
Green Hydrogels Composed of Sodium Mannuronate/Guluronate, Gelatin and Biointeractive Calcium Silicates/Dicalcium Phosphate Dihydrate Designed for Oral Bone Defects Regeneration

 ,
,  ,
, 
 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mineral-Filled Hydrogels Preparation
2.2. Calcium Release and Alkalizing Activity (pH of Soaking Water)
2.3. Solubility, Porosity, Water Sorption
2.4. Setting Time
2.5. Radiopacity
2.6. Surface Micromorphology and Apatite Nucleation in Hank Balanced Salt Solution
2.7. Cell Tests
2.7.1. Hydrogel Disks Sterilization
2.7.2. Cells Test: Direct Contact and Extract Test
2.7.3. CD31, ALP and OCN Gene Expression
2.8. Statistical Analysis
3. Results
3.1. Calcium Release and Alkalizing Activity
3.2. Solubility, Porosity, Water Absorption
3.3. Radiopacity
3.4. Setting Time
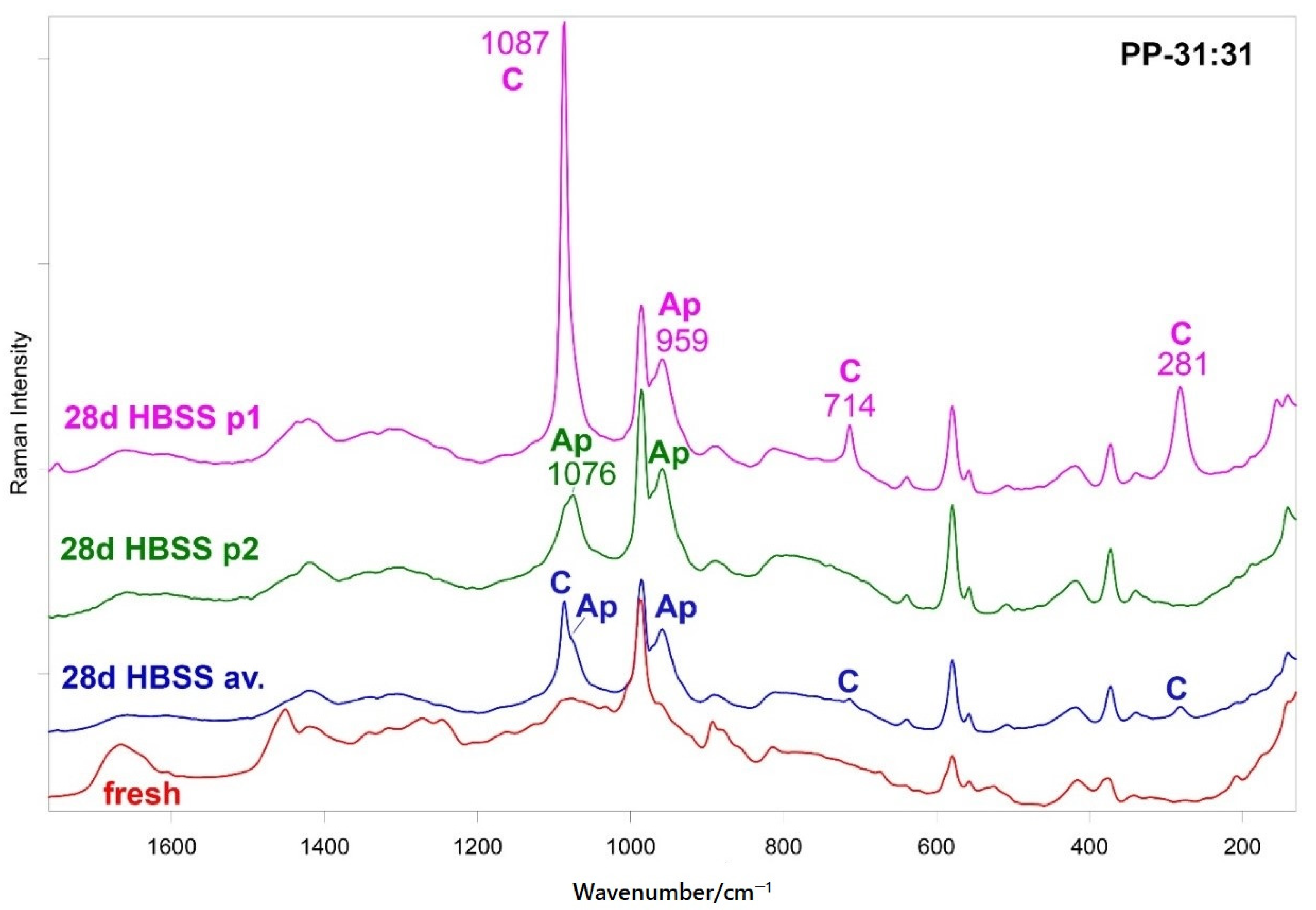
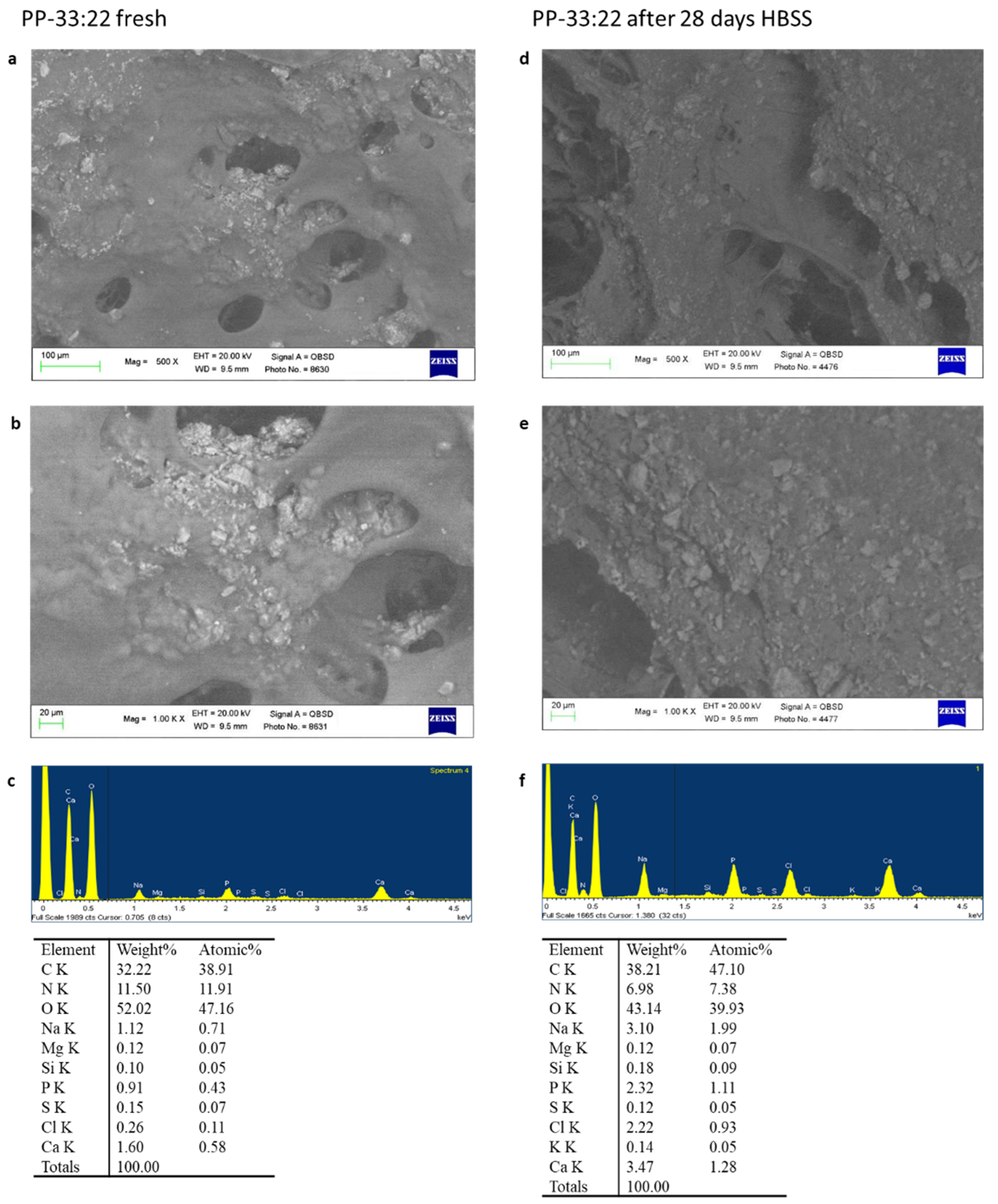
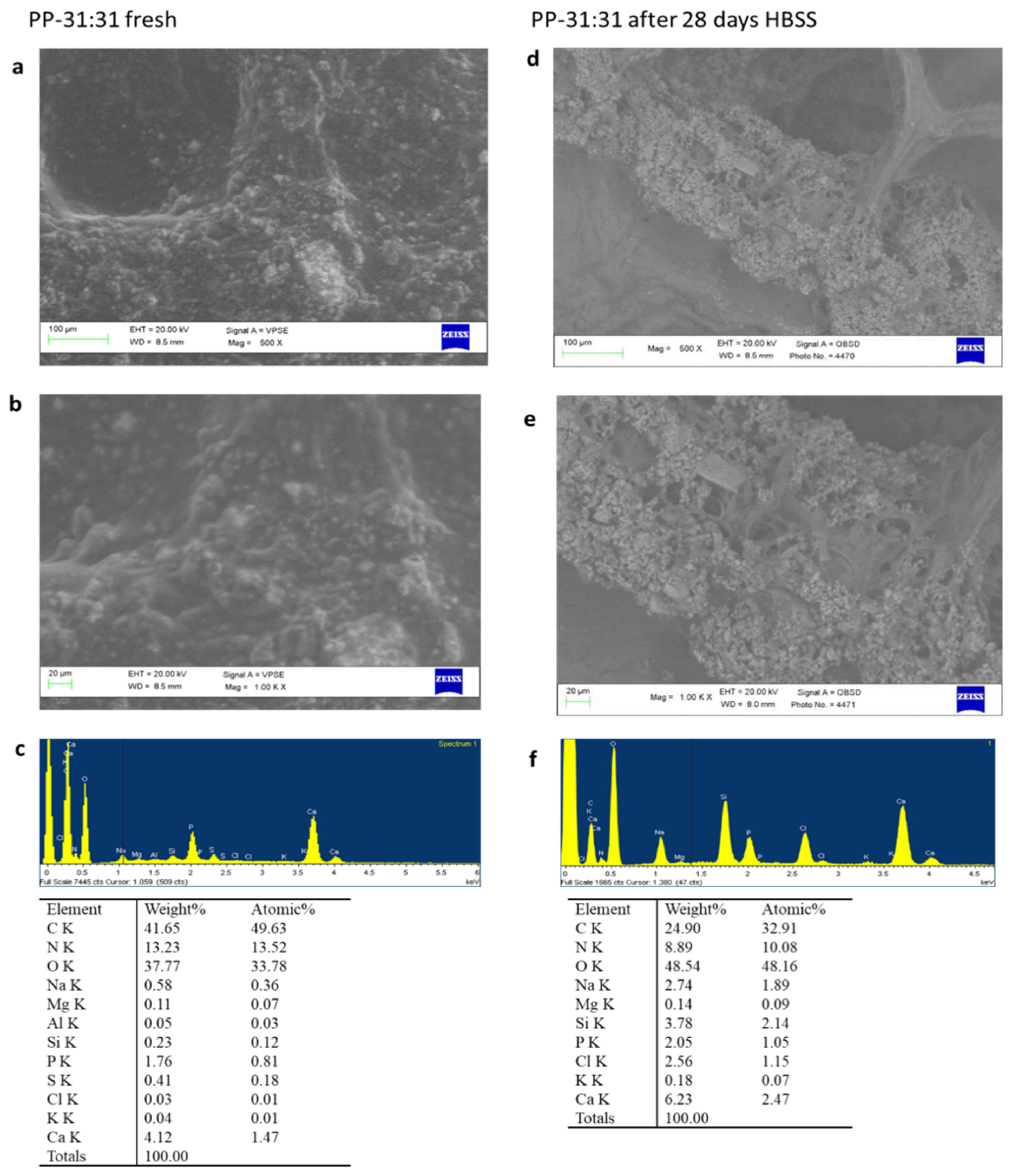
3.5. Surface Micromorphology and Apatite Nucleation in HBSS
