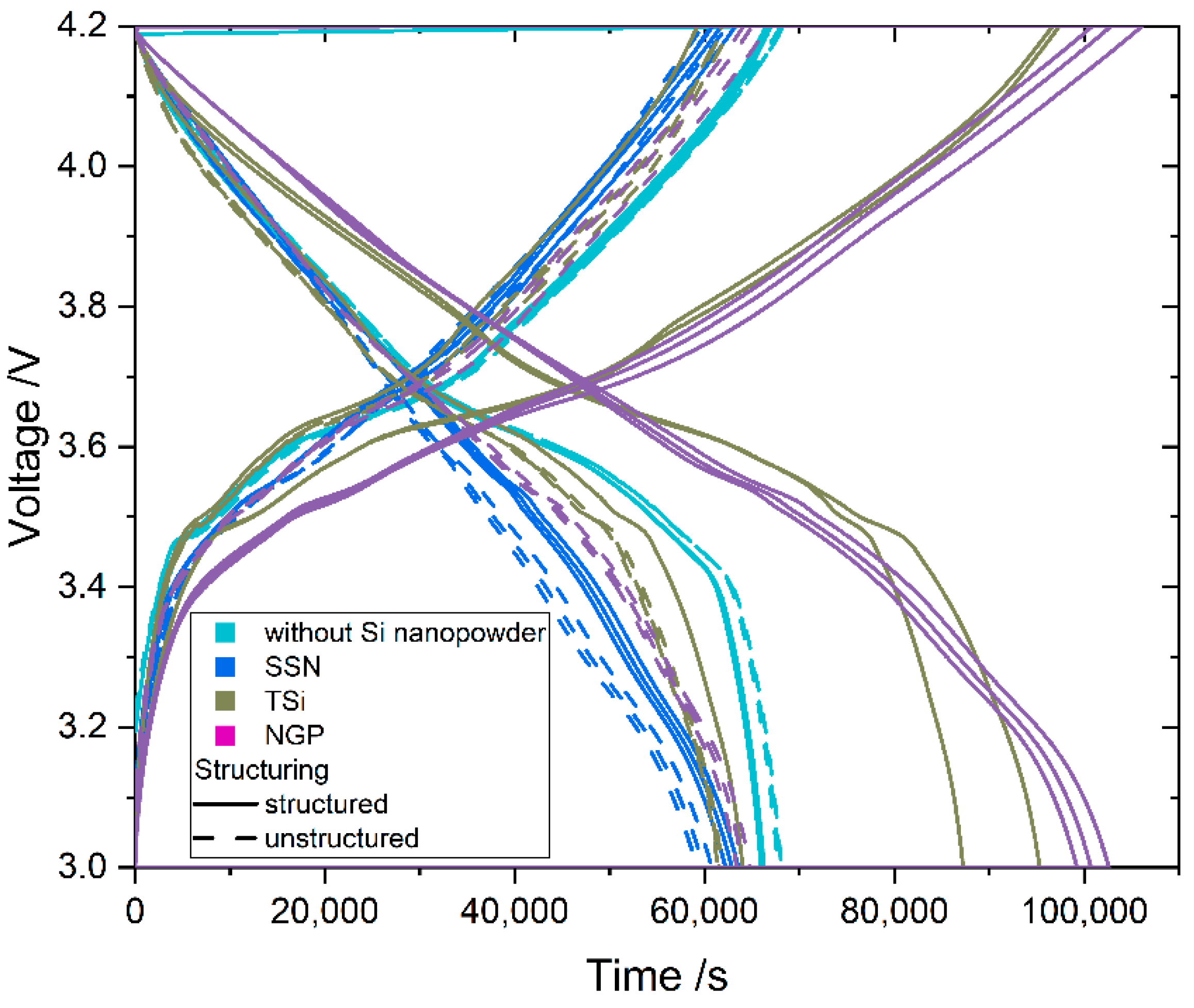
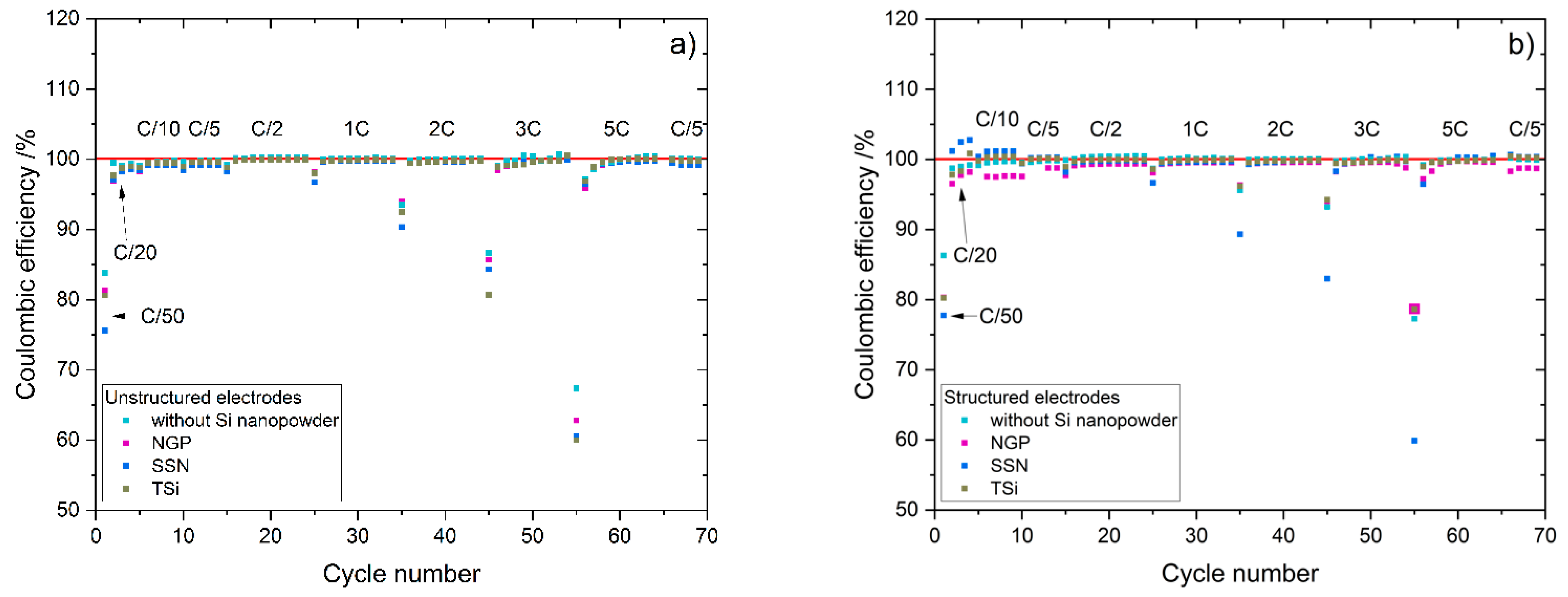
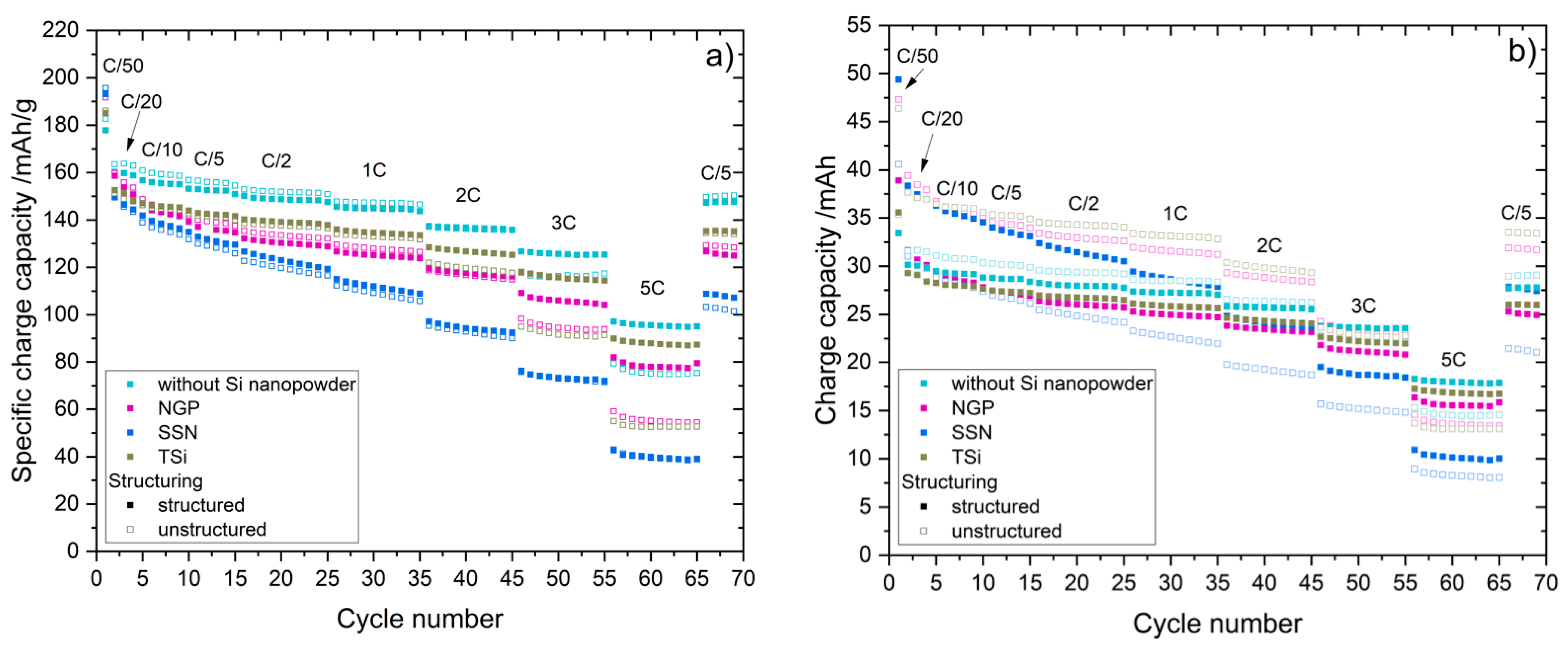
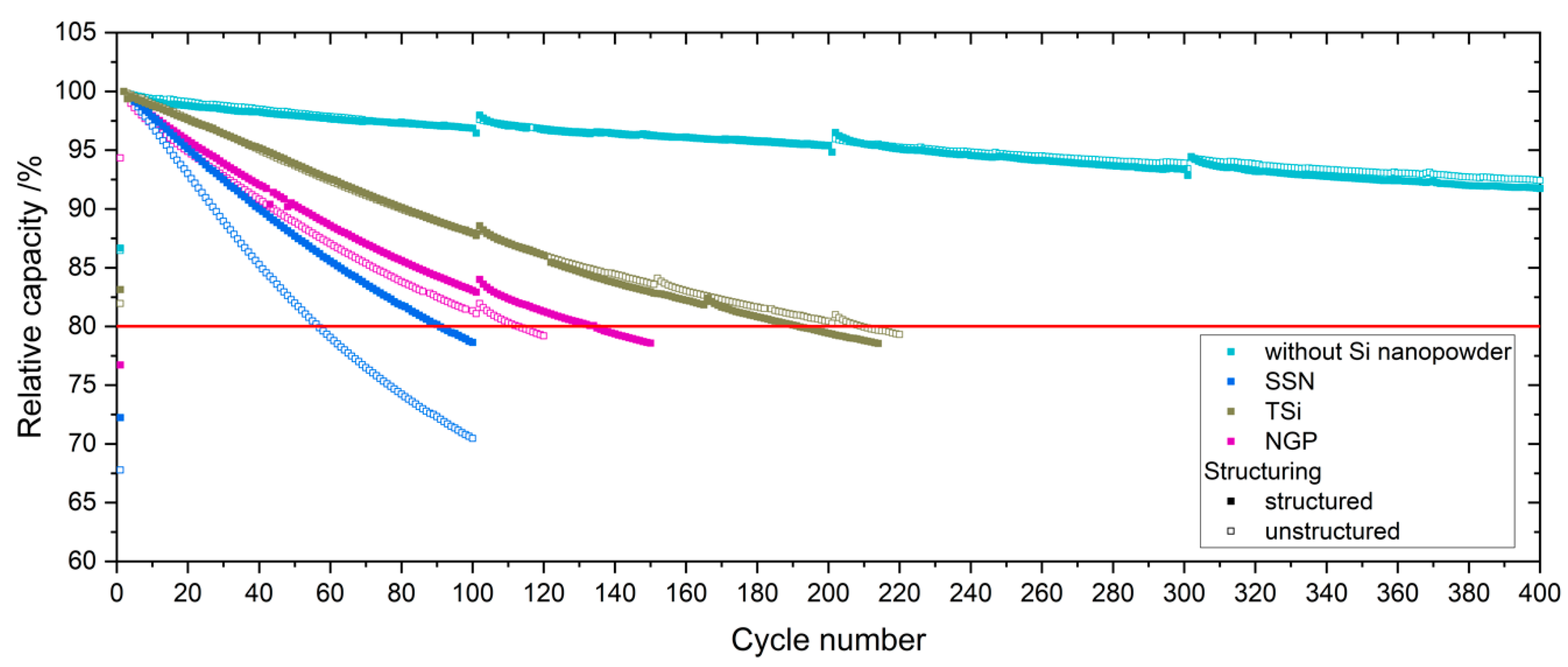
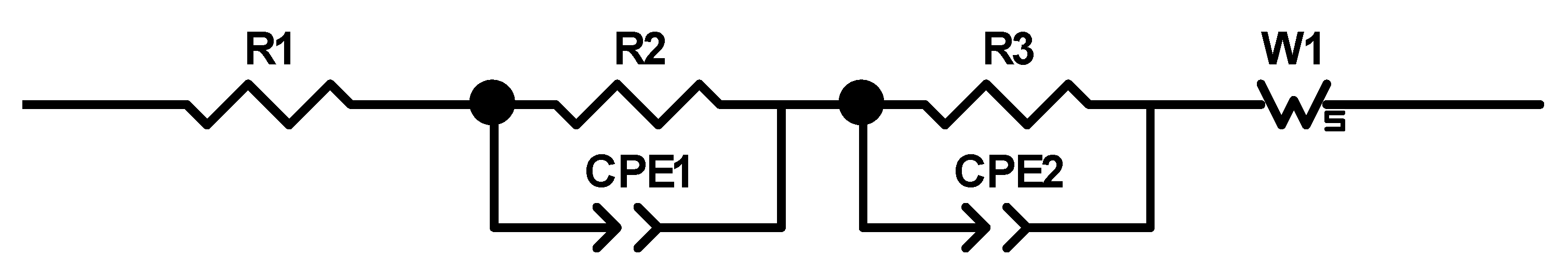
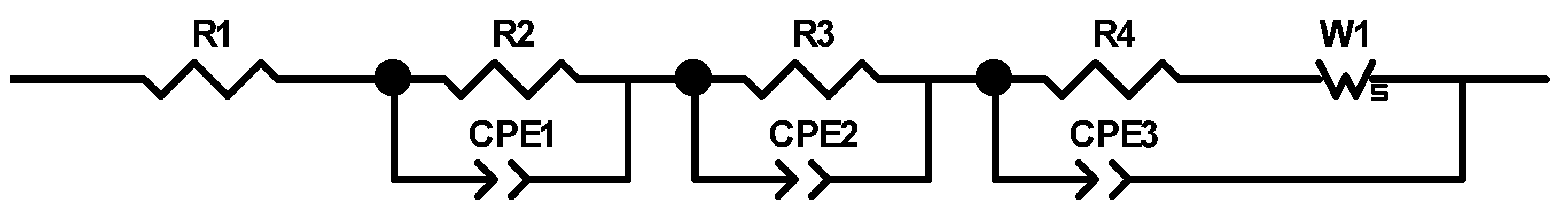
2.1. Electrode Preparation
Silicon/graphite anodes were prepared via ball milling. Three different grades of commercially available SiNPs were used from three different material suppliers, namely manufacturer 1 (TSi), Nanografi Nanotechnology (NGP, Ankara, Turkey), and SkySpring Nanomaterials (SSN, Houston, TX, USA). The grades of silicon varied in purity and grain size, which will be shown later.
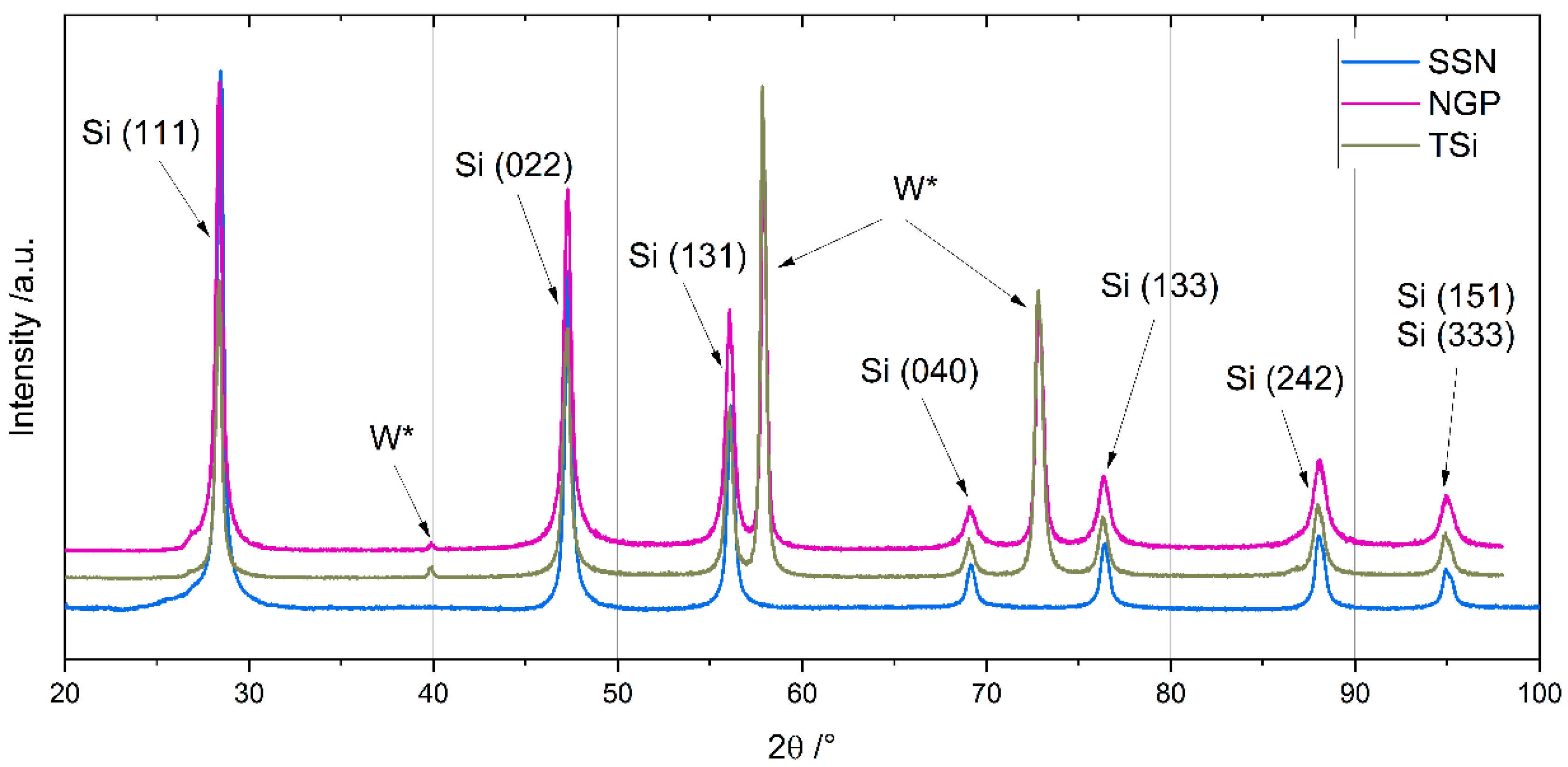
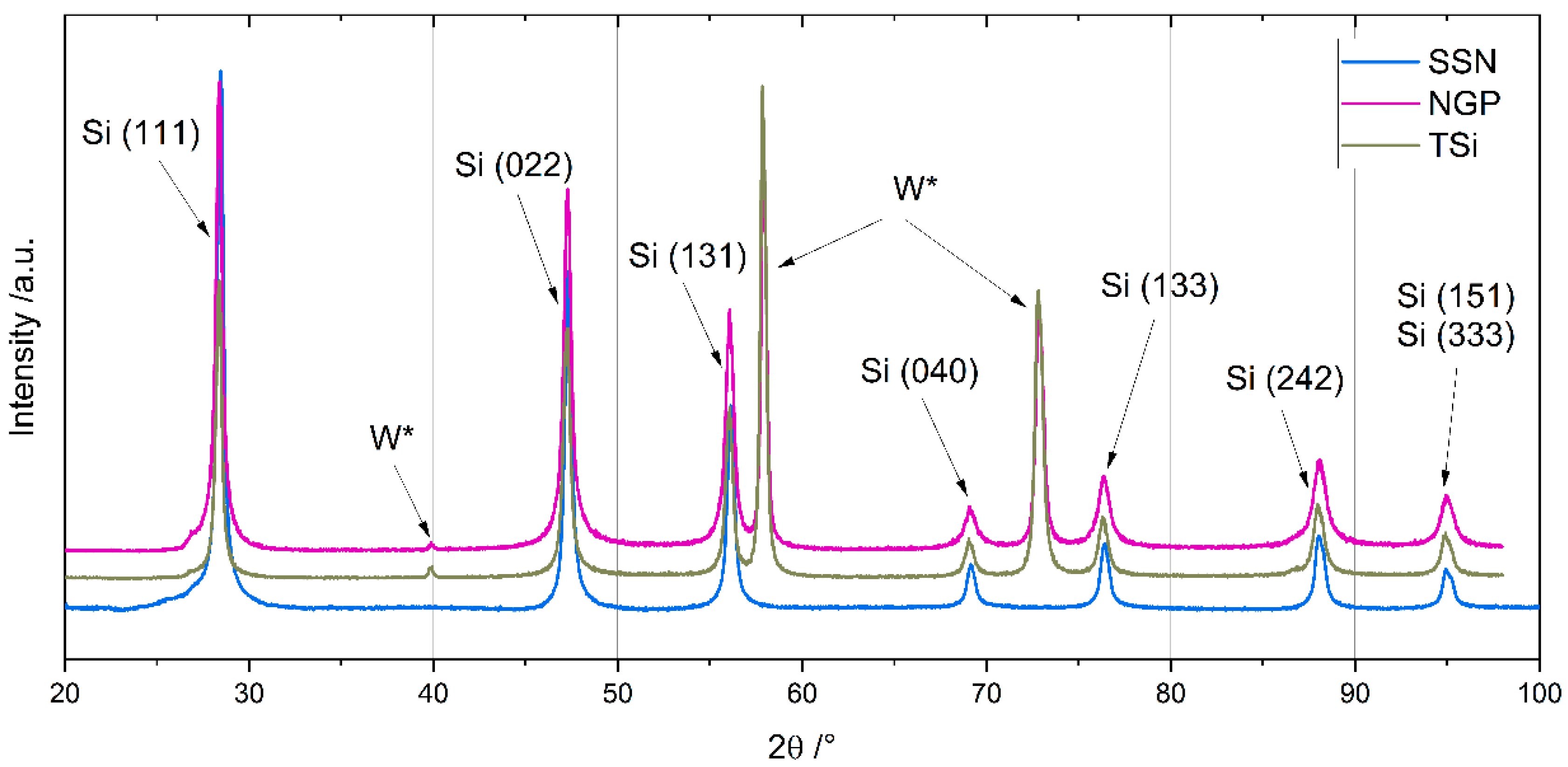
The chemical composition of the Si nanoparticles was measured with inductively coupled plasma optical emission spectroscopy (ICP-OES, iCAP 7600 DUO, Thermo-Fisher-Scientific, Waltham, MA, USA), carrier gas hot extraction (CGHE, G8 Galilei, Bruker AXS, Karlsruhe, Germany), and a carbon and sulfur analyzer (CS analyzer, TC600, LECO Instrumente, Mönchengladbach, Germany). The crystallinity was measured with X-ray diffraction (XRD, Empyrean, Malvern Panalytical, Malvern, UK) under an argon atmosphere in a non-ambient chamber (TTK 600, Malvern