
Structure–Activity Relationships between the State of Silver on Different Supports and Their I2 and CH3I Adsorption Properties



Abstract
1. Introduction
2. Materials and Methods
2.1. Parent Substrates and Silver Impregnation
2.1.1. Synthesis of Parent SBA-15
2.1.2. Other Substrates
2.1.3. Preparation of Silver-Impregnated Materials
2.2. Characterization Techniques
2.3. Iodine Adsorption Tests
2.3.1. I2 adsorption Tests
2.3.2. CH3I Adsorption Tests
3. Results and Discussion
3.1. Characterization of Silver-Impregnated Adsorbents
3.1.1. Silver Content
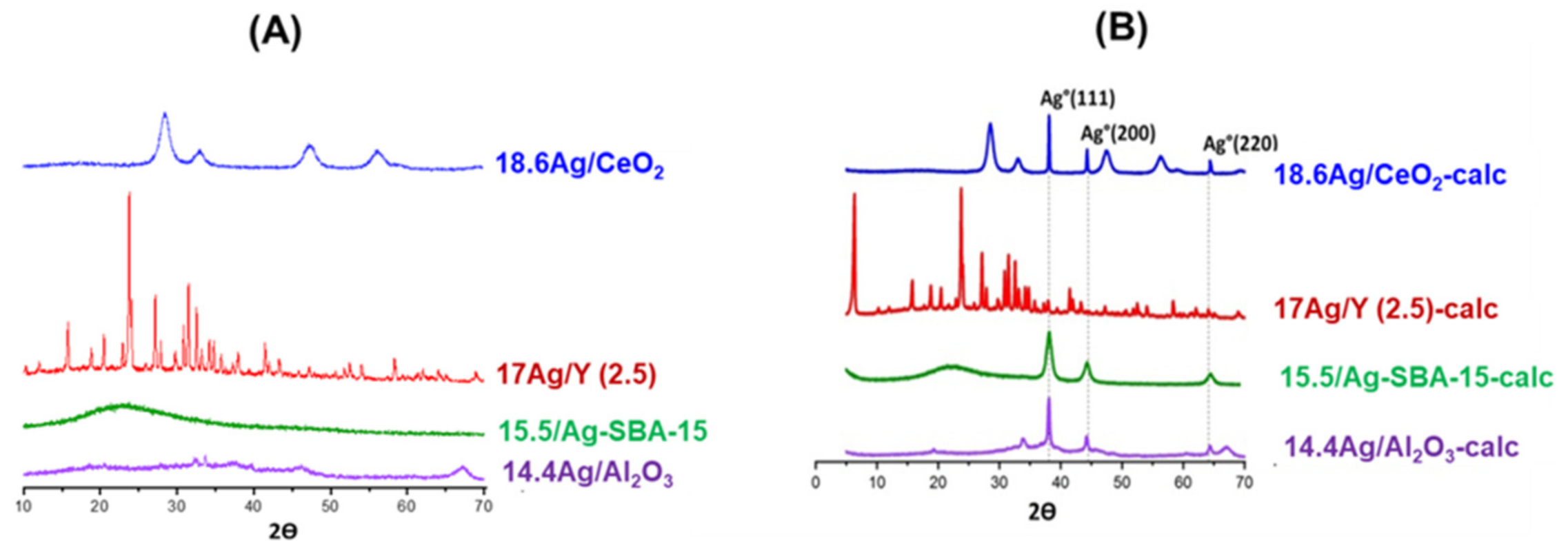
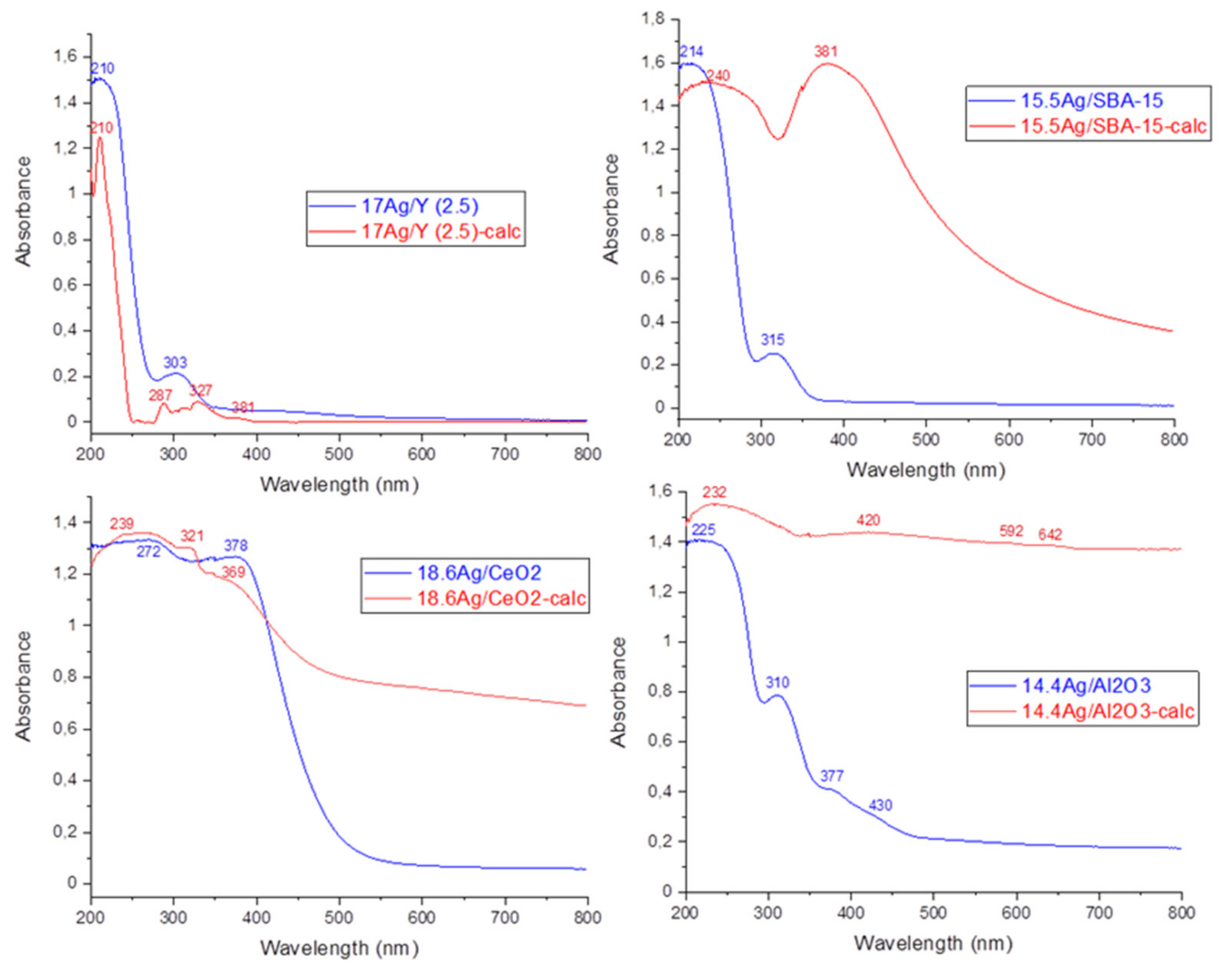
3.1.2. Study of Silver Speciation and Dispersion before and after Calcination by X-Ray Diffraction (XRD) and DR-UV-Visible Spectroscopy (DRS-UV-Vis)
3.2. Adsorption Properties for Molecular Iodine and Iodomethane
Comparison of Adsorption Capacities
3.3. Characterizations Performed on the Spent Adsorbents after I2 and CH3I Tests
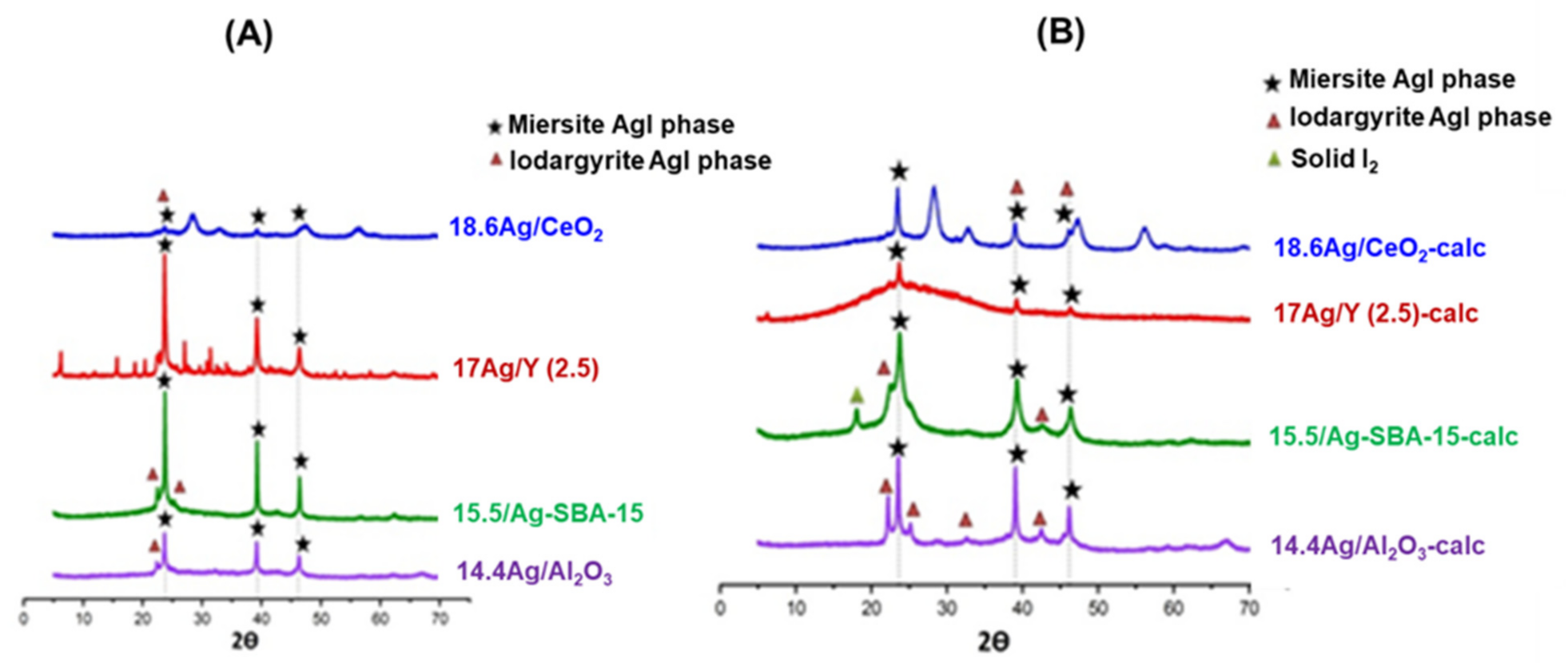
3.3.1. X-Ray Diffraction Analyses of AgI Precipitates
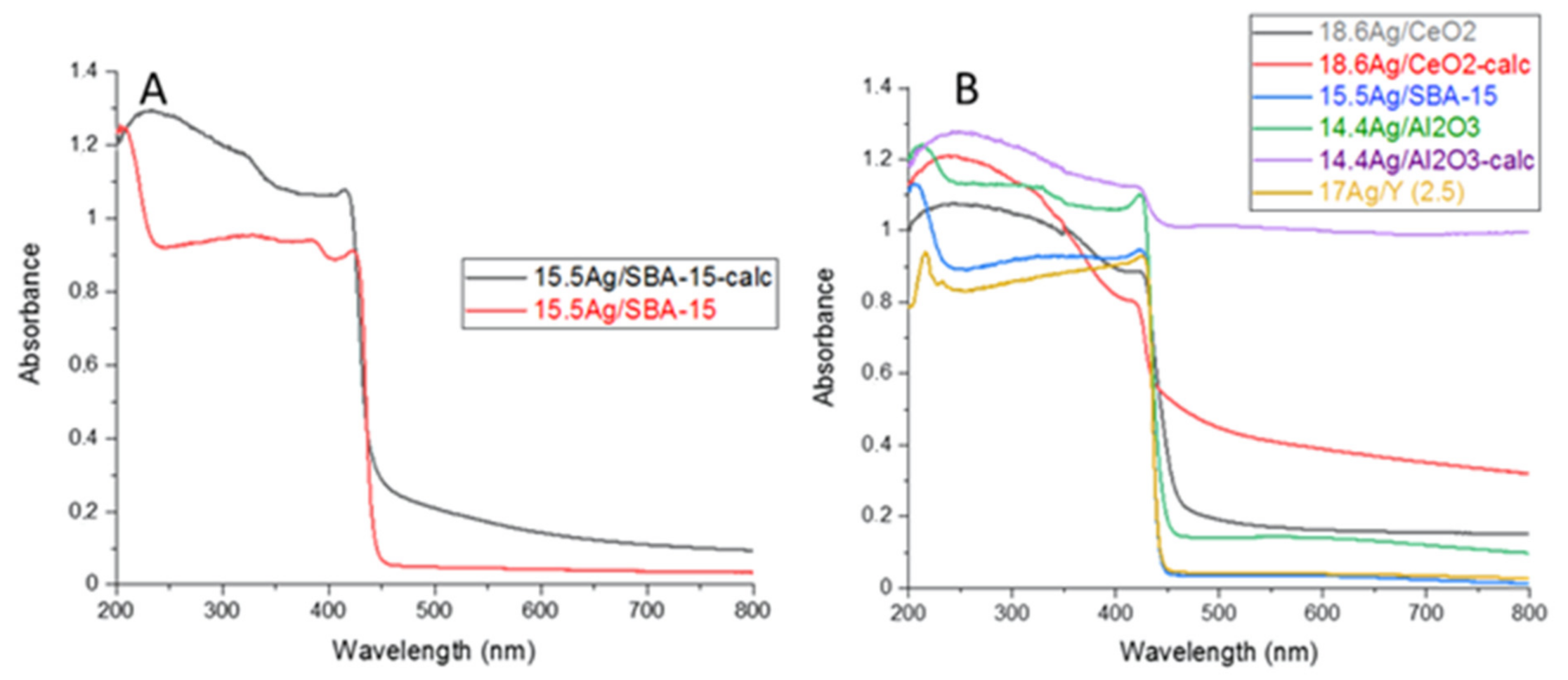
3.3.2. DRS-UV-Vis and TEM Analyses of AgI Precipitates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clement, B.; Cantrel, L.; Ducros, G.; Funke, F.; Herranz, L.; Rydl, A.; Weber, G.; Wren, C. State of the art report on iodine chemistry. In OECD Report, NEA/CSNI/R; OECD: Paris, France, 2007. [Google Scholar]
- Haefner, D.R.; Tranter, T.J. Methods of Gas Phase Capture of Iodine from Fuel Reprocessing Off-Gas: A Literature Survey. In INL/EXT-07-12299; Idaho National Laboratory: Idaho Falls, ID, USA, 2007. [Google Scholar]
