bellingham, WA, USA, 2014; Volume 8931. [Google Scholar]
- Lyles, Z.K.; Tarannum, M.; Mena, C.; Inada, N.M.; Bagnato, V.S.; Vivero-Escoto, J.L. Biodegradable Silica-Based Nanoparticles with Improved and Safe Delivery of Protoporphyrin IX for the In Vivo Photodynamic Therapy of Breast Cancer. Adv. Ther. 2020, 3, 2000022. [Google Scholar] [CrossRef]
- Juneja, R.; Lyles, Z.; Vadarevu, H.; Afonin, K.A.; Vivero-Escoto, J.L. Multimodal Polysilsesquioxane Nanoparticles for Combinatorial Therapy and Gene Delivery in Triple-Negative Breast Cancer. ACS Appl. Mater. Interfaces 2019, 11, 12308–12320. [Google Scholar] [CrossRef]
- Bacellar, I.O.; Tsubone, T.M.; Pavani, C.; Baptista, M.S. Photodynamic Efficiency: From Molecular Photochemistry to Cell Death. Int. J. Mol. Sci. 2015, 16, 523. [Google Scholar] [CrossRef] [Green Version]
- Reiners, J.J.; Agostinis, P.; Berg, K.; Oleinick, N.L.; Kessel, D.H. Assessing autophagy in the context of photodynamic therapy. Autophagy 2010, 6, 7–18. [Google Scholar] [CrossRef]
- Rubio, N.; Verrax, J.; Dewaele, M.; Verfaillie, T.; Johansen, T.; Piette, J.; Agostinis, P. p38(MAPK)-regulated induction of p62 and NBR1 after photodynamic therapy promotes autophagic clearance of ubiquitin aggregates and reduces reactive oxygen species levels by supporting Nrf2-antioxidant signaling. Free Radic. Biol. Med. 2014, 67, 292–303. [Google Scholar] [CrossRef]
- Mishchenko, T.A.; Balalaeva, I.V.; Vedunova, M.V.; Krysko, D.V. Ferroptosis and Photodynamic Therapy Synergism: Enhancing Anticancer Treatment. Trends Cancer 2021, 7, 484–487. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liao, H.; Li, F.; Ling, D. A mini-review and perspective on ferroptosis-inducing strategies in cancer therapy. Chin. Chem. Lett. 2019, 30, 847–852. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Pu, Y.; Li, S.; Gao, W.; He, B. PDT-Enhanced Ferroptosis by a Polymer Nanoparticle with pH-Activated Singlet Oxygen Generation and Superb Biocompatibility for Cancer Therapy. Biomacromolecules 2021, 22, 1167–1176. [Google Scholar] [CrossRef]
- Gao, M.; Deng, J.; Liu, F.; Fan, A.; Wang, Y.; Wu, H.; Ding, D.; Kong, D.; Wang, Z.; Peer, D.; et al. Triggered ferroptotic polymer micelles for reversing multidrug resistance to chemotherapy. Biomaterials 2019, 223, 119486. [Google Scholar] [CrossRef]
- Gomes, A.; Fernandes, E.; Lima, J.L.F.C. Fluorescence probes used for detection of reactive oxygen species. J. Biochem. Biophys. Methods 2005, 65, 45–80. [Google Scholar] [CrossRef]
- Aranda, A.; Sequedo, L.; Tolosa, L.; Quintas, G.; Burello, E.; Castell, J.V.; Gombau, L. Dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay: A quantitative method for oxidative stress assessment of nanoparticle-treated cells. Toxicol. Vitr. 2013, 27, 954–963. [Google Scholar] [CrossRef]
- Saxena, S.; Vekaria, H.; Sullivan, P.G.; Seifert, A.W. Connective tissue fibroblasts from highly regenerative mammals are refractory to ROS-induced cellular senescence. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Drummen, G.P.C.; van Liebergen, L.C.M.; Op den Kamp, J.A.F.; Post, J.A. C11-BODIPY581/591, an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic. Biol. Med. 2002, 33, 473–490. [Google Scholar] [CrossRef]