Panalytical, Malvern, UK) for diffraction angles between 20° and 100°. Scanning electron microscope (SEM, Phenom Pro, Thermo Fischer Scientific, Waltham, MA, USA) images were prepared to characterize the shape and size of the SiNPs and their agglomerates.
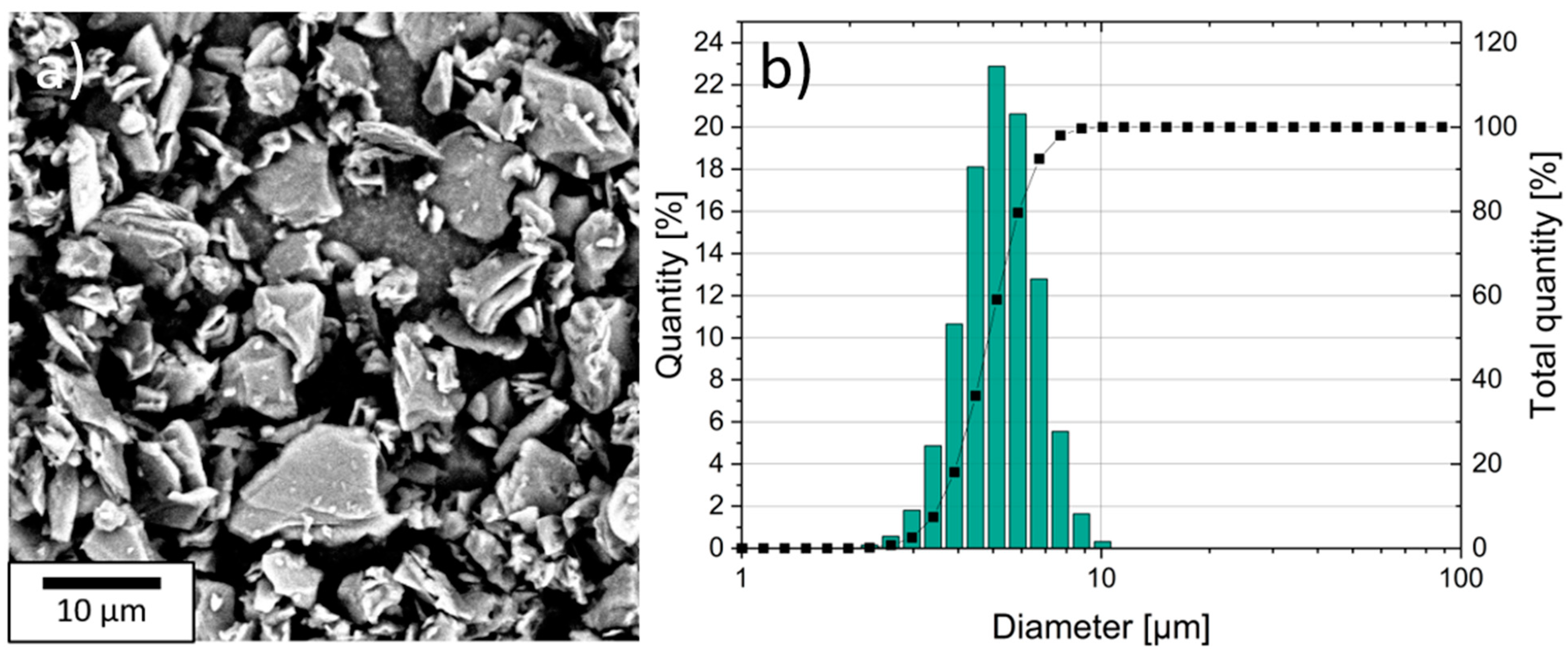
The particle size of the coke-based graphite (SPGPT808, Targray, Kirkland, QC, Canada) was measured with laser scattering (LA-950, Horiba Europe, Oberursel, Germany), and the Brunauer–Emmet–Teller (BET) surface area was determined using nitrogen adsorption (Gemini VII 2390, Micromeritics, Norcross, GA, USA). The graphite particles were also examined using a SEM (Phenom Pro, Thermo Fischer Scientific, Waltham, MA, USA) to characterize their shape and size.
The spatial distribution of the graphite, silicon, and conductive carbon black at the surface of the electrode was examined with a scanning electron microscope (SEM, Zeiss Merlin, Carl Zeiss SMT AG, Oberkochen, Germany) and the distribution of the elements was measured with energy dispersive X-ray spectroscopy (EDX).
The compositions of the cathode and anode slurries are summarized in
