
Deviation of Trypsin Activity Using Peptide Conformational Imprints


 ,
, 

Abstract
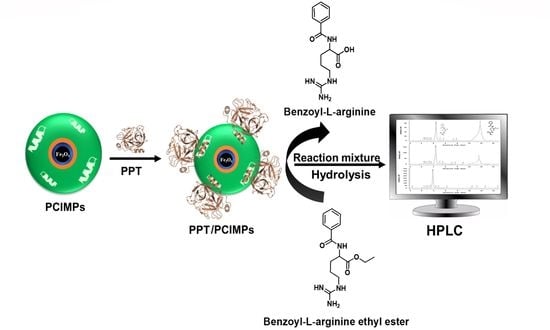
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
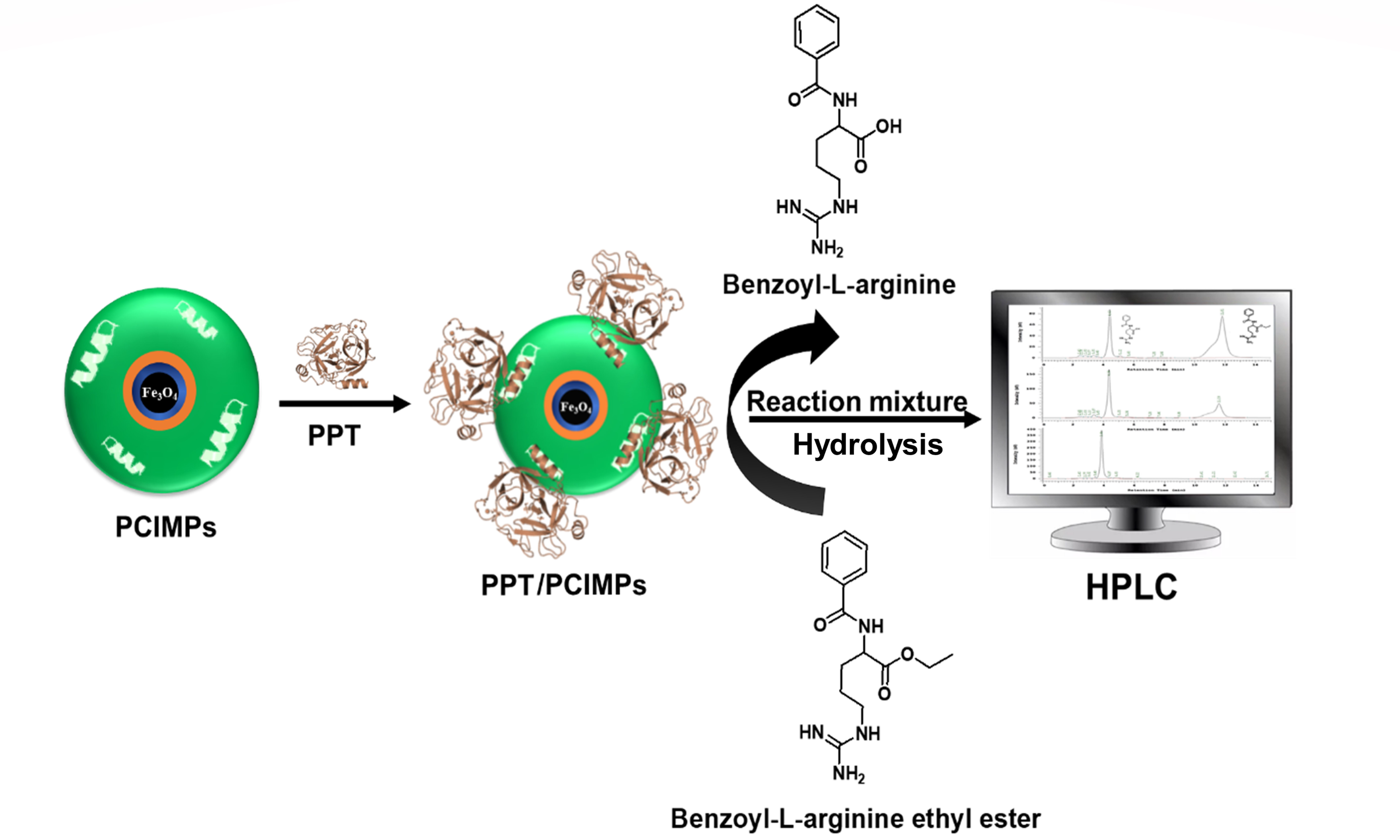
2.2. Template Synthesis
2.3. Preparation of PCIs on MPs
2.3.1. Construction of Fe3O4@APTMS-GA
2.3.2. Synthesis of Fe3O4@APTMS-GA-Acrylate
2.3.3. Preparation of PCIMPs
2.4. Determination of Binding Affinities of PCIMPs
2.5. Activity Assay of PPT and Immobilized PPT (PPT/PCIMPs)
2.6. PPT/PCIMPs Activity Assay
2.7. Determination of Kinetic Constants of PPT and PPT/PCIMPs
3. Results and Discussions
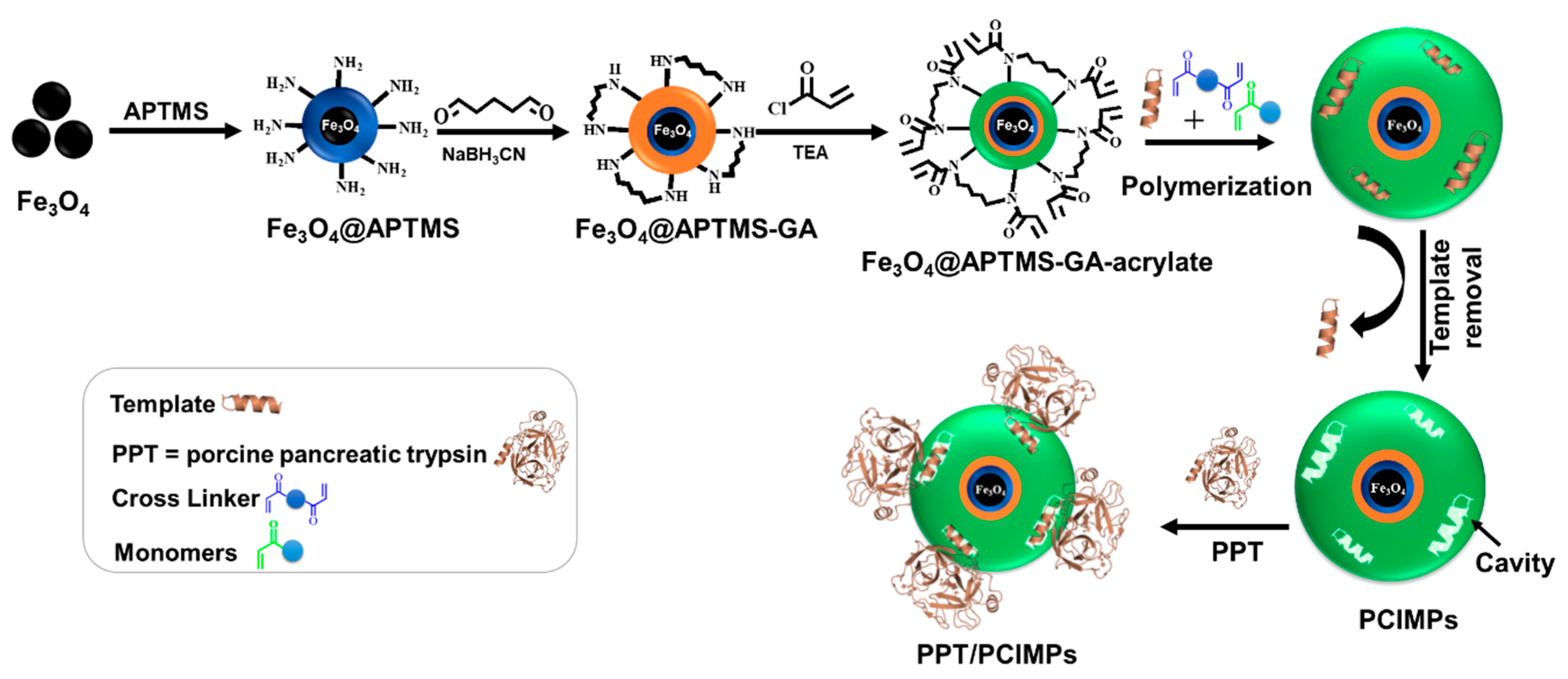
3.1. Rational Selection of the Template
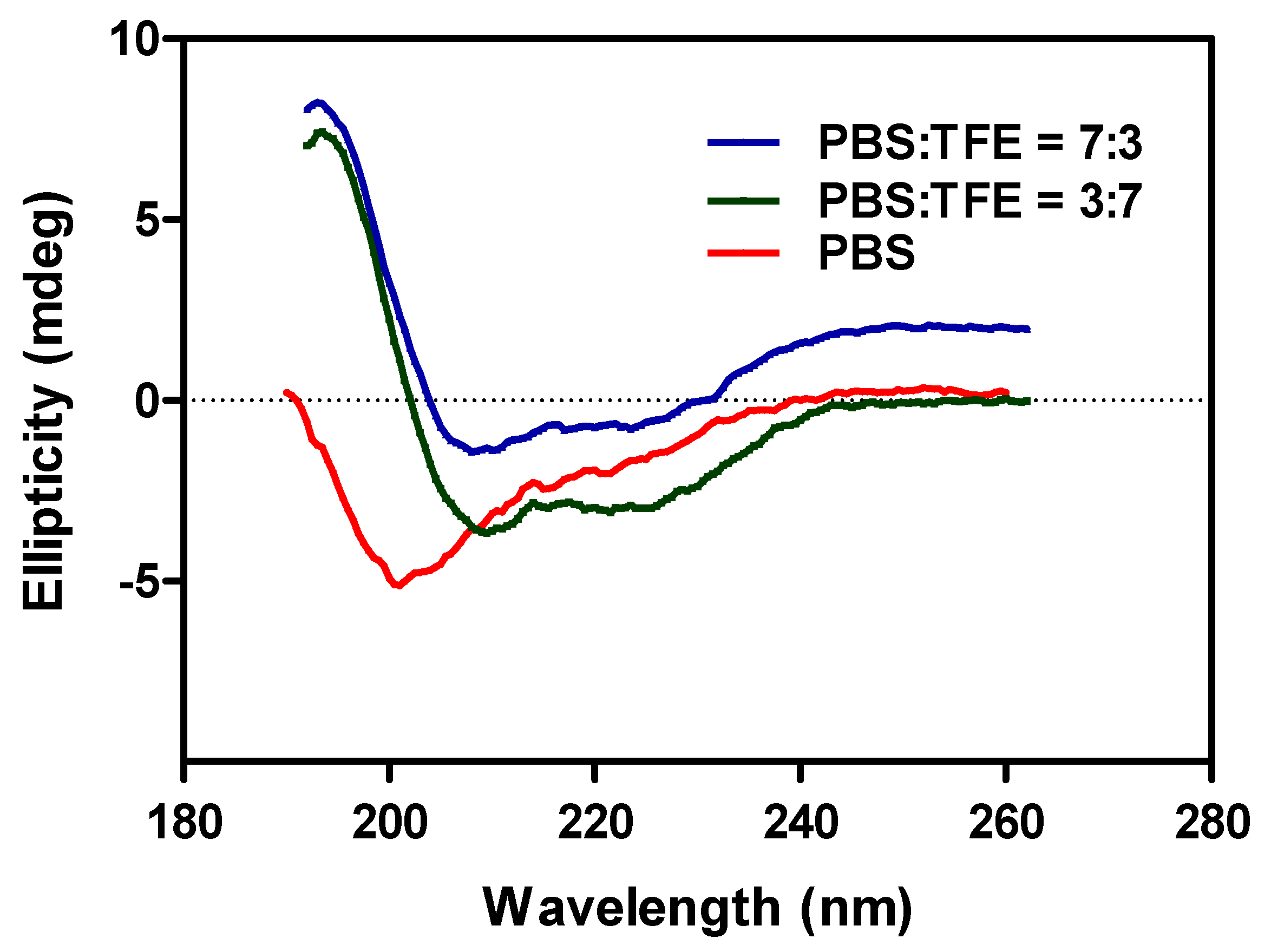
3.2. Analysis of the Template
3.3. Characterization of MPs and PCIMPs
3.3.1. FTIR Analysis
3.3.2. FE-SEM Analysis
3.4. Binding Studies of PCIMPs
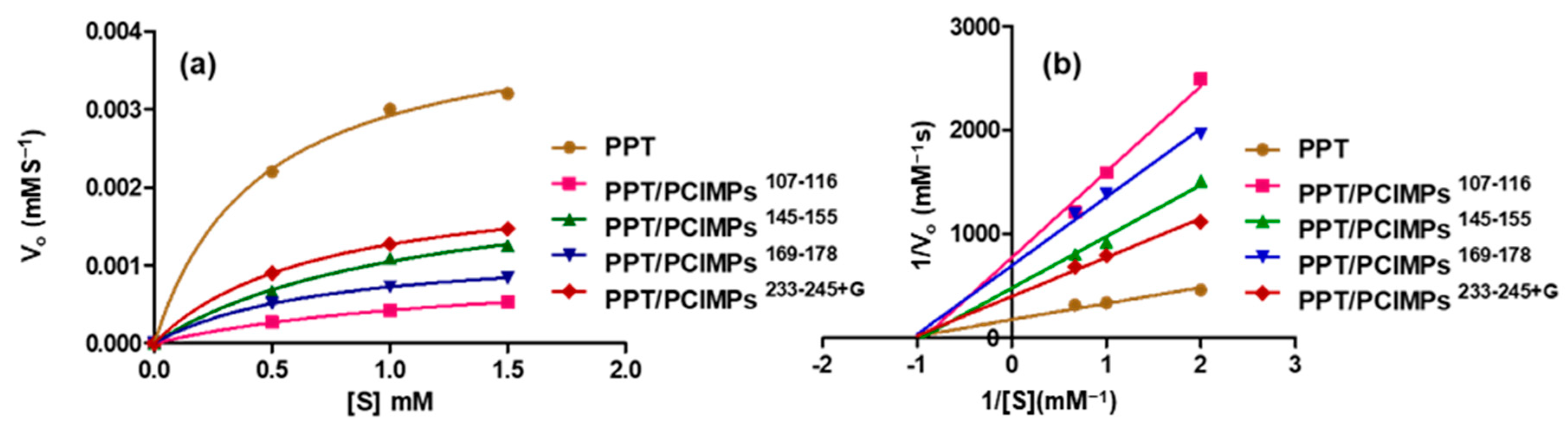
3.5. Kinetic Parameters of PPT and PPT/PCIMPs
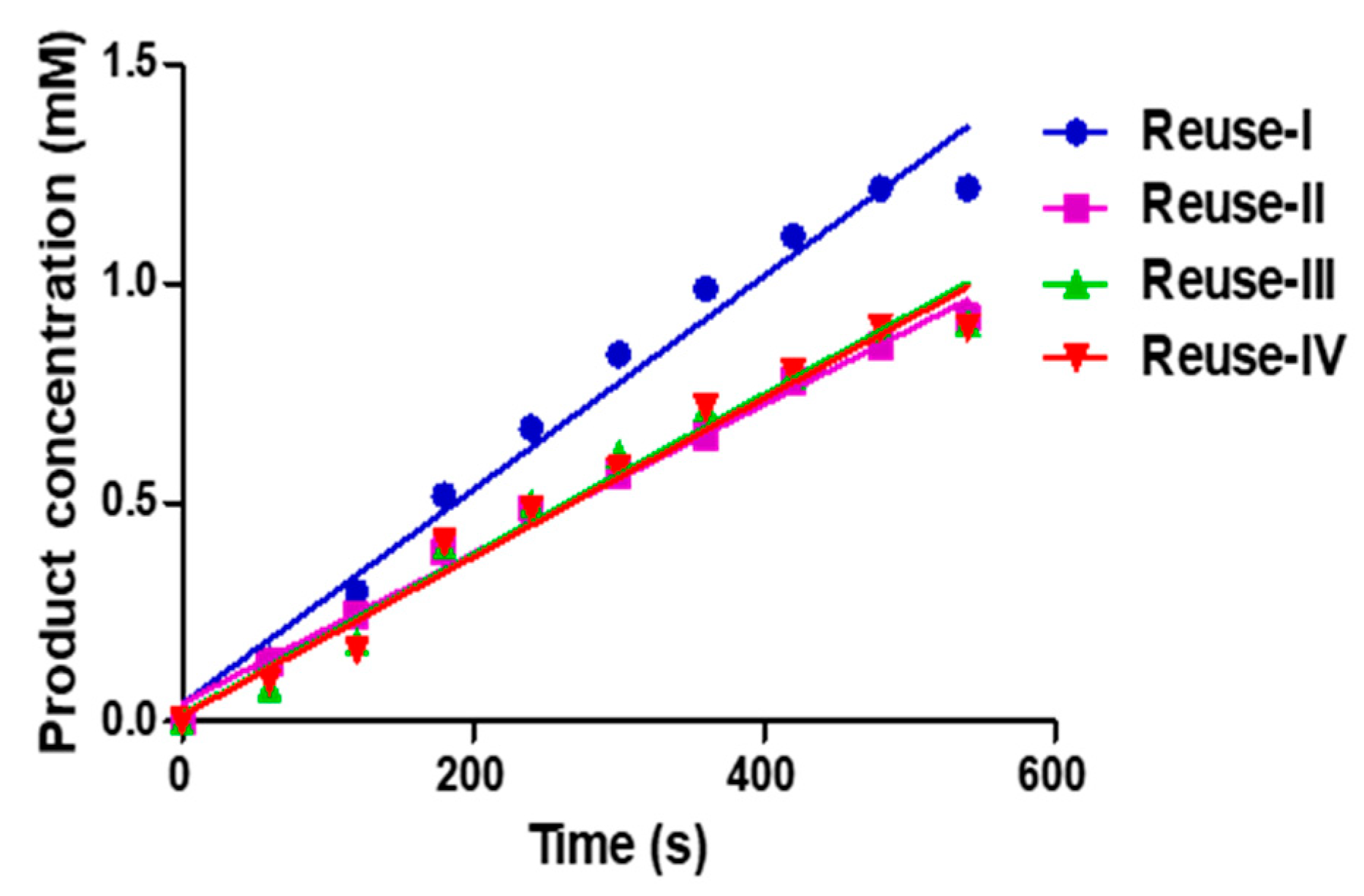
3.6. Reusability
3.7. Comparison Studies of the Proposed PPT/PCIMPs with Other Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Atacan, K.; Çakıroğlu, B.; Özacar, M. Covalent immobilization of trypsin onto modified magnetite nanoparticles and its application for casein digestion. Int. J. Biol. Macromol. 2017, 97, 148–155. [Google Scholar] [CrossRef]
- Perutka, Z.; Šebela, M. Pseudotrypsin: A Little-Known Trypsin Proteoform. Molecules 2018, 23, 2637. [Google Scholar] [CrossRef] [PubMed]
- Vorob’Ev, M.M. Proteolysis of β-lactoglobulin by Trypsin: Simulation by Two-Step Model and Experimental Verification by Intrinsic Tryptophan Fluorescence. Symmetry 2019, 11, 153. [Google Scholar] [CrossRef]