- Cantrel, L.; Herranz, L.E.; Guieu, S.; Albiol, T.; Collet, R.; Lind, T.; Karkela, T.; Mun, C.; Jacquemain, D.; Chebbi, M. Overview of ongoing and planned R&D works on delayed releases and FCVS efficiencies. In Proceedings of the ICAPP 2015, Nice, France, 3–6 May 2015. [Google Scholar]
- Ikemoto, T.; Magara, Y. Measures against impacts of nuclear disaster on drinking water supply systems in Japan. Water Pract. Technol. 2011, 6. [Google Scholar] [CrossRef]
- Maeck, W.J.; Pence, D.T.; Keller, J.H. A Highly Efficient Inorganic Adsorber for Airborne Iodine Species (Silver Zeolites Development Studies); Idaho Nuclear Corporation: Idaho Falls, ID, USA, 1969. [Google Scholar]
- Pence, D.T.; Duce, F.A.; Maeck, W.J.; First, M.W. Developments in the Removal of Airborne Iodine Species with Metal Substituted Zeolites. In Proceedings of the 12th AEC Air Cleaning Conference, Oak Ridge, TN, USA, 28–31 August 1972; AEC: Washington, DC, USA, 1972. [Google Scholar]
- Thomas, T.R.; Staples, B.A.; Murphy, L.P.; Nichols, J.T. Airborne Elemental Iodine Loading Capacities of Metal Exchanged Zeolites and a Method for Recycling Silver Zeolites; Idaho National Engineering Laboratory: Idaho Falls, ID, USA, 1977. [Google Scholar]
- Jubin, R.T. A Literature Survey of Methods to Remove Iodine from Off-Gas Streams Using Solid Sorbents; Report ORNL/TM-6607; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1979. [Google Scholar]
- Herranz, L.E.; Lind, T.; Dieschbourg, K.; Riera, E.; Morandi, S.; Rantanen, P.; Chebbi, M.; Losch, N. State of the Art Report: Technical Bases for Experimentation on Source Term Mitigation Systems. In Proceedings of the 10th International Topical Meeting on Nuclear Thermal Hydraulics, Operation and Safety (NUTHOS-10), Okinawa, Japan, 14–21 December 2014. [Google Scholar]
- Huve, J.; Ryzhikov, A.; Nouali, H.; Lalia, V.; Augé, G.; Daou, T.J. Porous sorbents for the capture of radioactive iodine compounds: A review. RSC Adv. 2018, 8, 29248–29273. [Google Scholar] [CrossRef]