Table 1. For the anodes, a water based 2.5 wt.% sodium carboxymethyl cellulose (CMC, MTI Corporation, Richmond, CA, USA) solution was prepared with a vacuum mixer (MSK-SFM-7, MTI Corporation, Richmond, CA, USA) and subsequently stirred for 24 h with a magnetic stirrer. The graphite, conductive carbon black (CB, C-nergy Super C65, Imerys G&C, Paris, France), silicon nanopowder, and CMC solution were then premixed with a centrifugal mixer (Speedmixer DAC 150 SP, Hauschild, Hamm, Germany). The slurry was mixed with a ball mill (PULVERISETTE 7 premium line, Fritsch, Idar-Oberstein, Germany) to achieve a homogeneous distribution of the silicon nanoparticles. Styrene butadiene rubber solution (SBR, 50% solid content, MTI Corporation, Richmond, CA, USA) was added and slowly stirred into the mixture with a centrifugal mixer. The solid content of the anode slurry was 33.33%. The slurry was tape casted on a copper current collector (9 µm thickness) and dried at room temperature. The doctor blade distance was 150 µm. The anodes were subsequently calendered to reach a porosity (
) of 40%, calculated with the following equation:
With being the thickness of the electrode, the weight of the electrode per area, the mass fractions of the materials, and their corresponding densities (2.20 g/cm³ graphite; 2.33 g/cm³ silicon; 1.60 g/cm³ CMC; 1.05 g/cm³ SBR; 1.94 g/cm³ CB; 2.24 g/cm³ KS6L; 1.77 g/cm³ PVDF; and 4.63 g/cm³ NMC 622).