- Indran, I.R.; Tufo, G.; Pervaiz, S.; Brenner, C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim. Biophys. Acta (BBA)—Bioenerg. 2011, 1807, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yang, L.; Peng, X.; Fan, Q.; Wei, S.; Yang, S.; Li, X.; Jin, H.; Wu, B.; Huang, M.; et al. Emerging mechanisms and applications of ferroptosis in the treatment of resistant cancers. Biomed. Pharmacother. 2020, 130, 110710. [Google Scholar] [CrossRef]
- Shishido, Y.; Amisaki, M.; Matsumi, Y.; Yakura, H.; Nakayama, Y.; Miyauchi, W.; Miyatani, K.; Matsunaga, T.; Hanaki, T.; Kihara, K.; et al. Antitumor Effect of 5-Aminolevulinic Acid Through Ferroptosis in Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2021, 28, 3996–4006. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Shi, L.; Yu, C.; Dong, Y.; Qiu, F.; Shen, L.; Qian, Q.; Zhou, G.; Zhu, X. Ferroptosis Promotes Photodynamic Therapy: Supramolecular Photosensitizer-Inducer Nanodrug for Enhanced Cancer Treatment. Theranostics 2019, 9, 3293–3307. [Google Scholar] [CrossRef] [PubMed]
- Siano, P.; Johnston, A.; Loman-Cortes, P.; Zhin, Z.; Vivero-Escoto, J.L. Evaluation of Polyhedral Oligomeric Silsesquioxane Porphyrin Derivatives on Photodynamic Therapy. Molecules 2020, 25, 4965. [Google Scholar] [CrossRef] [PubMed]
- Rocca, J.D.; Werner, M.E.; Kramer, S.A.; Huxford-Phillips, R.C.; Sukumar, R.; Cummings, N.D.; Vivero-Escoto, J.L.; Wang, A.Z.; Lin, W. Polysilsesquioxane nanoparticles for triggered release of cisplatin and effective cancer chemoradiotherapy. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.-R.; Chiu, S.-J.; Chou, H.-C.; Hu, T.-M. An efficient S-NO-polysilsesquioxane nano-platform for the co-delivery of nitric oxide and an anticancer drug. Chem. Commun. 2015, 51, 15649–15652. [Google Scholar] [CrossRef]
- Rocca, J.; Huxford, R.; Comstock-Duggan, E.; Lin, W. Polysilsesquioxane Nanoparticles for Targeted Platin-Based Cancer Chemotherapy by Triggered Release. Angew. Chem. (Int. Ed. Engl.) 2011, 50, 10330–10334. [Google Scholar] [CrossRef]
- Cai, J.J.; Zheng, Q.P.; Huang, H.F.; Li, B.H. 5-aminolevulinic acid mediated photodynamic therapy inhibits survival activity and promotes apoptosis of A375 and A431 cells. Photodiagnosis Photodyn. Ther. 2018, 21, 257–262. [Google Scholar] [CrossRef]
- Li, Z.; Wang, C.F.; Deng, H.H.; Wu, J.M.; Huang, H.; Sun, R.; Zhang, H.B.; Xiong, X.X.; Feng, M. Robust Photodynamic Therapy Using 5-ALA-Incorporated Nanocomplexes Cures Metastatic Melanoma through Priming of CD4(+)CD8(+) Double Positive T Cells. Adv. Sci. 2019, 6, 1802057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, S.M.; Oleinick, N.L. Dissociation of mitochondrial depolarization from cytochrome c release during apoptosis induced by photodynamic therapy. Br. J. Cancer 2001, 84, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.-Y.; Chiu, S.-M.; Oleinick, N.L. Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene 2001, 20, 3420–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessel, D. Promotion of PDT efficacy by a Bcl-2 antagonist. Photochem. Photobiol. 2008, 84, 809–814. [Google Scholar] [CrossRef] [Green Version]