- Siddiqui, I.; Husain, Q. Stabilization of polydopamine modified silver nanoparticles bound trypsin: Insights on protein hydrolysis. Colloids Surf. B Biointerfaces 2019, 173, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Sasai, Y.; Kanno, H.; Doi, N.; Yamauchi, Y.; Kuzuya, M.; Kondo, S.-I. Synthesis and Characterization of Highly Stabilized Polymer–Trypsin Conjugates with Autolysis Resistance. Catalysts 2016, 7, 4. [Google Scholar] [CrossRef]
- Aslani, E.; Abri, A.; Pazhang, M. Immobilization of trypsin onto Fe3O4@SiO2-NH2 and study of its activity and stability. Colloids Surf. B Biointerfaces 2018, 170, 553–562. [Google Scholar] [CrossRef]
- Sanchez, A.; Cruz, J.; Rueda, N.; Dos Santos, J.C.S.; Torres, R.; Ortiz, C.; Villalonga, R.; Fernandez-Lafuente, R. Inactivation of immobilized trypsin under dissimilar conditions produces trypsin molecules with different structures. RSC Adv. 2016, 6, 27329–27334. [Google Scholar] [CrossRef]
- Stolarow, J.; Heinzelmann, M.; Yeremchuk, W.; Syldatk, C.; Hausmann, R. Immobilization of trypsin in organic and aqueous media for enzymatic peptide synthesis and hydrolysis reactions. BMC Biotechnol. 2015, 15, 77. [Google Scholar] [CrossRef]
- Kankala, R.K.; Zhang, H.; Liu, C.; Kanubaddi, K.R.; Lee, C.; Wang, S.; Cui, W.; Santosaf, H.A.; Lin, K.; Chen, A. Metal Species–Encapsulated Mesoporous Silica Nanoparticles: Current Advancements and Latest Breakthroughs. Adv. Funct. Mater. 2019, 29, 1902652. [Google Scholar] [CrossRef]
- Wahab, R.A.; Elias, N.; Abdullah, F.; Ghoshal, S.K. On the taught new tricks of enzymes immobilization: An all-inclusive overview. React. Funct. Polym. 2020, 152, 104613. [Google Scholar] [CrossRef]
- Homaei, A.; Sariri, R.; Vianello, F.; Stevanato, R. Enzyme immobilization: An update. J. Chem. Biol. 2013, 6, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Reis, C.; Sousa, E.; Serpa, J.; Oliveira, R.; Santos, J. Design of immobilized enzyme biocatalysts: Drawbacks and opportunities. Quím. Nova 2019, 42, 768–783. [Google Scholar] [CrossRef]