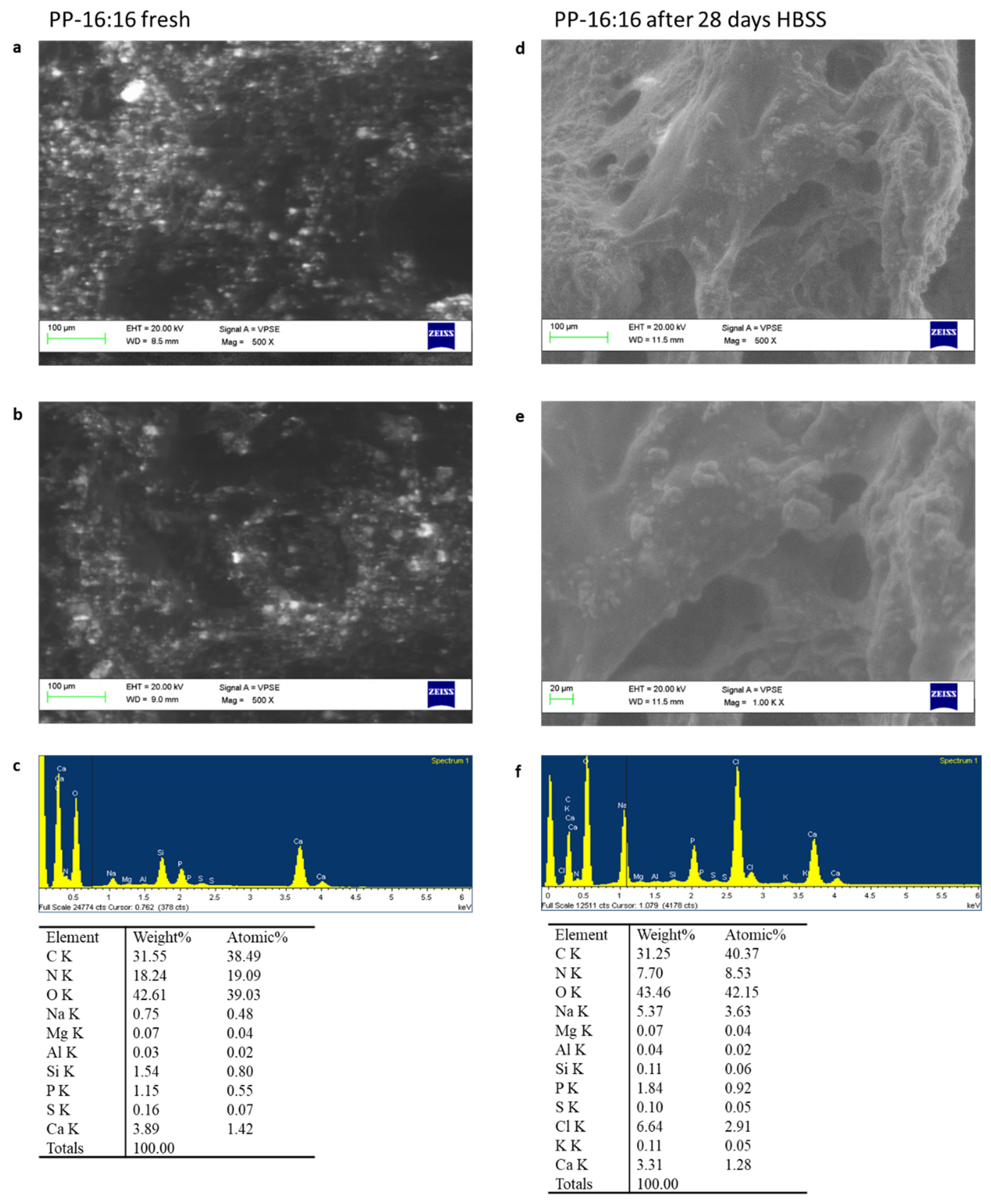
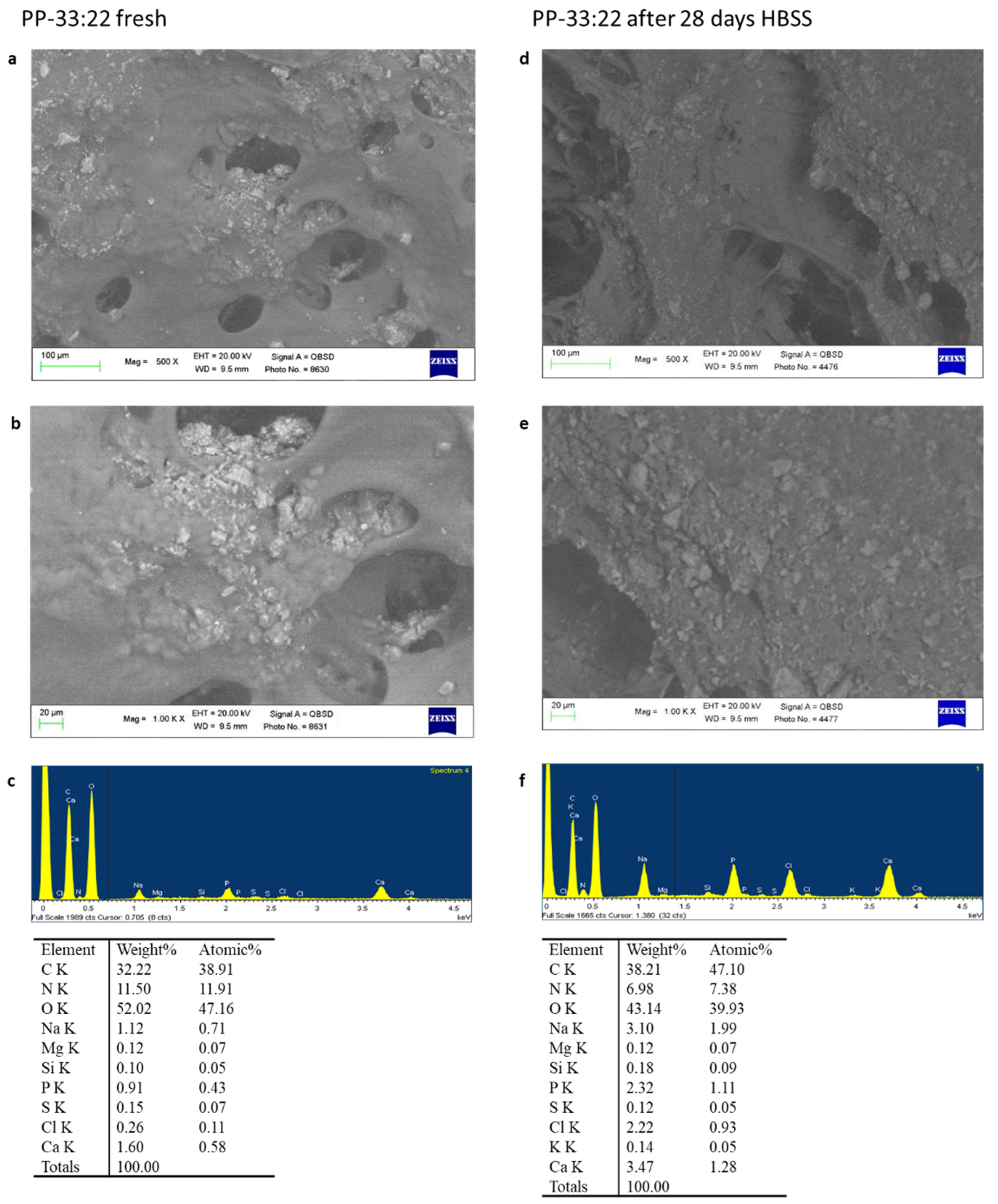
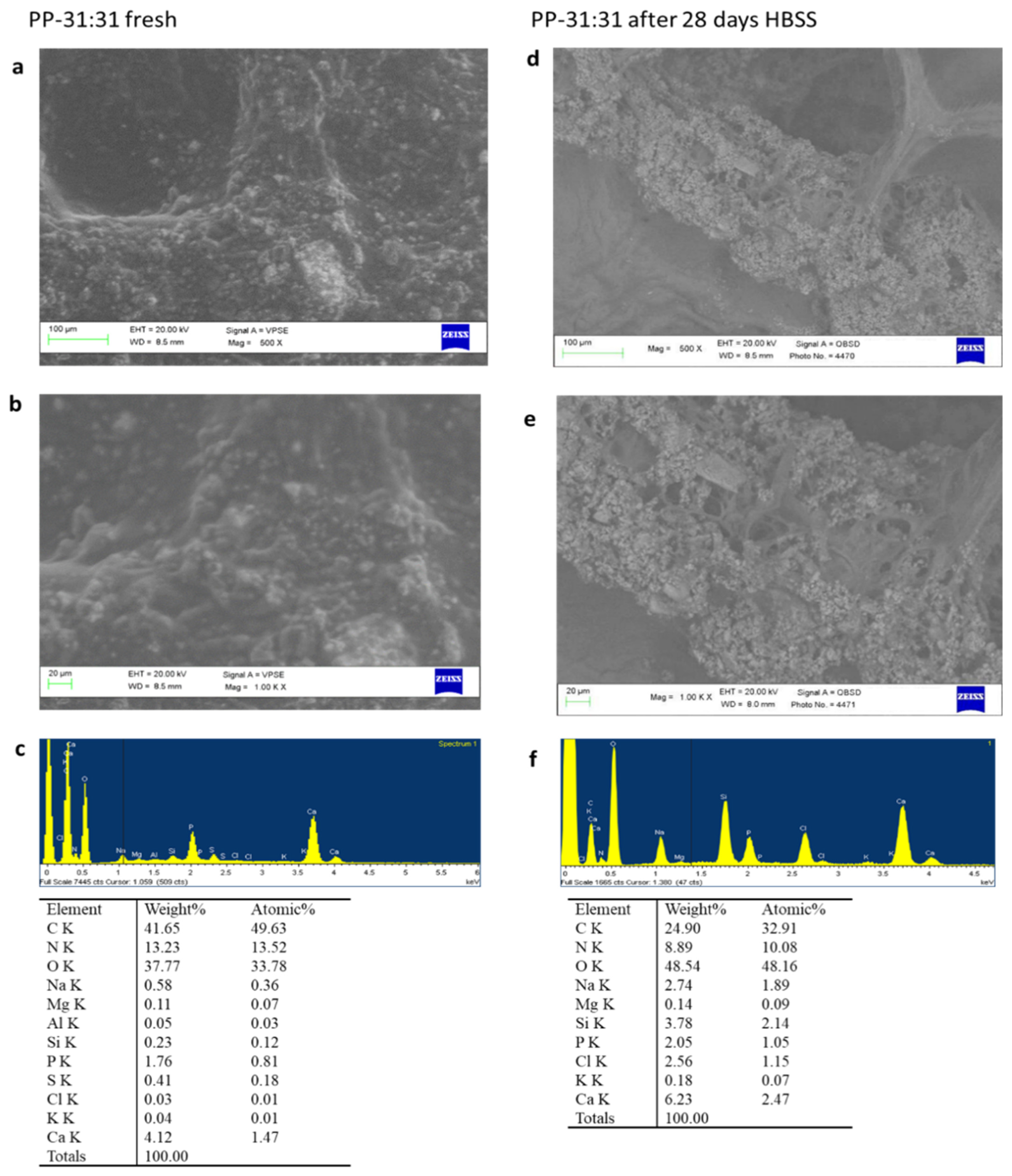
3.5.1. ESEM-EDX
PP-16:16
PP-33:22
PP-31:31
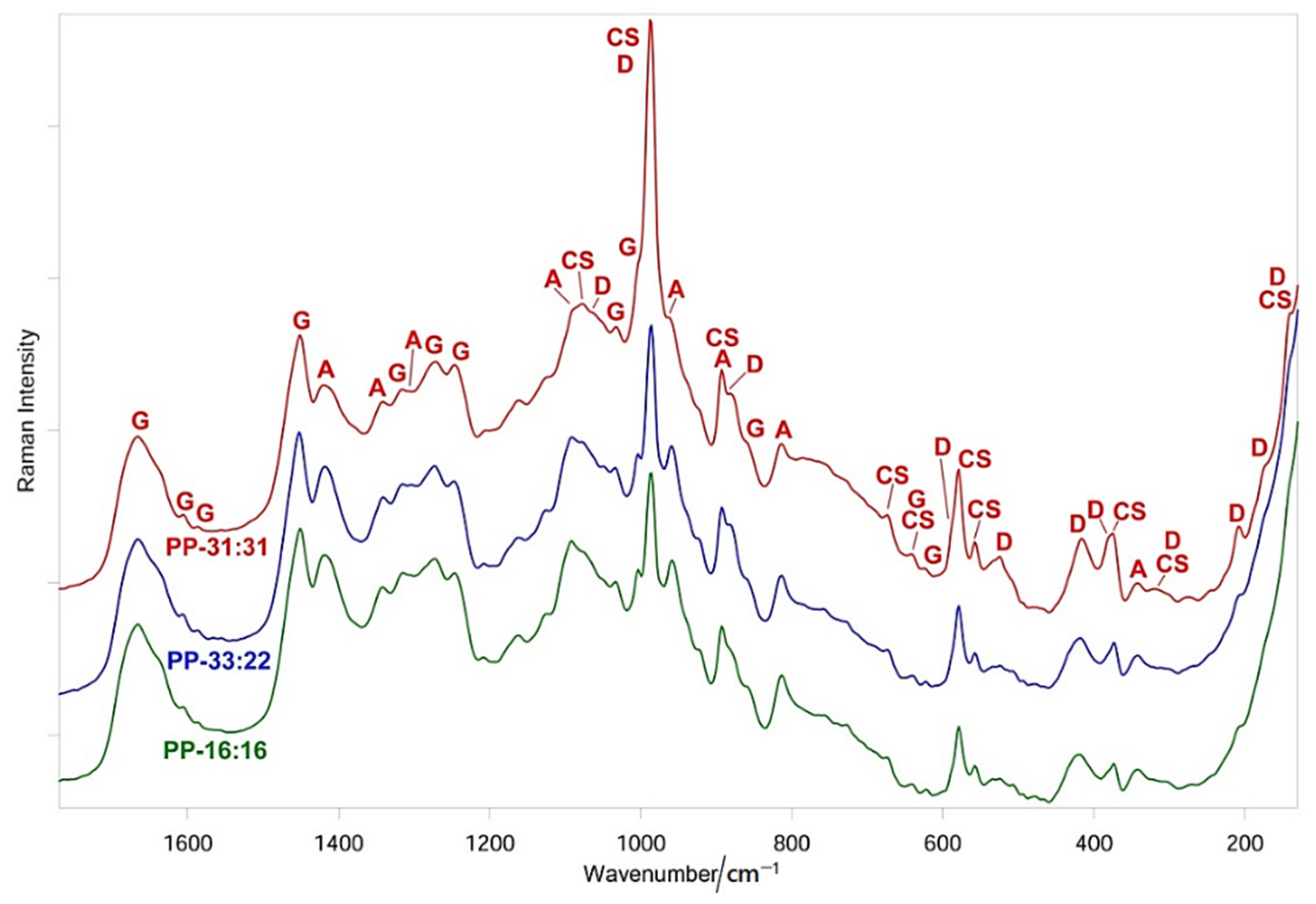
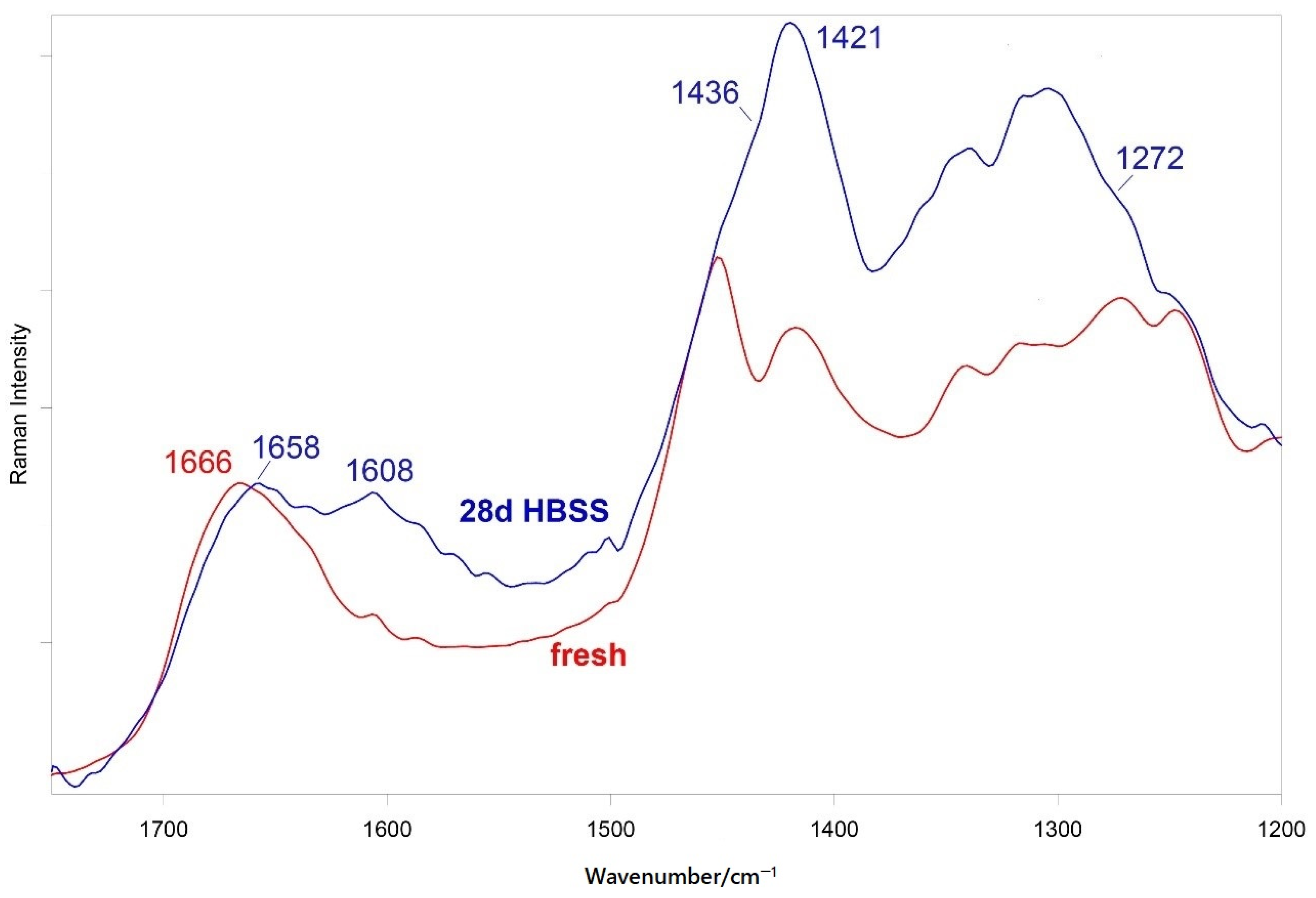
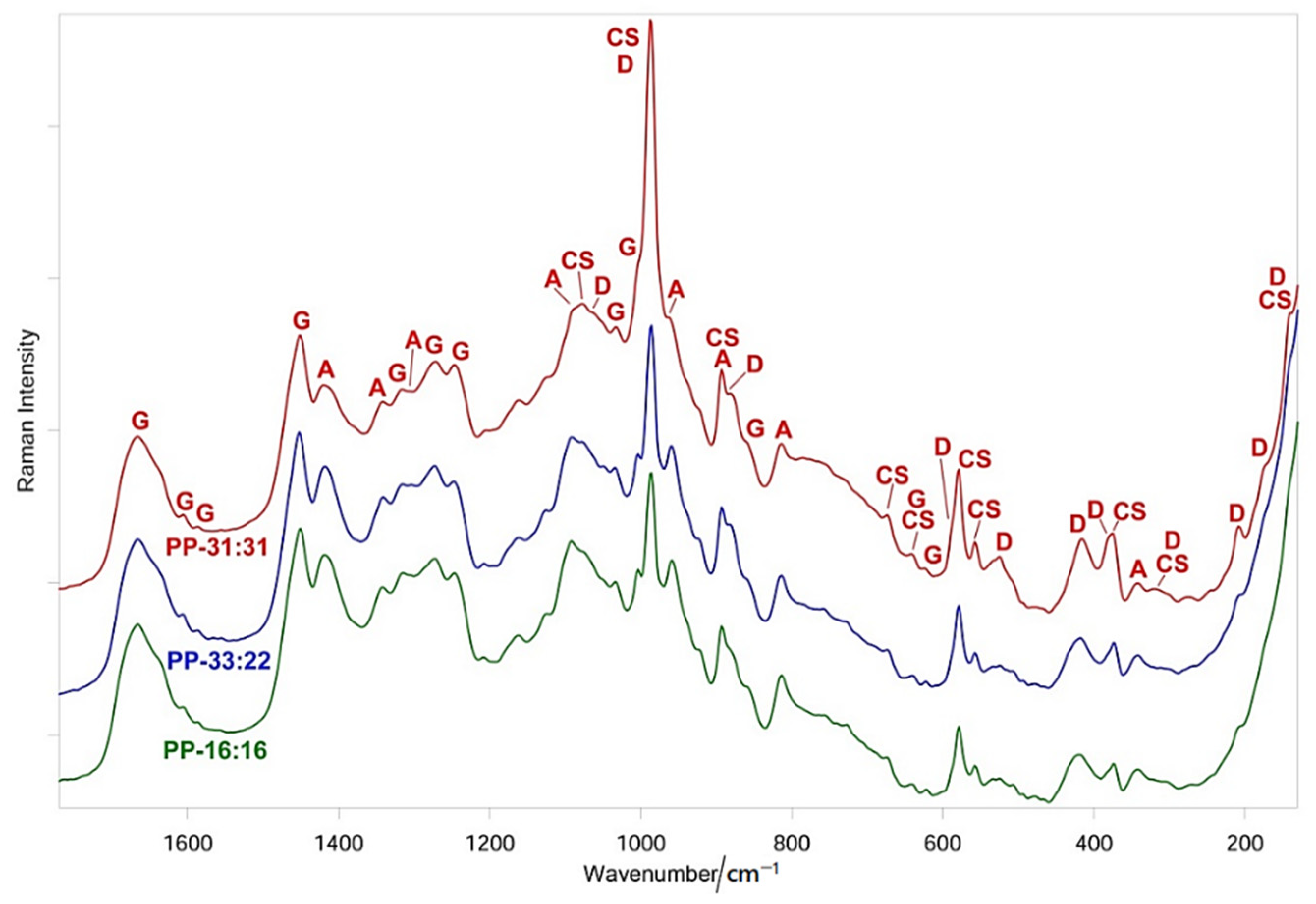
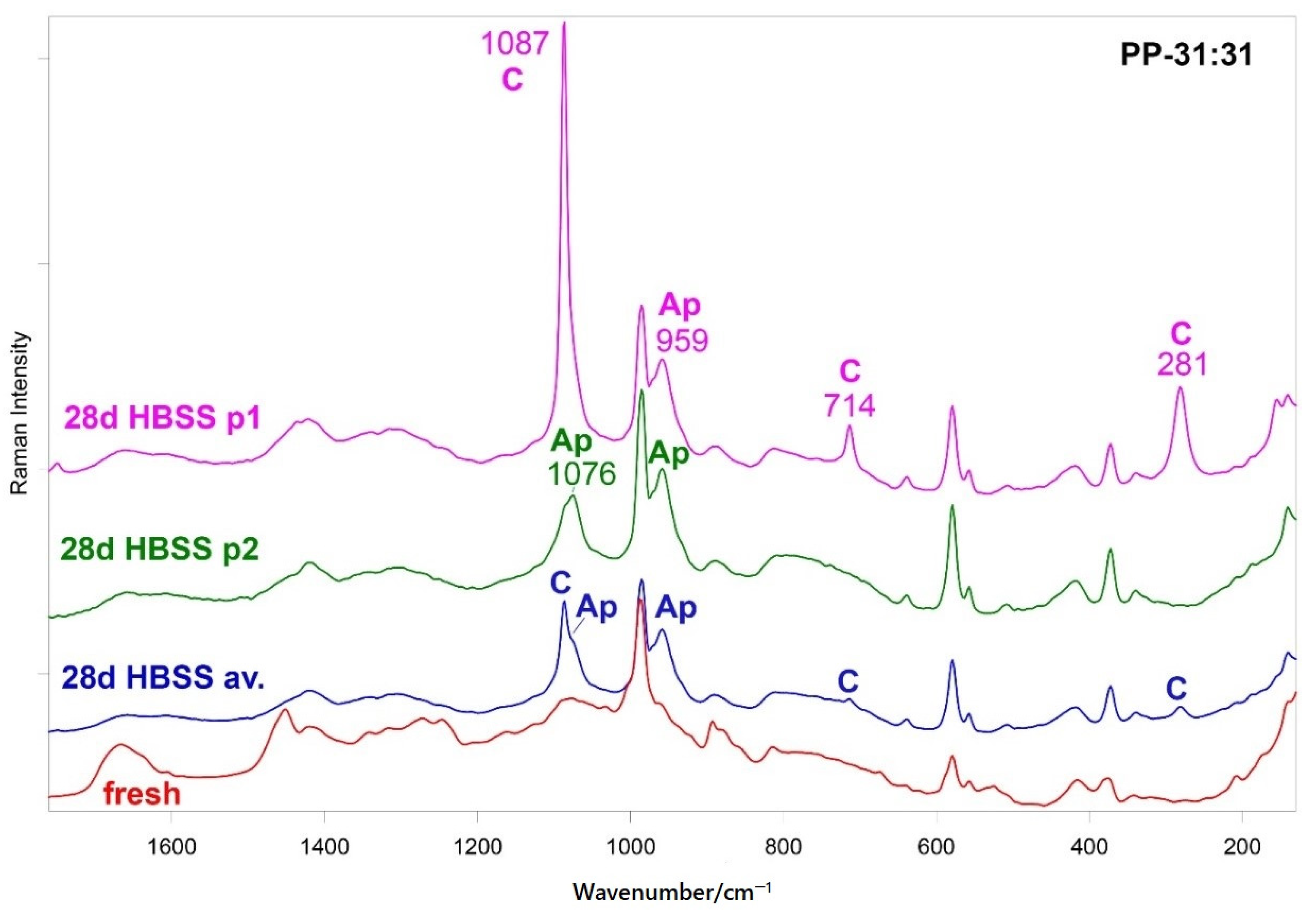
3.5.2. FT-Raman and Micro-Raman Analyses
3.6. Cell Tests
3.6.1. Biocompatibility
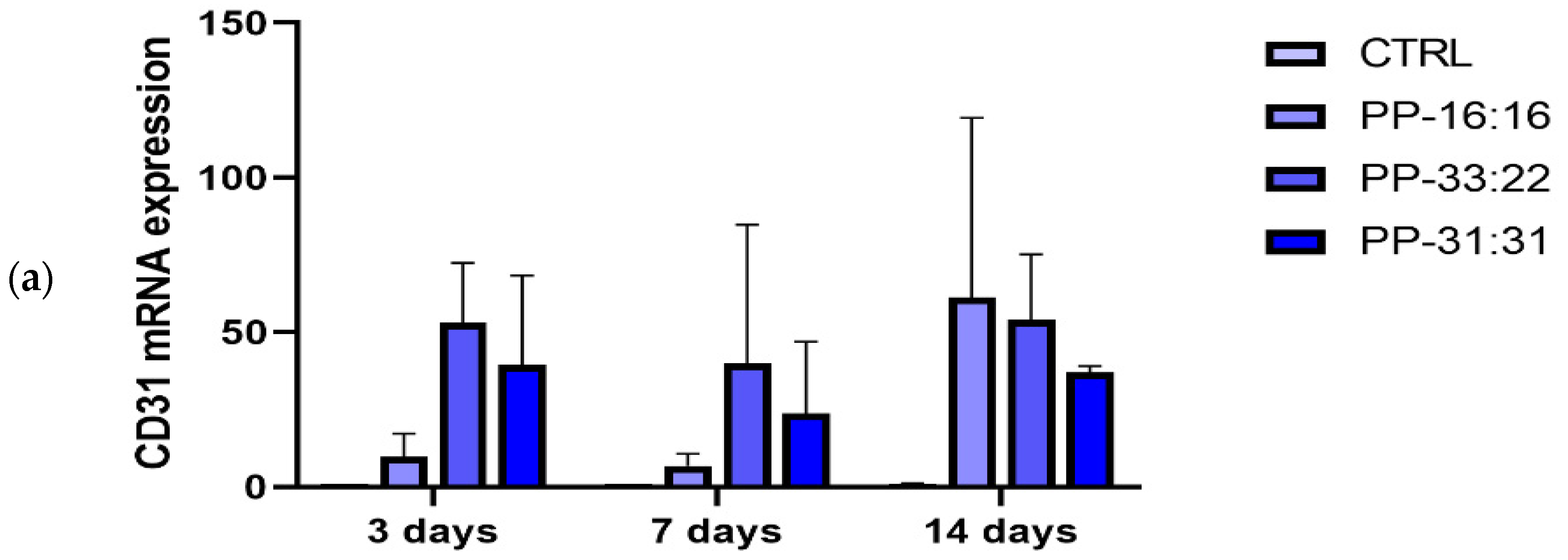