Single crystal NMC 622 (Targray, Kirkland, QC, Canada) cathodes were also prepared. PVDF (Solef 5130, Solvay GmbH, Hannover, Germany) and N-methyl-2-pyrrolidone solvent (NMP, BASF, Ludwigshafen, Germany) were premixed, then the NMC 622, conductive carbon black and conductive graphite were added. Additional NMP was used to adjust the viscosity. The solid content of the slurry was 66.67%. The cathode slurry was mixed in a centrifugal mixer and then tape casted on an aluminum current collector (20 µm thickness) with a doctor blade distance of 85 to 100 µm, depending on the required areal capacity to match the anodes with a cell balancing factor of 1.1 to 1.3. For the calculation of the cell balancing factor, a practical specific capacity of 172 mAh/g was used for the NMC 622, and 330 mAh/g was denoted for the graphite. In the applied voltage window between 3.0 and 4.2 V, a practical capacity of 2000 mAh/g was used for the silicon [
4,
25]. The NMC 622 electrodes were dried at 60 °C and subsequently calendered to reach a calculated porosity of 35%.
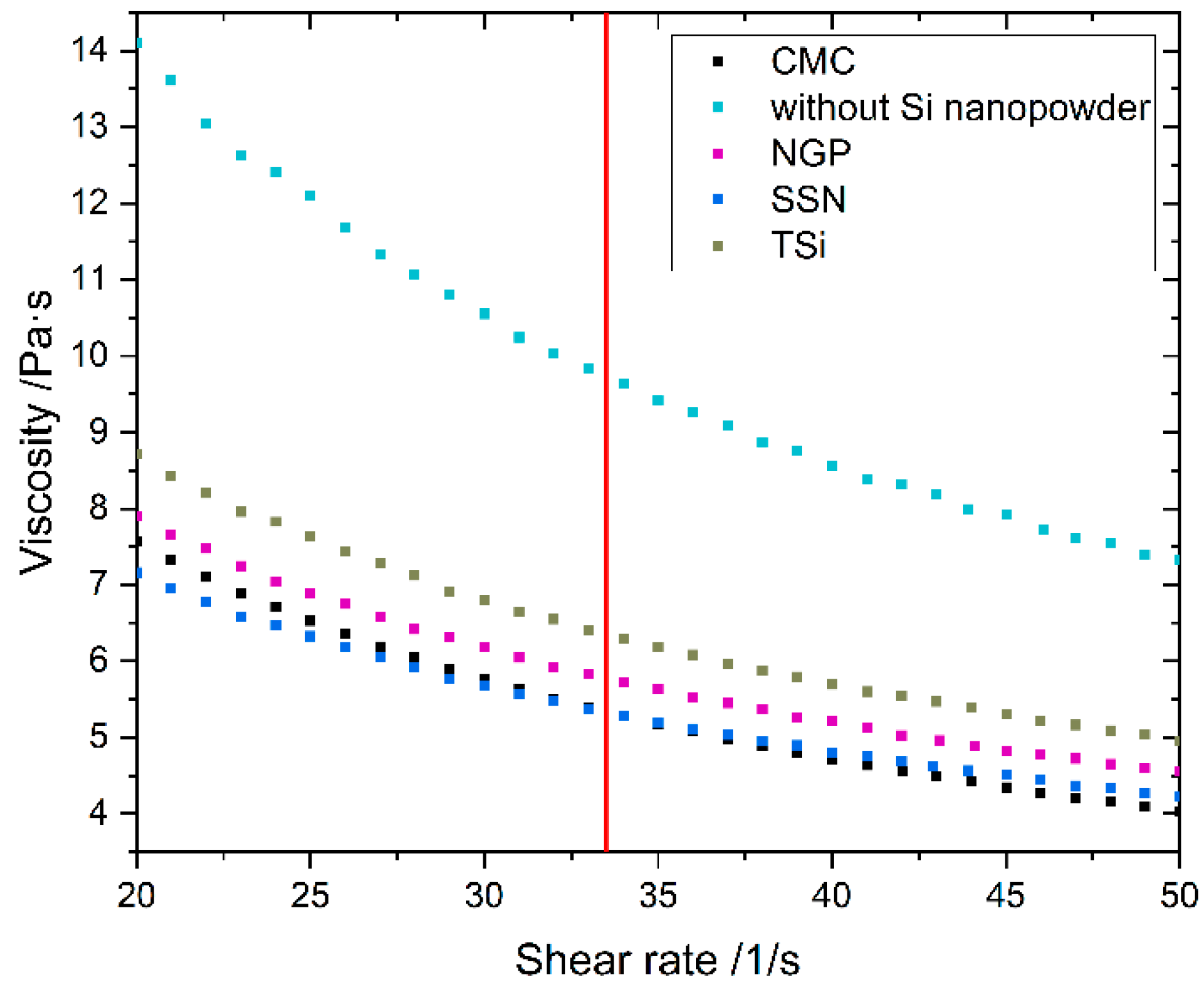
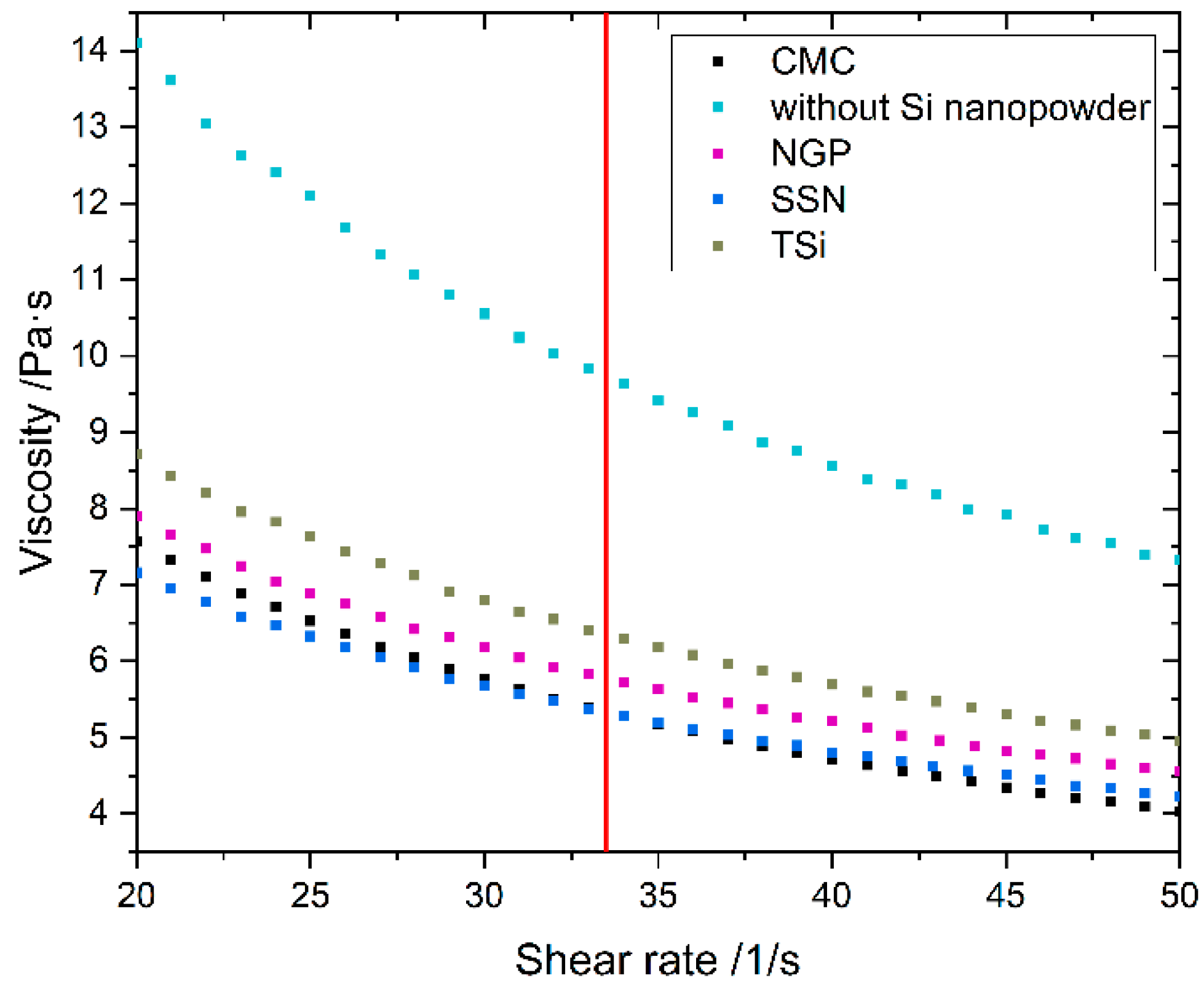
The dynamic viscosity of the anode slurry and the CMC solution was measured with a parallel plates viscometer (MCR72, Anton Paar, Graz, Austria) at 25 °C and a plate distance of 0.5 mm for shear rates from 1 to 100 s−1.