- Chebbi, M. Piégeage D’espèces Iodées Volatiles sur des Adsorbants Poreux de Type Zéolithique dans le Contexte d’un Accident Nucléaire Grave. Ph.D. Thesis, Université de Lorraine, Lorraine, France, 2016. [Google Scholar]
- Chebbi, M.; Azambre, B.; Cantrel, L.; Koch, A. A combined DRIFTS and DR-UV–Vis spectroscopic in situ study on the trapping of CH3I by silver-exchanged faujasite zeolite. J. Phys. Chem. C 2016, 120, 18694–18706. [Google Scholar] [CrossRef]
- Chebbi, M.; Chibani, S.; Paul, J.F.; Cantrel, L.; Badawi, M. Evaluation of volatile iodine trapping in presence of contaminants: A periodic DFT study on cation exchanged-faujasite. Microporous Mesoporous Mater. 2017, 239, 111–122. [Google Scholar] [CrossRef]
- Chebbi, M.; Azambre, B.; Cantrel, L.; Huvé, M.; Albiol, T. Influence of structural, textural and chemical parameters of silver zeolites on the retention of methyl iodide. Microporous Mesoporous Mater. 2017, 244, 137–150. [Google Scholar] [CrossRef]
- Azambre, B.; Chebbi, M. Evaluation of silver zeolites sorbents toward their ability to promote stable CH3I storage as AgI Precipitates. ACS Appl. Mater. Interfaces 2017, 9, 25194–25203. [Google Scholar] [CrossRef] [PubMed]
- Azambre, B.; Chebbi, M.; Leroy, O.; Cantrel, L. Effects of zeolitic parameters and irradiation on the retention properties of silver zeolites exposed to molecular iodine. Ind. Eng. Chem. Res. 2018, 57, 1468–1479. [Google Scholar] [CrossRef]
- Azambre, B.; Chebbi, M.; Hijazi, A. Effects of the cation and Si/Al ratio on CH3I adsorption by faujasite zeolites. Chem. Eng. J. 2020, 379, 122308. [Google Scholar] [CrossRef]