3.6.2. Vascular Differentiation (CD31 Gene Expression) of MSCs in Contact with the Mineral-Filled Hydrogels
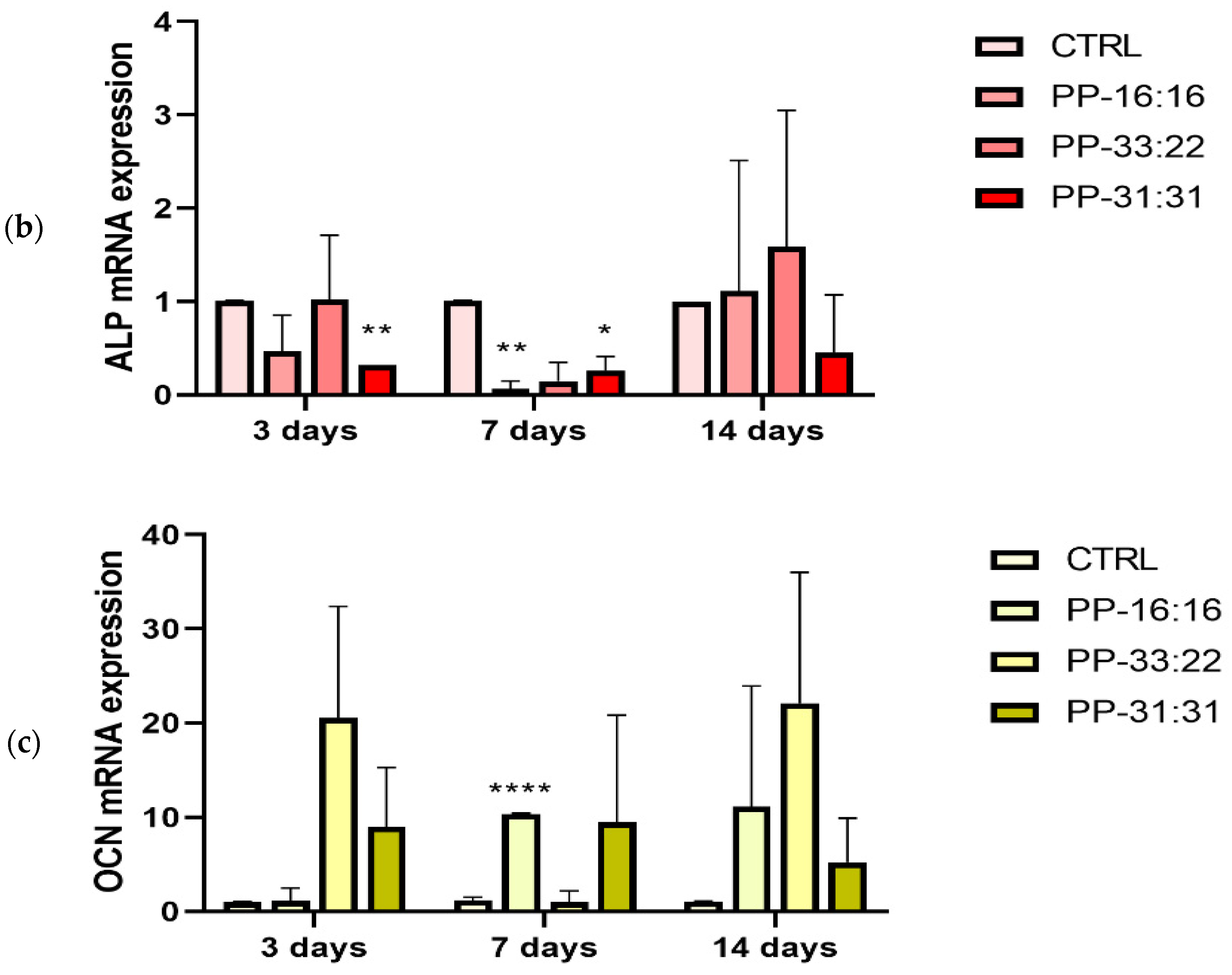
3.6.3. Osteogenic Differentiation (ALP and OCN Gene Expression) of MSCs in Contact with the Mineral-Filled Hydrogels
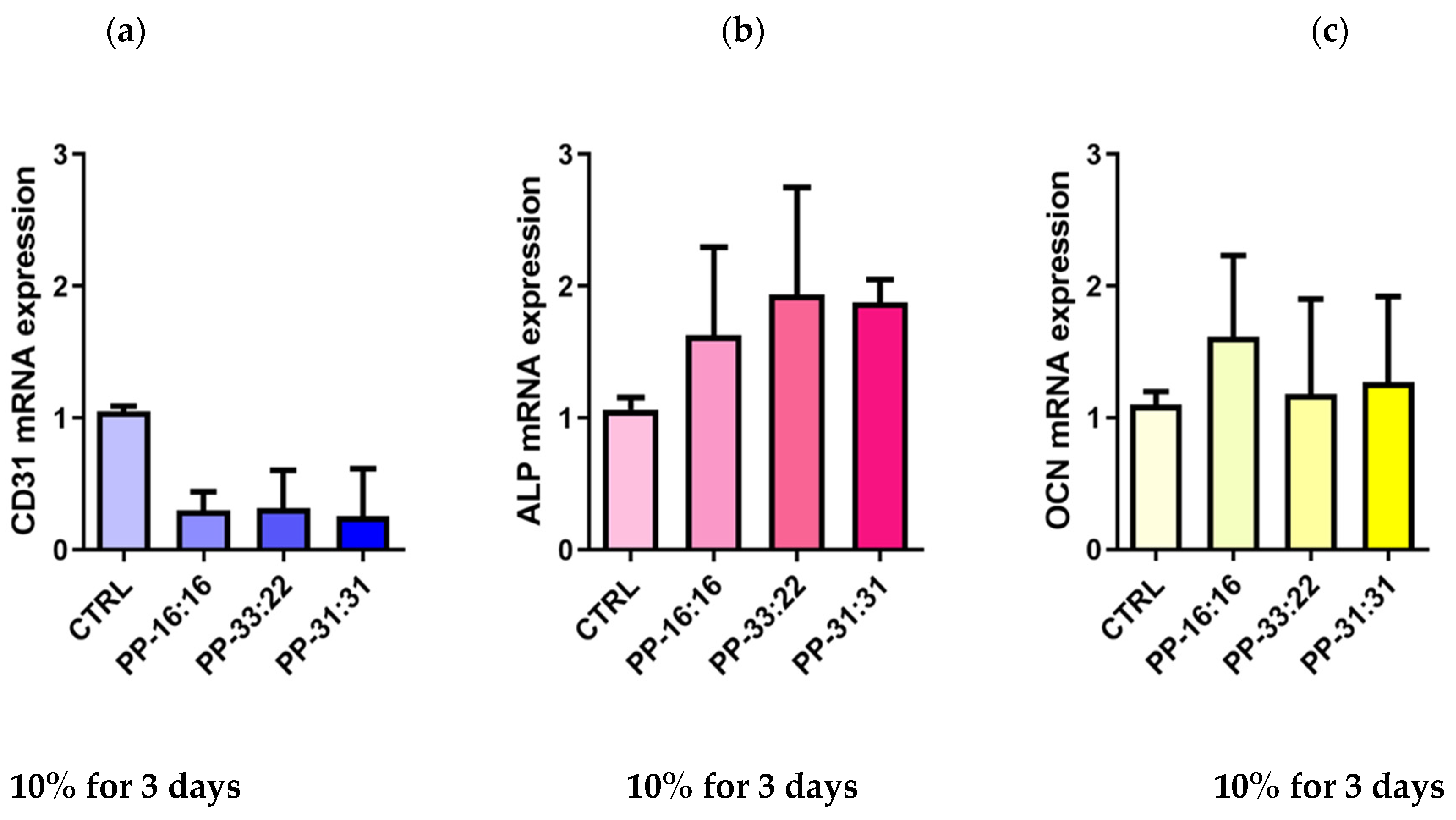
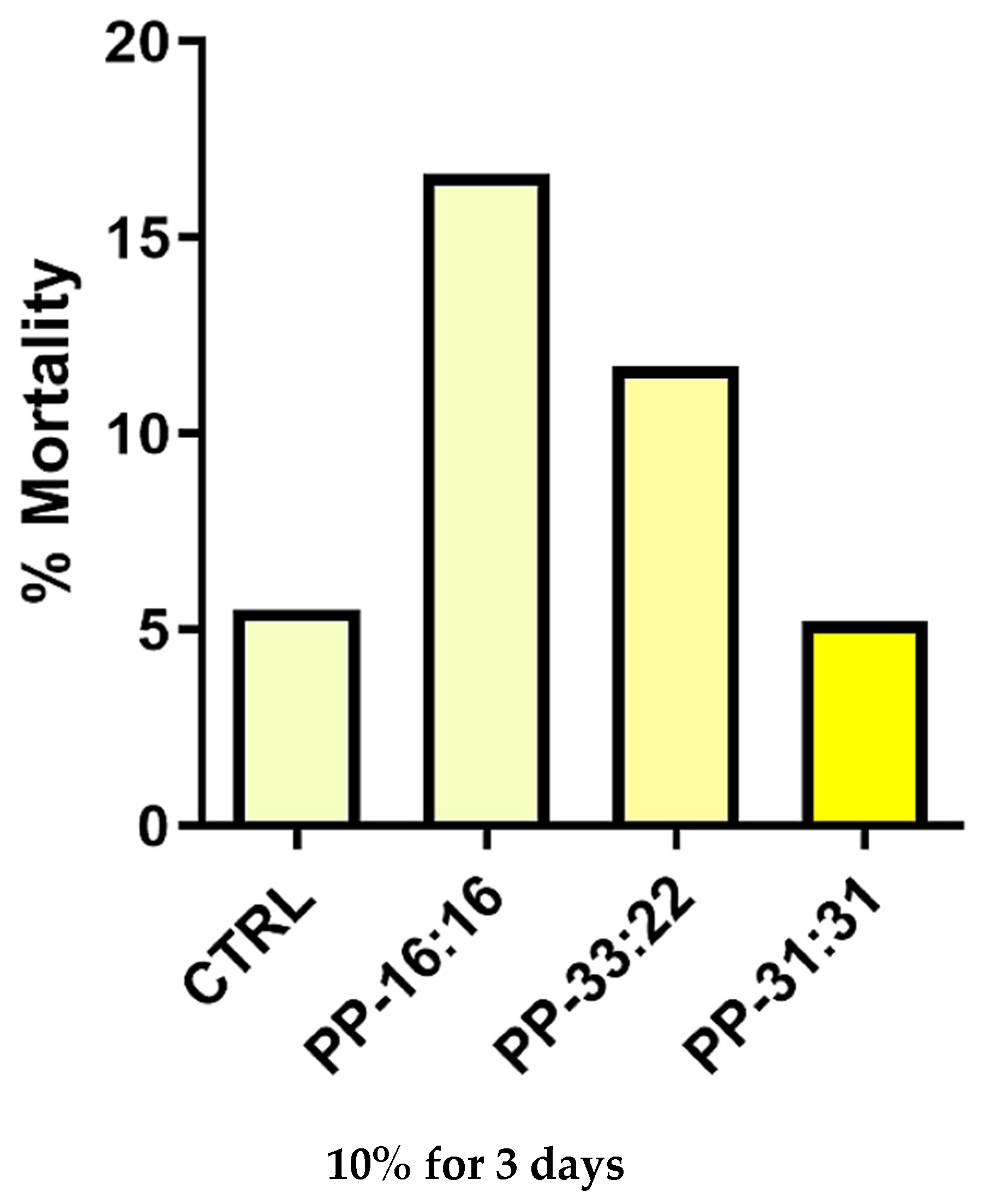
3.6.4. Vascular Differentiation (CD31 Gene Expression) of MSCs Cultured with 10% Hydrogel Extracts Added to Culture Medium