- Liu, C.; Saeki, D.; Matsuyama, H. A novel strategy to immobilize enzymes on microporous membranes via dicarboxylic acid halides. RSC Adv. 2017, 7, 48199–48207. [Google Scholar] [CrossRef]
- Xing, R.; Ma, Y.; Wang, Y.; Wen, Y.; Liu, Z. Specific recognition of proteins and peptides via controllable oriented surface imprinting of boronate affinity-anchored epitopes. Chem. Sci. 2019, 10, 1831–1835. [Google Scholar] [CrossRef]
- Ansell, R.J.; Mosbach, K. Magnetic molecularly imprinted polymer beads for drug radioligand binding assay. Analyst 1998, 123, 1611–1616. [Google Scholar] [CrossRef]
- Tan, C.J. and Y.W. Tong, Preparation of Superparamagnetic Ribonuclease A Surface-Imprinted Submicrometer Particles for Protein Recognition in Aqueous Media. Anal. Chem. 2007, 79, 299–306. [Google Scholar] [CrossRef]
- Jing, T.; Du, H.; Dai, Q.; Xia, H.; Niu, J.; Hao, Q.; Mei, S.; Zhou, Y. Magnetic molecularly imprinted nanoparticles for recognition of lysozyme. Biosens. Bioelectron. 2010, 26, 301–306. [Google Scholar] [CrossRef]
- Guerreiro, A.; Poma, A.; Karim, K.; Moczko, E.; Takarada, J.; De Vargas-Sansalvador, I.P.; Turner, N.; Piletska, E.; De Magalhães, C.S.; Glazova, N.; et al. Influence of Surface-Imprinted Nanoparticles on Trypsin Activity. Adv. Healthc. Mater. 2014, 3, 1426–1429. [Google Scholar] [CrossRef]
- Tai, D.-F.; Jhang, M.-H.; Chen, G.-Y.; Wang, S.-C.; Lu, K.-H.; Lee, Y.-D.; Liu, H.-T. Epitope-Cavities Generated by Molecularly Imprinted Films Measure the Coincident Response to Anthrax Protective Antigen and Its Segments. Anal. Chem. 2010, 82, 2290–2293. [Google Scholar] [CrossRef]
- Tai, D.-F.; Ho, Y.-F.; Wu, C.-H.; Lin, T.-C.; Lu, K.-H.; Lin, K.-S. Artificial-epitope mapping for CK-MB assay. Analyst 2011, 136, 2230–2233. [Google Scholar] [CrossRef]