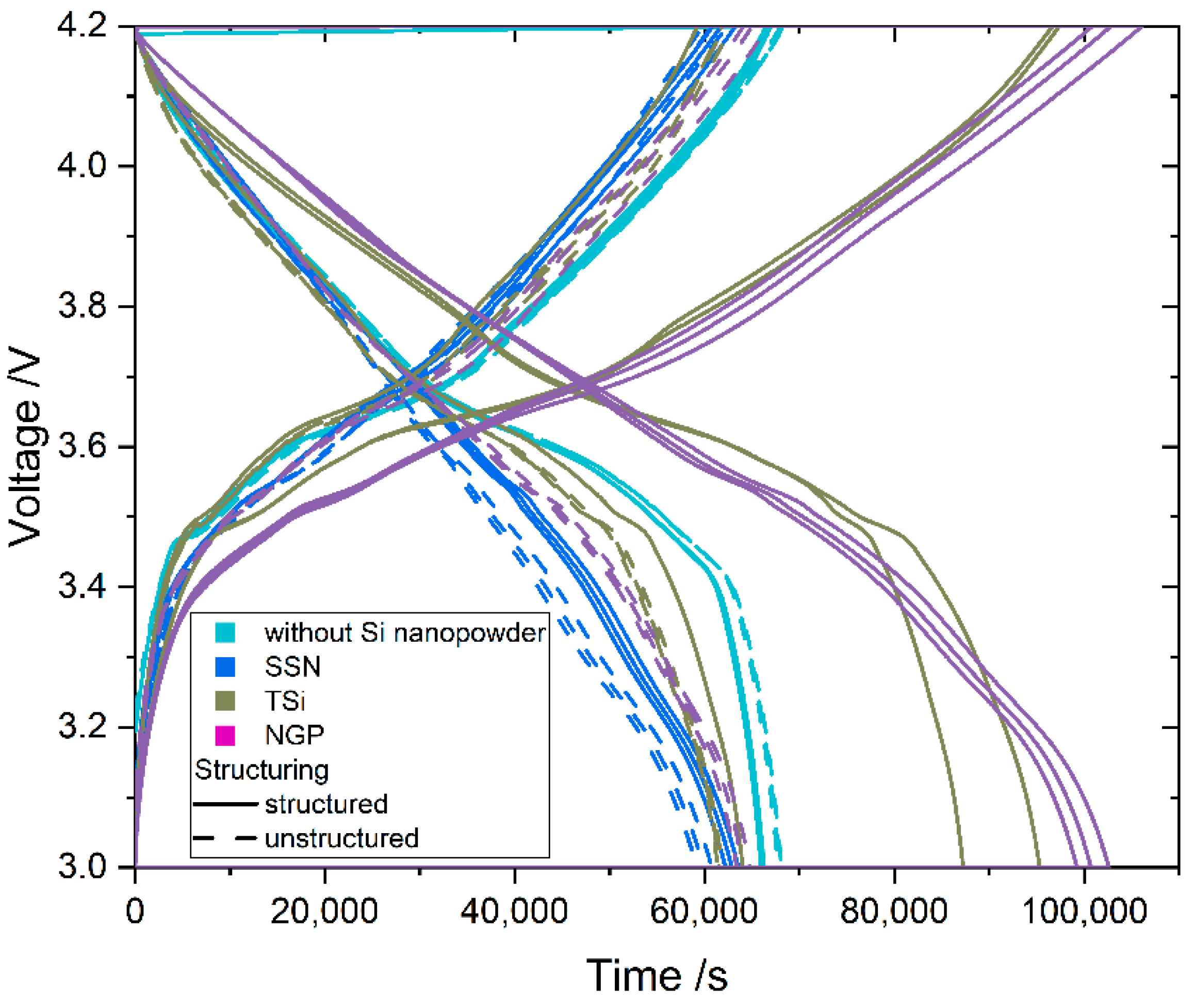
Laser patterning of the calendered anodes was performed with an ultrafast femtosecond (fs) fiber laser source (Tangerine, Amplitude Systèmes, Pessac, France) with a wavelength of 515 nm in ambient air. A repetition rate of 500 kHz and an average power of 1.5 W was applied to structure the electrodes. The scanning speed was kept constant at 500 mm/s. The process of laser ablation via scanning of the laser beam across the electrode surface was repeated several times until ablation up to the current collector was achieved. To assess the laser parameters and find the applicable number of scans, metallographic samples of the material were prepared and examined with a light microscope (Reicher-Jung MeF3, Leica Microsystems, Wetzlar, Germany). The reference electrodes remained unstructured. The laser structure consisted of a line pattern with a pitch of 200 µm. Electrodes for coin cells (CR2032) and pouch cells were laser cut. For the coin cells, circles with a diameter of 12 mm were cut. The pouch cells had a footprint area of 2492.3 mm².
2.2. Cell Assembly and Electrochemical Analysis
The electrodes for the coin cells were heated to 100 °C under vacuum for 24 h to remove excess moisture. The coin cells were assembled as half cells versus metallic lithium (Merck, Darmstadt, Germany) in an argon-filled glove box (argon 6.0, H
2O < 0.1 ppm; O