- Chebbi, M.; Azambre, B.; Monsanglant-Louvet, C.; Marcillaud, B.; Roynette, A.; Cantrel, L. Effects of water vapour and temperature on the retention of radiotoxic CH3I by silver faujasite zeolites. J. Hazard. Mater. 2021, 409, 124947. [Google Scholar] [CrossRef] [PubMed]
- Jacquemain, D.; Guentay, S.; Basu, S.; Sonnenkalb, M.; Lebel, L.; Allelein, H.J.; Liebana, B.; Eckardt, B.; Ammirabile, L. Status Report on Filtered Containment Venting. In OECD/NEA/CSNI, Report NEA/ CSNI/R; OECD: Paris, France, 2014. [Google Scholar]
- Herrmann, F.J.; Herrmann, B.; Hoeflich, V.; Beyer, C.H.; Furrer, J. Removal Efficiency of Silver Impregnated Filter Materials and Performance of Iodine Filters in of the Off-Gases of the Karlsruhe Reprocessing Plant WAK. In Proceedings of the 24th DOE/NRC Nuclear Air Cleaning and Treatment Conference, Portland, OR, USA, 15–18 July 1996. [Google Scholar]
- Fukasawa, T.; Funabashi, K.; Kondo, Y. Influences of impurities on iodine removal efficiency. In Proceedings of the 24th DOE/NRC Nuclear Air Cleaning Conference and Air Treatment, Portland, OR, USA, 15–18 July 1996. [Google Scholar]
- Wilhelm, J.G.; Furrer, J. Head-end iodine removal from a reprocessing plant with a solid sorbent. In Proceedings of the ERDA 14th Air Cleaning Conference, CONF720823, Springfield, VA, USA, 28–31 August 1977. [Google Scholar]
- IAEA Report. Treatment, Conditioning and Disposal of Iodine-129; Technical Reports Series No. 276; International Atomic Energy Agency: Vienna, Austria, 1987. [Google Scholar]
- Matyáš, J.; Fryxell, G.E.; Busche, B.J.; Wallace, K.; Fifield, L.S. Ceramic Materials for Energy Applications: Ceramic Engineering and Science; Lin, H.-T., Katoh, Y., Fox, K.M., Belharouak, I., Widjaja, S., Singh, D., Eds.; Wiley-American Ceramic Society: Daytona Beach, FL, USA, 2011; pp. 23–33. [Google Scholar]