- Chou, C.-Y.; Lin, C.-Y.; Wu, C.-H.; Tai, D.-F. Sensing HIV Protease and Its Inhibitor Using “Helical Epitope”—Imprinted Polymers. Sensors 2020, 20, 3592. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.M.; Porter, K.A.; Singh, S.K.; Vanier, G.S. High-Efficiency Solid Phase Peptide Synthesis (HE-SPPS). Org. Lett. 2014, 16, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhu, Y.; Luo, B.; Lan, F.; Wu, Y.; Gu, Z. pH-Responsive magnetic nanospheres for the reversibly selective capture and release of glycoproteins. J. Mater. Chem. B 2017, 5, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Xing, Y.; Radosz, M.; Shen, Y. Magnetic Nanoparticle Supported Catalyst for Atom Transfer Radical Polymerization. Macromolecules 2006, 39, 6399–6405. [Google Scholar] [CrossRef]
- Ellwanger, A.; Karlsson, L.; Owens, P.K.; Berggren, C.; Crecenzi, C.; Ensing, K.; Bayoudh, S.; Cormack, P.A.; Sherrington, D.; Sellergren, B. Evaluation of methods aimed at complete removal of template from molecularly imprinted polymers. Analyst 2001, 126, 784–792. [Google Scholar] [CrossRef]
- Lorenzo, R.A.; Carro, A.M.; Alvarez-Lorenzo, C.; Concheiro, A. To remove or not to remove? The challenge of extracting the template to make the cavities available in Molecularly Imprinted Polymers (MIPs). Int. J. Mol. Sci. 2011, 12, 4327–4347. [Google Scholar] [CrossRef]
- Gerdon, A.E.; Wright, D.W.; Cliffel, D.E. Quartz Crystal Microbalance Detection of Glutathione-Protected Nanoclusters Using Antibody Recognition. Anal. Chem. 2005, 77, 304–310. [Google Scholar] [CrossRef]
- Diltemiz, S.E.; Hür, D.; Ersöz, A.; Denizli, A.; Say, R. Designing of MIP based QCM sensor having thymine recognition sites based on biomimicking DNA approach. Biosens. Bioelectron. 2009, 25, 599–603. [Google Scholar] [CrossRef]
- Tai, D.-F.; Lin, Y.-F.; Lu, K.-H.; Chen, G.-Y.; Shu, H.-C. A Direct Immersion System for Peptide Enrichment. J. Chin. Chem. Soc. 2012, 59, 338–344. [Google Scholar] [CrossRef]
- Bossi, A.M.; Sharma, P.S.; Montana, L.; Zoccatelli, G.; Laub, O.; Levi, R. Fingerprint-Imprinted Polymer: Rational Selection of Peptide Epitope Templates for the Determination of Proteins by Molecularly Imprinted Polymers. Anal. Chem. 2012, 84, 4036–4041. [Google Scholar] [CrossRef]