- Juneja, R.; Vadarevu, H.; Halman, J.; Tarannum, M.; Rackley, L.; Dobbs, J.; Marquez, J.; Chandler, M.; Afonin, K.; Vivero-Escoto, J.L. Combination of Nucleic Acid and Mesoporous Silica Nanoparticles: Optimization and Therapeutic Performance In Vitro. ACS Appl. Mater. Interfaces 2020, 12, 38873–38886. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Salvati, A.; Boya, P. Lysosome-dependent cell death and deregulated autophagy induced by amine-modified polystyrene nanoparticles. Open Biol. 2018, 8, 170271. [Google Scholar] [CrossRef] [Green Version]
- Akter, S.; Inai, M.; Saito, S.; Honda, N.; Hazama, H.; Nishikawa, T.; Kaneda, Y.; Awazu, K. Photodynamic therapy by lysosomal-targeted drug delivery using talaporfin sodium incorporated into inactivated virus particles. Laser Ther. 2019, 28, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girotti, A.W. Photosensitized oxidation of membrane lipids: Reaction pathways, cytotoxic effects, and cytoprotective mechanisms. J. Photochem. Photobiol. B-Biol. 2001, 63, 103–113. [Google Scholar] [CrossRef]
- Farmer, E.E.; Mueller, M.J. ROS-Mediated Lipid Peroxidation and RES-Activated Signaling. Annu. Rev. Plant Biol. 2013, 64, 429–450. [Google Scholar] [CrossRef]
- Suryo Rahmanto, A.; Pattison, D.I.; Davies, M.J. Photo-oxidation-induced inactivation of the selenium-containing protective enzymes thioredoxin reductase and glutathione peroxidase. Free Radic. Biol. Med. 2012, 53, 1308–1316. [Google Scholar] [CrossRef]
- Sakharov, D.V.; Elstak, E.D.R.; Chernyak, B.; Wirtz, K.W.A. Prolonged lipid oxidation after photodynamic treatment. Study with oxidation-sensitive probe C11-BODIPY581/591. FEBS Lett. 2005, 579, 1255–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, T.; Kobayashi, S.; Fujii, J. Induction of ferroptosis by singlet oxygen generated from naphthalene endoperoxide. Biochem. Biophys. Res. Commun. 2019, 518, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 2016, 11, 977–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vadarevu, H.; Juneja, R.; Lyles, Z.; Vivero-Escoto, J.L. Light-Activated Protoporphyrin IX-Based Polysilsesquioxane Nanoparticles Induce Ferroptosis in Melanoma Cells. Nanomaterials 2021, 11, 2324. https://doi.org/10.3390/nano11092324
Vadarevu H, Juneja R, Lyles Z, Vivero-Escoto JL. Light-Activated Protoporphyrin IX-Based Polysilsesquioxane Nanoparticles Induce Ferroptosis in Melanoma Cells. Nanomaterials. 2021; 11(9):2324. https://doi.org/10.3390/nano11092324
Chicago/Turabian StyleVadarevu, Hemapriyadarshini, Ridhima Juneja, Zachary Lyles, and Juan L. Vivero-Escoto. 2021. "Light-Activated Protoporphyrin IX-Based Polysilsesquioxane Nanoparticles Induce Ferroptosis in Melanoma Cells" Nanomaterials 11, no. 9: 2324. https://doi.org/10.3390/nano11092324
APA StyleVadarevu, H., Juneja, R., Lyles, Z., & Vivero-Escoto, J. L. (2021). Light-Activated Protoporphyrin IX-Based Polysilsesquioxane Nanoparticles Induce Ferroptosis in Melanoma Cells. Nanomaterials, 11(9), 2324. https://doi.org/10.3390/nano11092324