2 < 0.1 ppm). A polypropylene (PP) separator sheet (Celgard, Charlotte, NC, USA) with a thickness of 25 µm and a diameter of 19 mm was applied. As electrolyte, 120 µL of LP57 (EC:EMC 3:7, 1.3 M LiPF6) with 5 wt.% FEC (Gotion, Fremont, CA, USA) was utilized. It was previously shown by [
26,
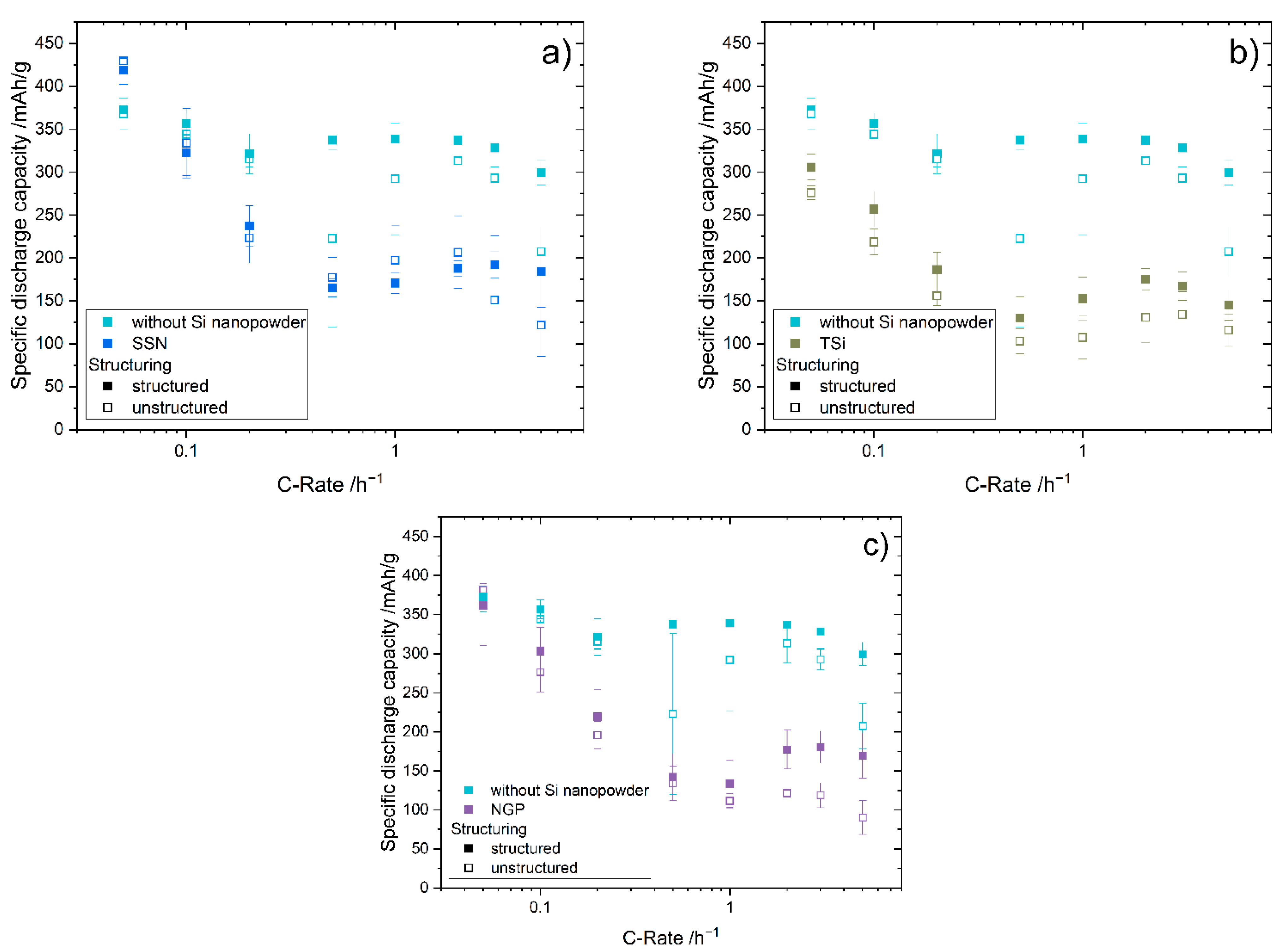
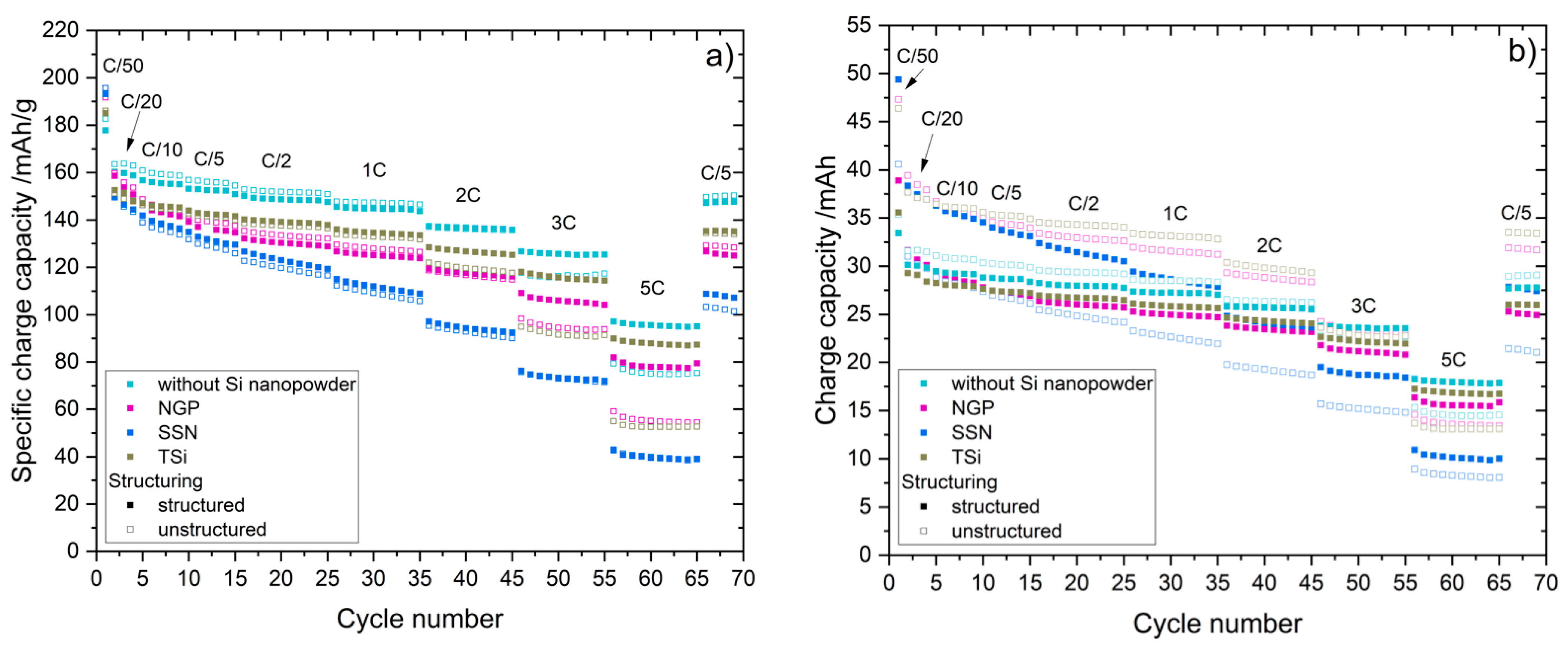
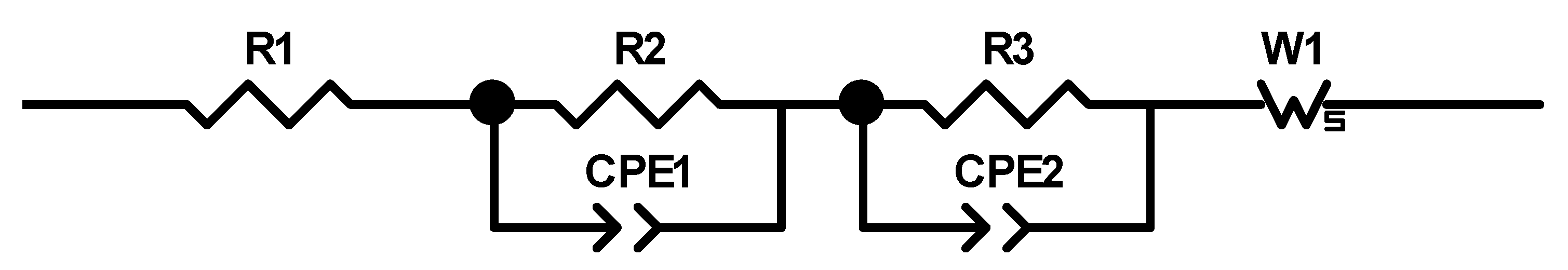
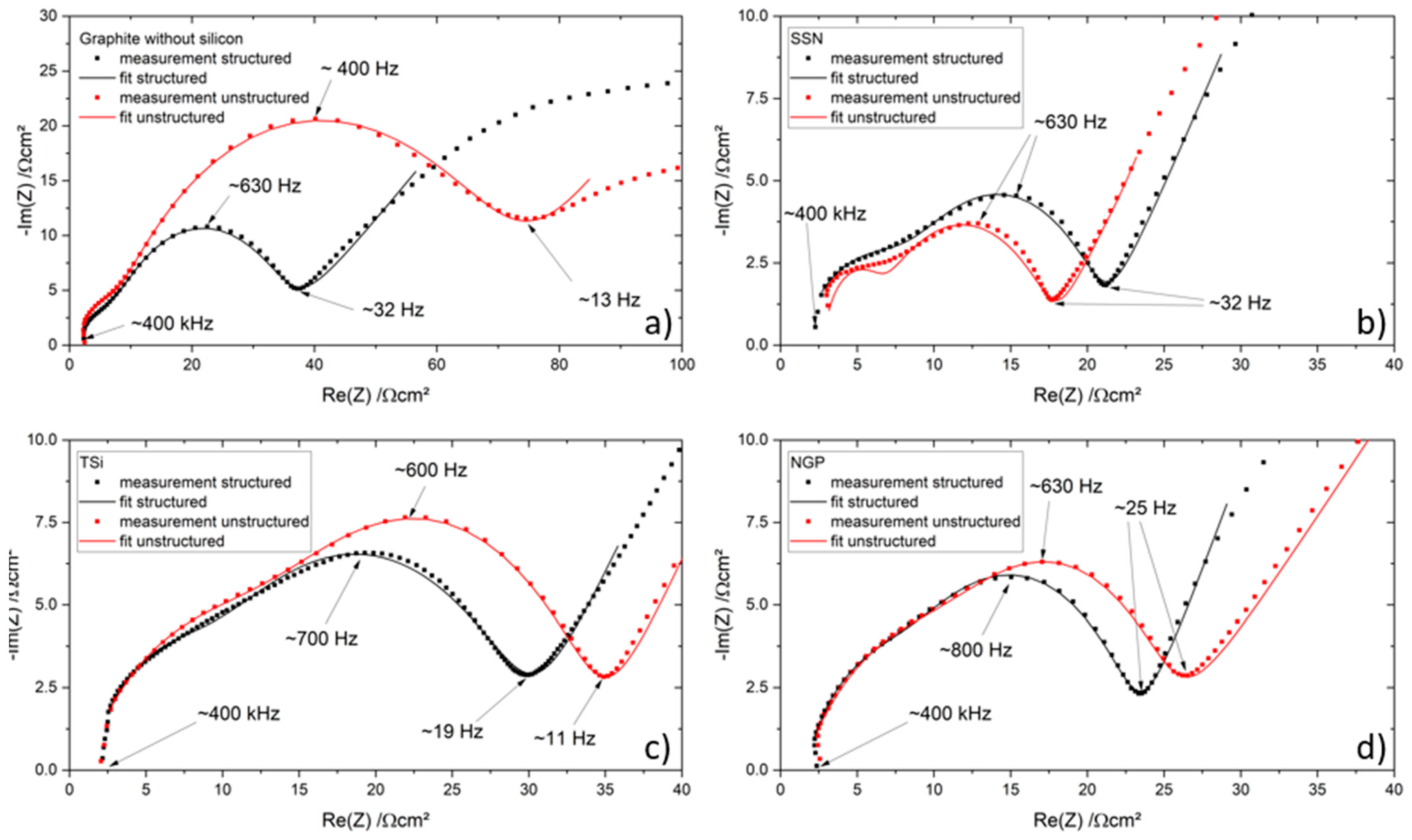
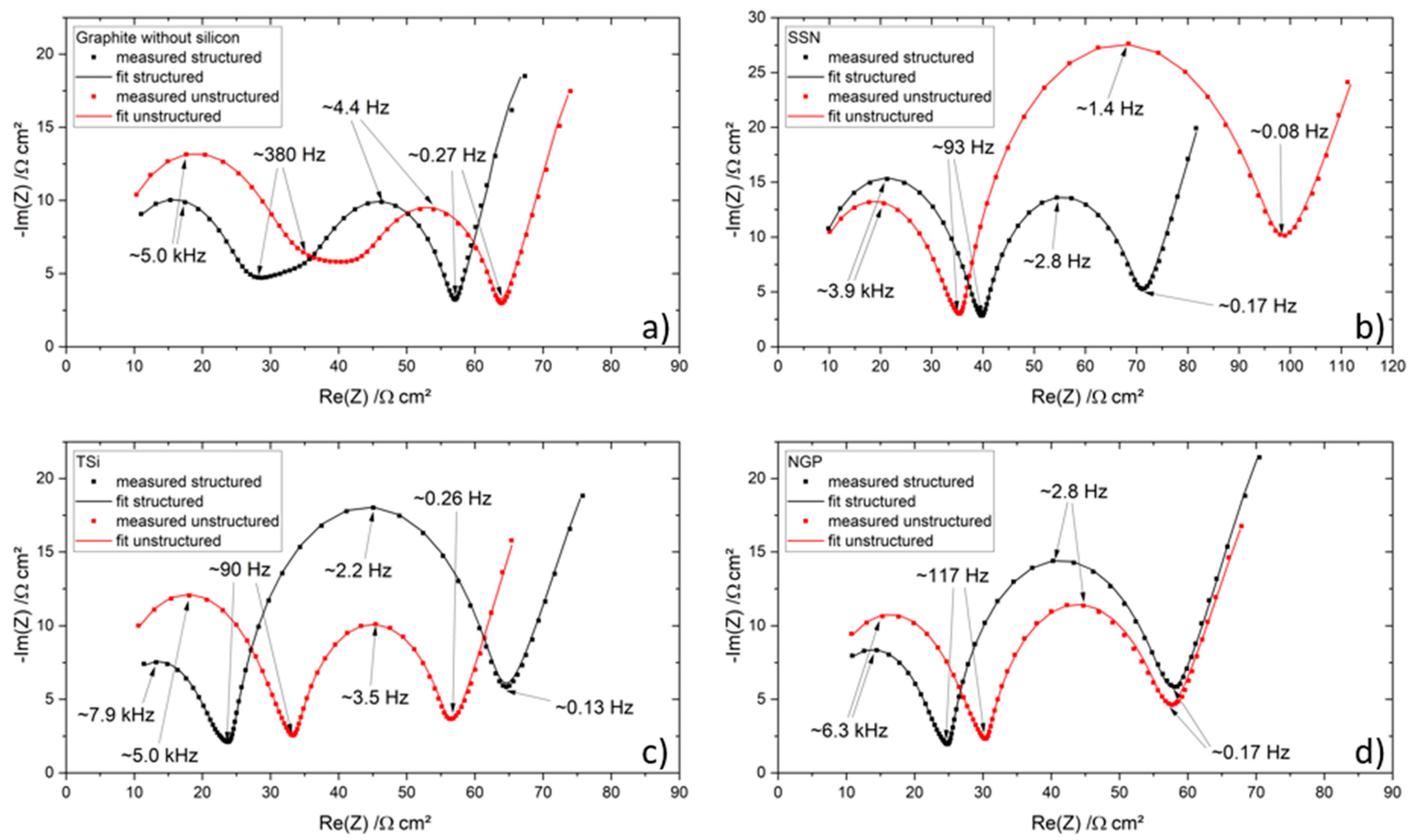
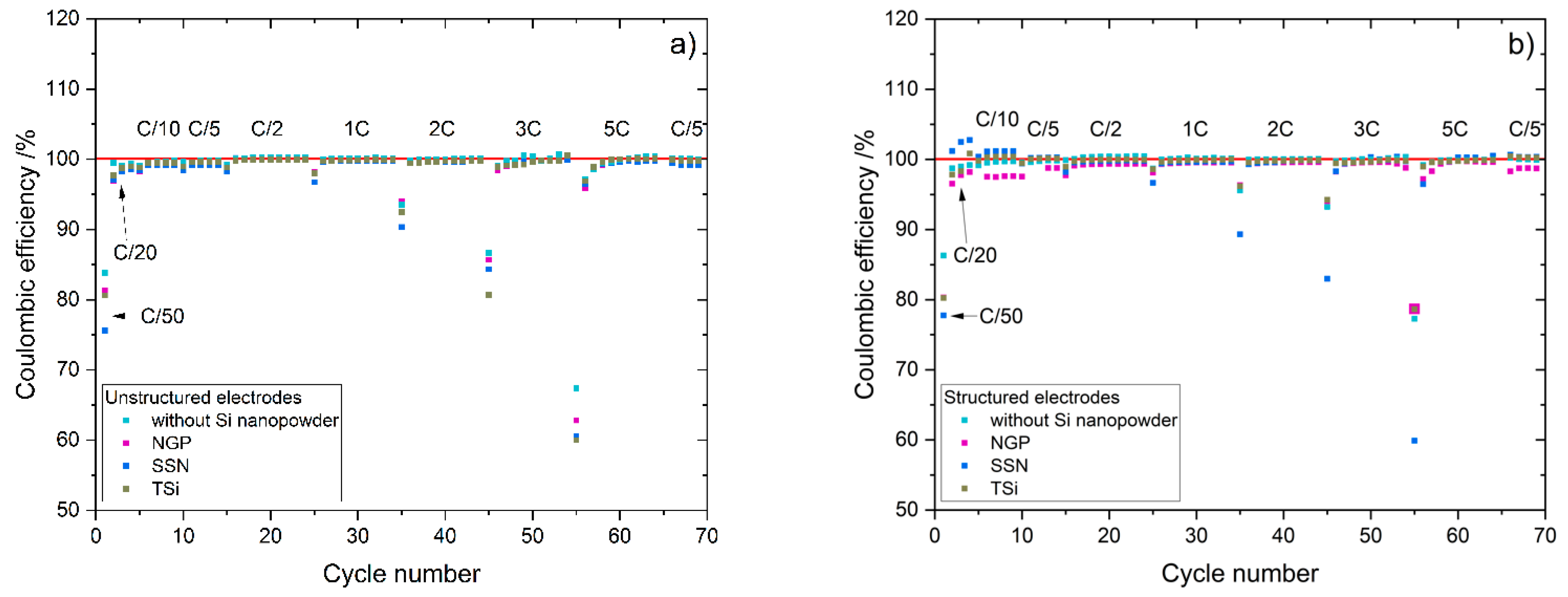
27] that the addition of up to 5 wt.% FEC improves the performance of silicon/graphite electrodes. After stacking, the cells were sealed with an electric crimper (MSK-160D, MTI, Richmond, CA, USA) and stored at room temperature for 20 h to allow for a homogeneous wetting of the components with liquid electrolyte. The cells were then galvanostatically characterized using a battery cycler (Arbin Instruments, College Station, TX, USA). The voltage window was adjusted depending on the electrode composition: 0.01 to 1.5 V (anodes without silicon) or 0.06 to 1.2 V (anodes with silicon). The formation followed a constant current-constant voltage protocol. The cells that contained silicon were cycled with a C-rate of C/50 for one cycle, where the cut-off current in the constant voltage phase was set to C/100. After that, three more cycles with C/20 and a cut-off current at CV of C/50 followed. The formation of the other electrodes omitted the first formation cycle. After the formation step, the impedance of the cells was measured by electrochemical impedance spectroscopy (EIS) using a battery cycler (BCS810, Biologic, Seyssinet-Pariset, France) at 0% state of charge (SoC) between 30 Hz and 10 MHz with a voltage amplitude of 10 mV. Ten measurements per decade with logarithmic spacing and three measurements per frequency were carried out. Subsequently, the rate capability of the cells was measured with a constant current-constant voltage (CCCV) measurement protocol, the parameters of which are shown in
Table 2. The currents were calculated based on the theoretical capacities of the active materials and the mass of the electrodes. Since the coin cells were cycled in a voltage window of 0.01 to 1.5 V/0.06 to 1.2 V, a practical specific capacity of 330 mAh/g was used for the graphite, and 3560 mAh/g for the silicon [
4]. After the rate capability test, the cells were again cycled at C/5 for five cycles to characterize the capacity retention.
The electrodes that were to be assembled in pouch cells were dried inside the heated airlock of the glove box (M. Braun, Germany, argon 6.0, H
2O < 0.1 ppm; O
2 < 0.1 ppm) at 60 °C under vacuum for 24 h to remove the excess moisture. A polypropylene separator (25 µm thickness, Celgard, USA) with dimensions of 60 × 60 mm
2 was used. After dry stacking, 3 mL LP57 with 5 wt.% FEC (as above) was added. The electrodes were soaked for 30 min, then the excess electrolyte was removed. After the cells were sealed, they were stored at room temperature for 20 h, until homogeneous wetting with the liquid electrolyte was