- Van Rosmalen, M.; Krom, M.; Merkx, M. Tuning the Flexibility of Glycine-Serine Linkers To Allow Rational Design of Multidomain Proteins. Biochemistry 2017, 56, 6565–6574. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Sharma, M.; Zhou, H.-X.; Cross, T.A. Glycines: Role in α-Helical Membrane Protein Structures and a Potential Indicator of Native Conformation. Biochemistry 2012, 51, 4779–4789. [Google Scholar] [CrossRef] [PubMed]
- Transue, T.R.; Krahn, J.M.; Gabel, S.A.; Derose, A.E.F.; London, R.E. X-ray and NMR Characterization of Covalent Complexes of Trypsin, Borate, and Alcohols. Biochemistry 2004, 43, 2829–2839. [Google Scholar] [CrossRef] [PubMed]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Thyparambil, A.A.; Latour, R.A. Protein helical structure determination using CD spectroscopy for solutions with strong background absorbance from 190 to 230 nm. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2014, 1844, 2331–2337. [Google Scholar] [CrossRef] [PubMed]
- Farjadian, F.; Ghasemi, S.; Mohammadi-Samani, S.; Ghasemia, S. Hydroxyl-modified magnetite nanoparticles as novel carrier for delivery of methotrexate. Int. J. Pharm. 2016, 504, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yi, Y.; Zhao, J.; Xia, Y.; Jiang, M.; Cao, F.; Zhou, H.; Wei, P.; Jia, H.; Yong, X. Co-immobilization of laccase and TEMPO onto amino-functionalized magnetic Fe3O4 nanoparticles and its application in acid fuchsin decolorization. Bioresour. Bioprocess. 2018, 5, 27. [Google Scholar] [CrossRef]
- Kurtan, U.; Baykal, A. Fabrication and characterization of Fe3O4@APTES@PAMAM-Ag highly active and recyclable magnetic nanocatalyst: Catalytic reduction of 4-nitrophenol. Mater. Res. Bull. 2014, 60, 79–87. [Google Scholar] [CrossRef]