- Mnasri, N.; Charnay, C.; Ménoval, L.; Moussaoui, Y.; Elaloui, E.; Zajac, J. Silver nanoparticle-containing submicron-in-size mesoporous silica-based systems for iodine entrapment and immobilization from gas phase. Microporous Mesoporous Mater. 2014, 196, 305–313. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, A.; Azambre, B.; Finqueneisel, G.; Vibert, F.; Blin, J.L. High iodine adsorption by polyethyleneimine impregnated nanosilica sorbents. Microporous Mesoporous Mater. 2019, 288, 109586. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic Properties of Ceria and CeO2-Containing Materials. Catal. Rev. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Flanigen, E.M. Introduction to Zeolite Science and Practice, Studies in Surface Science and Catalysis; Van Bekkum, H., Flanigen, E.M., Jansen, J.C., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; p. 58. [Google Scholar]
- Bartolomeu, R.; Azambre, B.; Westermann, A.; Fernandes, A.; Bertolo, R.; Issa Hamoud, H.; Henriques, C.; Da Costa, P. Investigation of the nature of silver species on different Ag-containing NOx reduction catalysts: On the effect of the support. Appl. Catal. B Environ. 2014, 150–151, 204–217. [Google Scholar] [CrossRef]
- Salles, N. Etude des Différents Polymorphes de L’Alumine et des Phases Transitoires Apparaissant lors des Premiers Stades D’Oxydation de L’Aluminium: Simulation à L’Échelle Atomique par un Modèle à Charges Variables en Liaisons Fortes. Ph.D. Thesis, Université de Bourgogne, Dijon, France, 2014. [Google Scholar]
- Kanipandian, N.; Kannan, S.; Ramesh, R.; Subramanian, P.; Thirumurugan, R. Characterization, antioxidant and cytotoxicity evaluation of green synthesized silver nanoparticles using Cleistanthus collinus extract as surface modifier. Mater. Res. 2014, 49, 494–502. [Google Scholar] [CrossRef]