achieved. They were cycled in a voltage window between 3 and 4.2 V at C/50 (CCCV, CV limit C/100) for one cycle, then degassed. For cells without silicon/graphite composite anodes, C/20 was chosen for the first formation cycle (CV limit C/50). The formation continued with three cycles of C/20 (CCCV, CV limit C/50). After a relaxation time of 24 h at 100% SoC, EIS measurements were performed between 10 Hz and 10 MHz with a voltage amplitude of 10 mV. Ten measurements per decade with logarithmic spacing and three measurements per frequency were carried out, followed by the symmetric rate capability test. The parameters of the rate capability test were identical to the parameters for the half cells, and are shown in
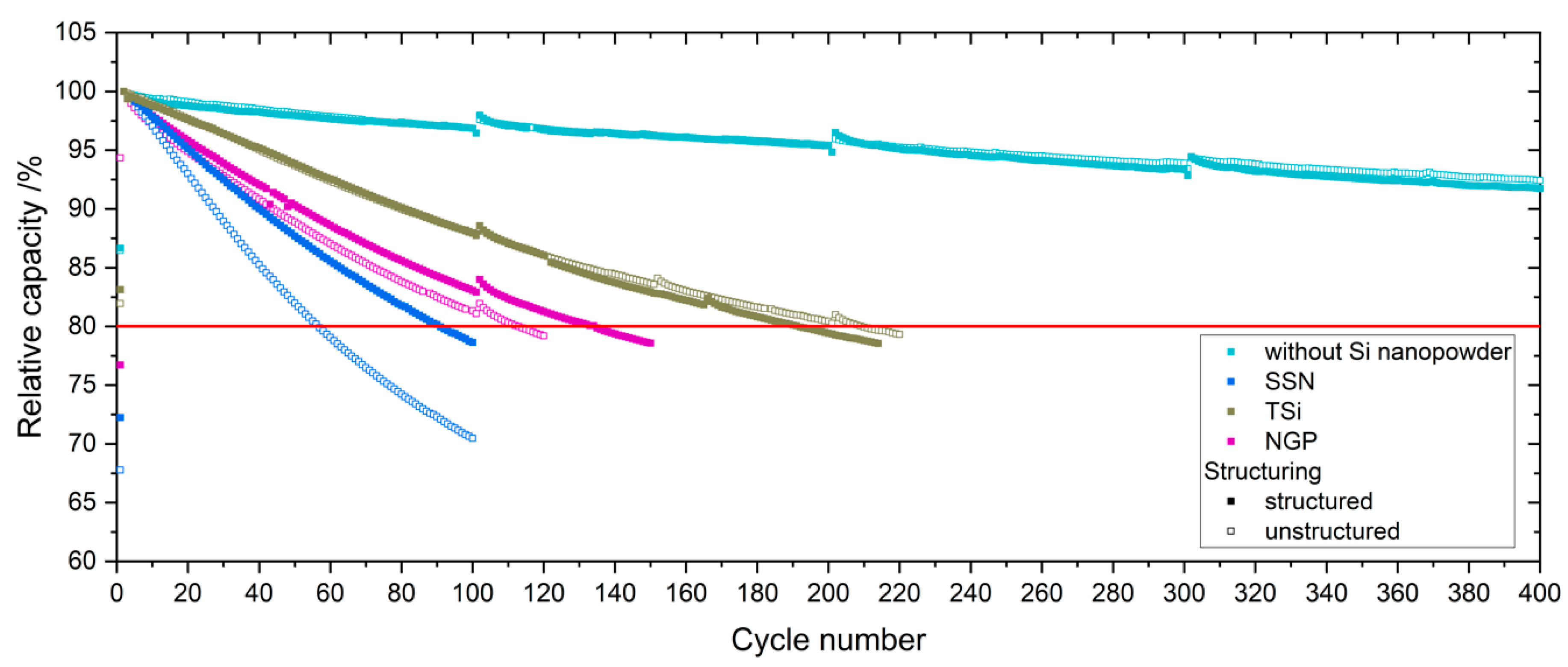
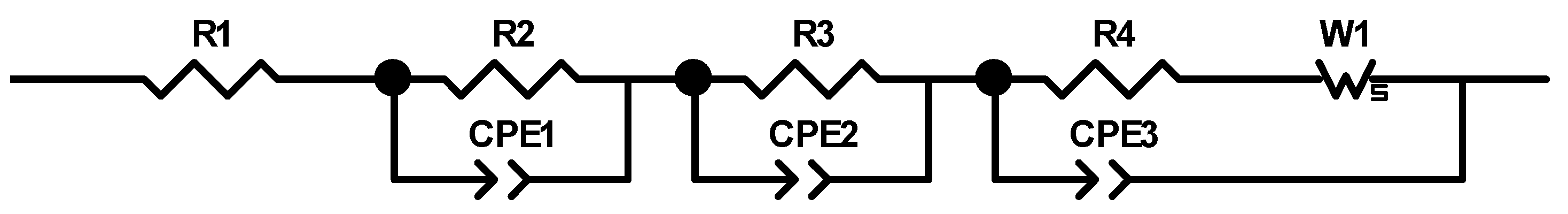
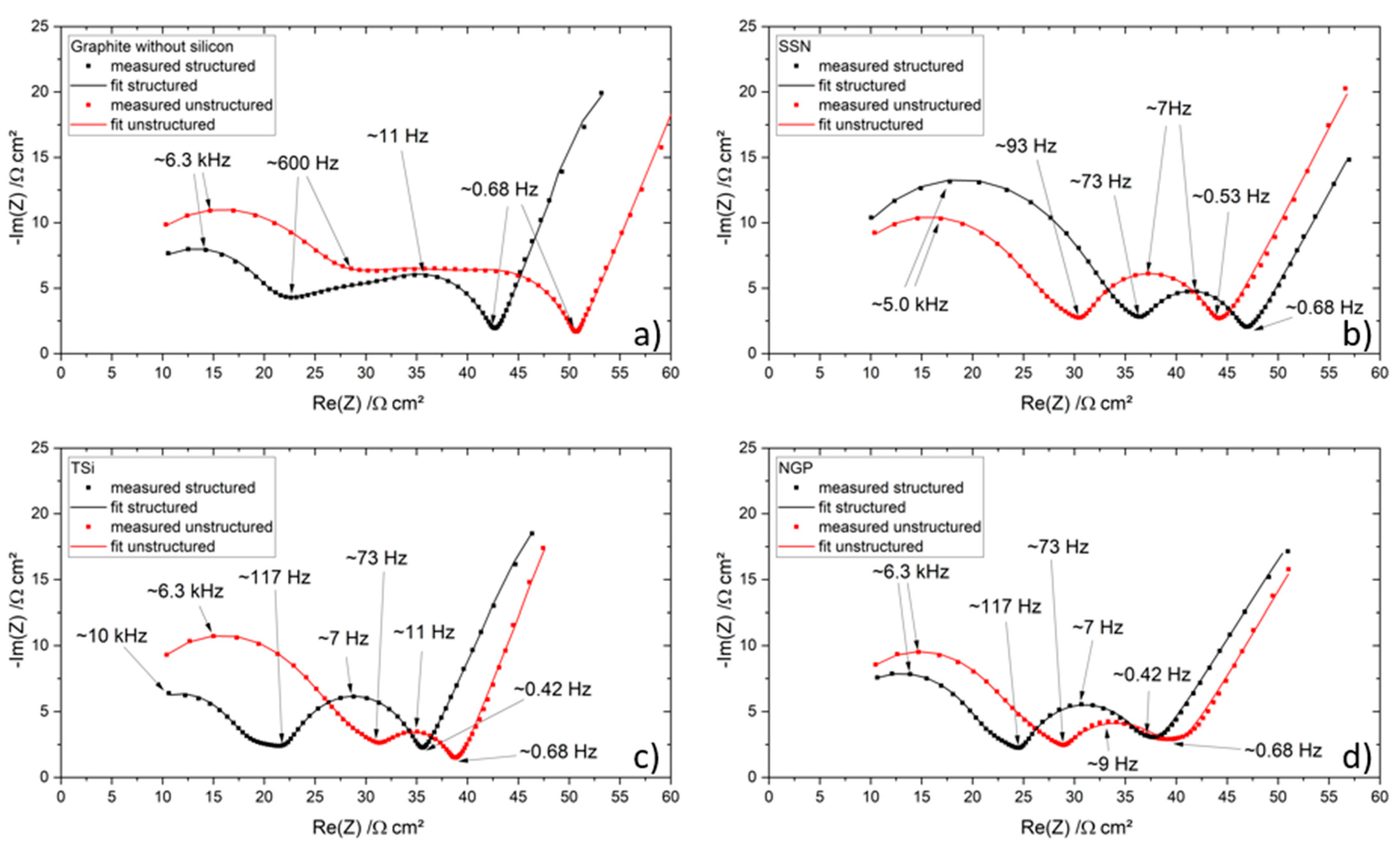
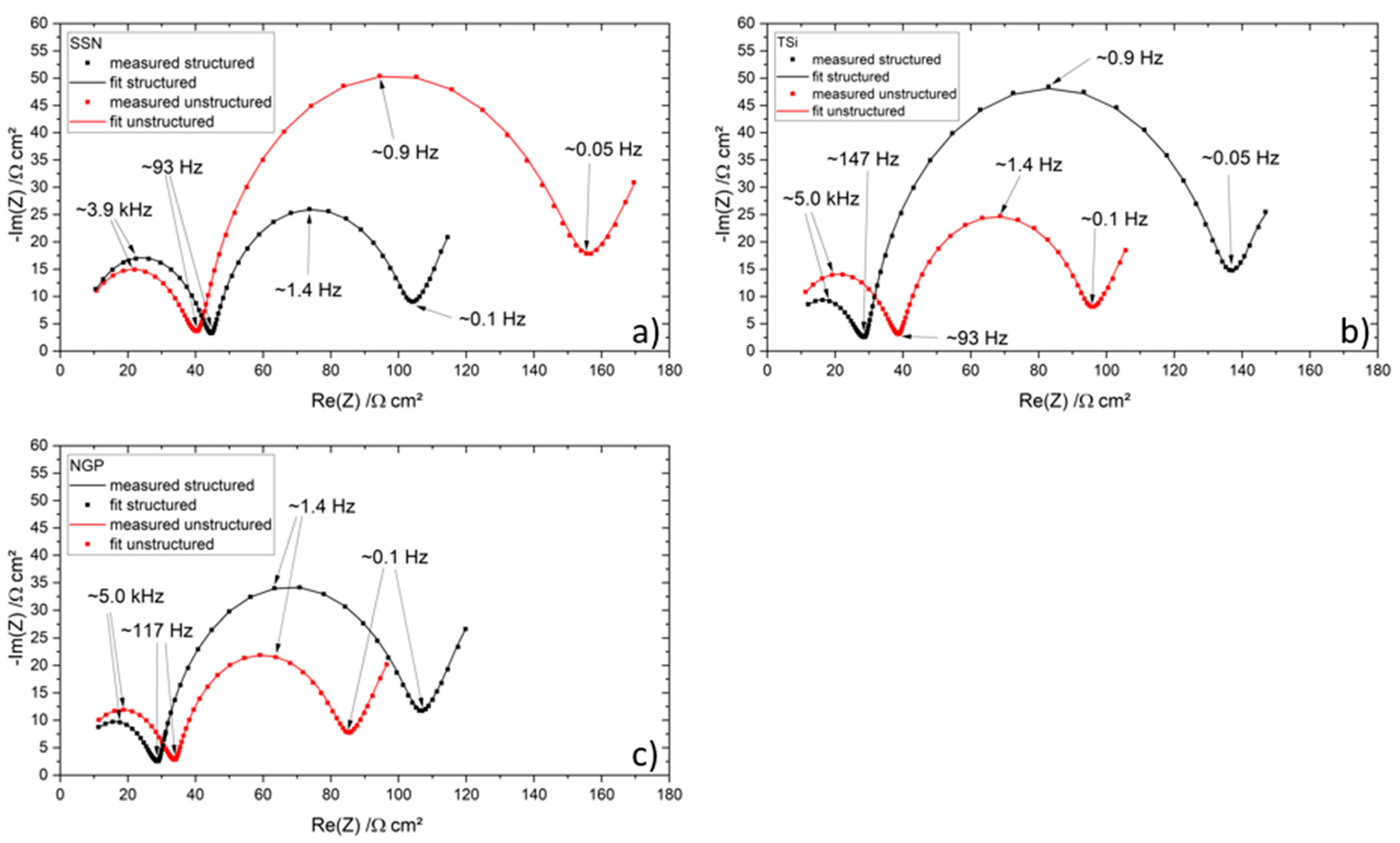
Table 2. The cells were cycled in a voltage window between 3 and 4.2 V. After the rate capability test, the cells were again cycled at C/5 for five cycles to characterize the capacity retention. Another EIS measurement was performed at 100% SoC after the rate capability test; then, the long-term test with CCCV measuring protocol at a C-rate of 1C was performed. The C-rate was adjusted depending on the specific discharge capacity (SDC) of the cell at the fifth cycle of the rate capability test at 1C. After a minimum of 100 cycles, the EIS measurement was repeated.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}