- Farjadian, F.; Hosseini, M.; Ghasemi, S.; Tamami, B. Phosphinite-functionalized silica and hexagonal mesoporous silica containing palladium nanoparticles in Heck coupling reaction: Synthesis, characterization, and catalytic activity. RSC Adv. 2015, 5, 79976–79987. [Google Scholar] [CrossRef]
- Iijima, M.; Kamiya, H. Surface Modification for Improving the Stability of Nanoparticles in Liquid Media. KONA Powder Part. J. 2009, 27, 119–129. [Google Scholar] [CrossRef]
- Boitard, C.; Lamouri, A.; Menager, C.; Griffete, N. Whole Protein Imprinting over Magnetic Nanoparticles Using Photopolymerization. ACS Appl. Polym. Mater. 2019, 1, 928–932. [Google Scholar] [CrossRef]
- Xu, J.; Prost, E.; Haupt, K.; Bui, B.T.S. Direct and sensitive determination of trypsin in human urine using a water-soluble signaling fluorescent molecularly imprinted polymer nanoprobe. Sens. Actuators B Chem. 2018, 258, 10–17. [Google Scholar] [CrossRef]
- Lin, H.; Zhang, C.; Lin, Y.; Chang, Y.; Crommen, J.; Wang, Q.; Jiang, Z.; Guo, J. A strategy for screening trypsin inhibitors from traditional Chinese medicine based on a monolithic capillary immobilized enzyme reactor coupled with offline liquid chromatography and mass spectrometry. J. Sep. Sci. 2019, 42, 1980–1989. [Google Scholar] [CrossRef] [PubMed]
- Bayramoglu, G.; Çelikbıçak, Ö.; Arica, Y.; Salih, B. Trypsin Immobilized on Magnetic Beads via Click Chemistry: Fast Proteolysis of Proteins in a Microbioreactor for MALDI-ToF-MS Peptide Analysis. Ind. Eng. Chem. Res. 2014, 53, 4554–4564. [Google Scholar] [CrossRef]
- Zdarta, J.; Antecka, K.; Jędrzak, A.; Synoradzki, K.; Łuczak, M.; Jesionowski, T. Biopolymers conjugated with magnetite as support materials for trypsin immobilization and protein digestion. Colloids Surf. B Biointerfaces 2018, 169, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Arica, M.Y.; Şenel, S.; Alaeddinoğlu, N.G.; Patir, S.; Denizli, A. Invertase immobilized on spacer-arm attached poly(hydroxyethyl methacrylate) membrane: Preparation and properties. J. Appl. Polym. Sci. 2000, 75, 1685–1692. [Google Scholar] [CrossRef]