- Sayah, E.; Brouri, D.; Wu, Y.; Musi, A.; Da Costa, P.; Massiani, P. A comparative in situ TEM and UV—Visible spectroscopic study of the thermal evolution of Ag species dispersed on Al2O3 and NaX zeolite supports. Appl. Catal. A Gen. 2011, 406, 94–101. [Google Scholar] [CrossRef]
- Aspromonte, S.G.; Mizrahi, M.D.; Schneeberger, F.A.; Lopez, J.M.R.; Boix, A.V. Study of the nature and location of Silver in Ag-exchanged mordenite catalysts. Characterization by spectroscopic techniques. J. Phys. Chem. C 2013, 117, 25433–25442. [Google Scholar] [CrossRef]
- Shibata, J.; Takada, Y.; Shichi, A.; Satokawa, S.; Satsuma, A.; Hattori, T. Influence of zeolite support on activity enhancement by addition of hydrogen for SCR of NO by propane over Ag-zeolites. Appl. Catal. B Environ. 2004, 54, 137–144. [Google Scholar] [CrossRef]
- Baker, M.D.; Ozin, G.A.; Godber, J. Far-infrared studies of silver atoms, silver ions, and silver clusters in zeolites A and Y. J. Phys. Chem. 1985, 89, 305–311. [Google Scholar] [CrossRef]
- Funke, F.; Greger, G.-U.; Bleier, A.; Hellmann, S.; Morell, W. The Reaction between Iodine and Silver under Severe PWR Accident Conditions. An Experimental Parameter Study. In PSI-97-02, 28035729 Report; IAEA: Vienna, Austria, 1996. [Google Scholar]
- Andryushechkin, B.V.; Zhidomirov, G.M.; Eltsov, K.N.; Hladchanka, Y.V.; Korlyukov, A.A. Local structure of the Ag(100) surface reacting with molecular iodine: Experimental and theoretical study. Phys. Rev. B 2009, 80, 125409. [Google Scholar] [CrossRef]
- Bruffey, S.H.; Jubin, R.T.; Jordan, J.A. Capture of elemental and organic iodine from dilute gas streams by silver-exchanged mordenite. Proc. Chem. 2016, 21, 293–299. [Google Scholar] [CrossRef]