- Homaei, A. Enhanced activity and stability of papain immobilized on CNBr-activated sepharose. Int. J. Biol. Macromol. 2015, 75, 373–377. [Google Scholar] [CrossRef]
- Shojaei, F.; Homaei, A.; Taherizadeh, M.R.; Kamrani, E. Characterization of biosynthesized chitosan nanoparticles from Penaeus vannamei for the immobilization of P. vannamei protease: An eco-friendly nanobiocatalyst. Int. J. Food Prop. 2017, 20 (Suppl. 2), 1413–1423. [Google Scholar]
- Atacan, K.; Çakıroğlu, B.; Özacar, M. Improvement of the stability and activity of immobilized trypsin on modified Fe3O4 magnetic nanoparticles for hydrolysis of bovine serum albumin and its application in the bovine milk. Food Chem. 2016, 212, 460–468. [Google Scholar] [CrossRef]
- Bayramoglu, G.; Yılmaz, M.; Şenel, A. Ülkü; Arıca, M.Y. Preparation of nanofibrous polymer grafted magnetic poly(GMA-MMA)-g-MAA beads for immobilization of trypsin via adsorption. Biochem. Eng. J. 2008, 40, 262–274. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, J.; Zhang, H.; Zhang, Y. Efficient Synthesis of Molecularly Imprinted Polymers with Enzyme Inhibition Potency by the Controlled Surface Imprinting Approach. ACS Macro Lett. 2013, 2, 566–570. [Google Scholar] [CrossRef]
- Blat, Y. Non-Competitive Inhibition by Active Site Binders. Chem. Biol. Drug Des. 2010, 75, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Bayramoglu, G.; Özalp, V.C.; Arica, M.Y. Magnetic Polymeric Beads Functionalized with Different Mixed-Mode Ligands for Reversible Immobilization of Trypsin. Ind. Eng. Chem. Res. 2013, 53, 132–140. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MPs | PCIMPs107–116 | PCIMPs145–155 | PCIMPs169–178 | PCIMPs233–245+G |
|---|---|---|---|---|
| Residue | 10 | 11 | 10 | 14 |
| [Kd] μM | 0.65 | 0.38 | 0.55 | 0.21 |