- Zhou, X.-L.; Solymosi, F.; Blass, P.M.; Cannon, K.C.; White, J.M. Interactions of methyl halides (Cl, Br, and I) with Ag(111). Surf. Sci. 1989, 219, 294–316. [Google Scholar] [CrossRef]
- Chapman, K.W.; Chupas, P.J.; Nenoff, T.M. Radioactive iodine capture in silver-containing mordenites through nanoscale silver iodide formation. J. Am. Chem. Soc. 2010, 132, 8897–8899. [Google Scholar] [CrossRef] [PubMed]
- Kodaira, T.; Ikeda, T.; Takeo, H. Optical and X-ray diffraction study of AgI clusters incorporated into zeolite LTA. Eur. Phys. J. D 1999, 9, 601–604. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbent | %Ag | Silver Speciation |
|---|---|---|
| 14.4 Ag/Al2O3 | 14.4 | Ag+, Agnδ+, Ag°m, traces of Ag° |
| 14.4 Ag/Al2O3-calc | 14.4 | Ag° (27 nm) |
| 15.5 Ag/SBA-15 | 15.5 | Ag+, Agnδ+, Ag°m, traces of Ag° |
| 15.5Ag/SBA-15-calc | 15.5 | Ag° (9 nm) |
| 17 Ag/Y (2.5) | 17 | Ag+, Agnδ+, Ag°m, traces of Ag° |
| 17Ag/Y (2.5)-calc | 17 | Ag+, Agnδ+, Ag°m |
| 18.6 Ag/CeO2 | 18.6 | n.d. |
| 18.6Ag/CeO2-calc | 18.6 | Ag° (106 nm) |
| Adsorbent | QI2 (mg/g) | I/Ag | Color after I2 Exposure | Detected Phases by XRD | D AgI Miersite Phase (nm) |
|---|---|---|---|---|---|
| 14.4 Ag/Al2O3 | 177 | 1.04 | Yellow | AgI Miersite AgI iodargyrite | 37 |
| 14.4 Ag/Al2O3-calc | 196 | 1.16 | Black | AgI Miersite AgI iodargyrite | 49 |
| 15.5 Ag/SBA-15 | 159 | 0.86 | Yellow | AgI Miersite AgI iodargyrite | 64 |
| 15.5Ag/SBA-15-calc | 172 | 0.94 | Yellow | AgI Miersite AgI iodargyrite | 16 |
| 17 Ag/Y (2.5) | 205 | 1.02 | Pale yellow | AgI Miersite | 37 |
| 17Ag/Y (2.5)-calc | 208 | 1.03 | Pale yellow | AgI Miersite | 35 |
| 18.6 Ag/CeO2 | 224 | 1.02 | Yellow | Small peaks AgI Miersite AgI iodargyrite | 16 |
| 18.6Ag/CeO2-calc | 253 | 1.15 | Grey | AgI Miersite AgI iodargyrite | 26 |
| Adsorbent | QCH3I (mg/g) | I/Ag | Detected Phases by XRD | D AgI Miersite Phase (nm) | D Ag° (nm) |
|---|---|---|---|---|---|
| 14.4 Ag/Al2O3 | 122 | 0.64 | AgI Miersite AgI iodargyrite | 44 | **** |
| 14.4 Ag/Al2O3-calc | 38 | 0.2 | Ag° nanoparticles | Small AgI peaks | 28 |
| 15.5 Ag/SBA-15 | 143 | 0.7 | AgI Miersite AgI iodargyrite | 73 | **** |
| 15.5 Ag/SBA-15-calc | 61 | 0.3 | Ag° nanoparticles | Small AgI peaks | 30 |
| 17 Ag/Y (2.5) | 150 | 0.67 | AgI Miersite | 73 | **** |
| 17 Ag/Y (2.5)-calc | 144 | 0.64 | AgI Miersite | 58 | **** |
| 18.6 Ag/CeO2 | 189 | 0.77 | AgI Miersite AgI iodargyrite | 39 | **** |
| 18.6 Ag/CeO2-calc | 22 | 0.09 | Ag° nanoparticles | **** | 29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azambre, B.; Chebbi, M.; Ibrahim, N. Structure–Activity Relationships between the State of Silver on Different Supports and Their I2 and CH3I Adsorption Properties. Nanomaterials 2021, 11, 1300. https://doi.org/10.3390/nano11051300
Azambre B, Chebbi M, Ibrahim N. Structure–Activity Relationships between the State of Silver on Different Supports and Their I2 and CH3I Adsorption Properties. Nanomaterials. 2021; 11(5):1300. https://doi.org/10.3390/nano11051300
Chicago/Turabian StyleAzambre, Bruno, Mouheb Chebbi, and Nagham Ibrahim. 2021. "Structure–Activity Relationships between the State of Silver on Different Supports and Their I2 and CH3I Adsorption Properties" Nanomaterials 11, no. 5: 1300. https://doi.org/10.3390/nano11051300
APA StyleAzambre, B., Chebbi, M., & Ibrahim, N. (2021). Structure–Activity Relationships between the State of Silver on Different Supports and Their I2 and CH3I Adsorption Properties. Nanomaterials, 11(5), 1300. https://doi.org/10.3390/nano11051300