| [Bmax] nM | 0.75 | 1.11 | 0.95 | 1.11 |
| Bmax/Kd | 1.15 | 2.92 | 1.73 | 5.29 |
| MPs | PPT | PPT/PCIMPs107–116 | PPT/PCIMPs145–155 | PPT/PCIMPs169–178 | PPT/PCIMPs233–245+G |
|---|---|---|---|---|---|
| Vmax (mM s−1) | 3.2 × 10−3 | 0.53 × 10−3 | 1.25 × 10−3 | 0.84 × 10−3 | 1.47 × 10−3 |
| [Km] mM | 0.36 | 0.52 | 0.46 | 0.44 | 0.42 |
| kcat (s−1) | 2.6 | 0.62 | 0.99 | 0.78 | 1.16 |
| kcat/Km (mM−1 s−1) | 7.32 | 1.19 | 2.15 | 1.77 | 2.79 |
| Trypsin/Immobilized Trypsin | Km | Vmax | kcat (s−1) | kcat/Km (mM−1 s−1) | Reference |
|---|---|---|---|---|---|
| BPT/Immobilized BPT | 5.1/7.88 mM | 23/18.3 mM min−1 | - | - | [42] |
| BPT/Immobilized BPT | 9.7/13.6 mM | 5890/3946 U/mg | - | 607/290 | [43] |
| BPT/Immobilized BPT | 9.3/16.8 mM | 7345/5115 U/mg | - | - | [44] |
| PPT and PPT/PCIMPs233–245+G | 0.36/0.42 mM | 3.2 × 10−3/1.47 × 10−3 mM s−1 | 2.6/>1.16 | 7.32/2.79 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanubaddi, K.R.; Huang, P.-Y.; Chang, Y.-L.; Wu, C.H.; Li, W.; Kankala, R.K.; Tai, D.-F.; Lee, C.-H. Deviation of Trypsin Activity Using Peptide Conformational Imprints. Nanomaterials 2021, 11, 334. https://doi.org/10.3390/nano11020334
Kanubaddi KR, Huang P-Y, Chang Y-L, Wu CH, Li W, Kankala RK, Tai D-F, Lee C-H. Deviation of Trypsin Activity Using Peptide Conformational Imprints. Nanomaterials. 2021; 11(2):334. https://doi.org/10.3390/nano11020334
Chicago/Turabian StyleKanubaddi, Kiran Reddy, Pei-Yu Huang, Ya-Lin Chang, Cheng Hsin Wu, Wei Li, Ranjith Kumar Kankala, Dar-Fu Tai, and Chia-Hung Lee. 2021. "Deviation of Trypsin Activity Using Peptide Conformational Imprints" Nanomaterials 11, no. 2: 334. https://doi.org/10.3390/nano11020334
APA StyleKanubaddi, K. R., Huang, P.-Y., Chang, Y.-L., Wu, C. H., Li, W., Kankala, R. K., Tai, D.-F., & Lee, C.-H. (2021). Deviation of Trypsin Activity Using Peptide Conformational Imprints. Nanomaterials, 11(2), 334. https://doi.org/10.3390/nano11020334