3.6.5. Osteogenic Differentiation (ALP, OCN Gene Expression) of MSCs Cultured with 10% Hydrogel Extracts Added to Culture Medium
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Oryan, A.; Alidadi, S.; Moshiri, A.; Maffulli, N. Bone regenerative medicine: Classic options, novel strategies, and future directions. J. Orthop. Surg. Res. 2014, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Gao, M.; Syed, S.; Zhuang, J.; Xu, X.; Zhang, X.-Q. Bioactive hydrogels for bone regeneration. Bioact. Mater. 2018, 3, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Jahangirian, H.; Lemraski, E.G.; Rafiee-Moghaddam, R.; Webster, T.J. A review of using green chemistry methods for biomaterials in tissue engineering. Int. J. Nanomed. 2018, 13, 5953–5969. [Google Scholar] [CrossRef] [Green Version]
- Roi, A.; Ardelean, L.C.; Roi, C.I.; Boia, E.-R.; Boia, S.; Rusu, L.-C. Oral Bone Tissue Engineering: Advanced Biomaterials for Cell Adhesion, Proliferation and Differentiation. Materials 2019, 12, 2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerecht-Nir, S.; Cohen, S.; Ziskind, A.; Itskovitz-Eldor, J. Three-dimensional porous alginate scaffolds provide a conducive environment for generation of well-vascularized embryoid bodies from human embryonic stem cells. Biotechnol. Bioeng. 2004, 88, 313–320. [Google Scholar] [CrossRef]
- Gong, Y.; Han, G.T.; Zhang, Y.M.; Zhang, J.F.; Jiang, W.; Tao, X.W.; Gao, S.C. Preparation of alginate membrane for tissue engineering. J. Polym. Eng. 2016, 36, 363–370. [Google Scholar] [CrossRef]
- Degli Esposti, M.; Chiellini, F.; Bondioli, F.; Morselli, D.; Fabbri, P. Highly porous PHB-based bioactive scaffolds for bone tissue engineering by in situ synthesis of hydroxyapatite. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 100, 286–296. [Google Scholar] [CrossRef]
- Saeed, M.; Beigi-Boroujeni, S.; Rajabi, S.; Ashteiani, G.R.; Dolatfarahi, M.; Özcan, M. A simple, green chemistry technology for fabrication of tissue-engineered scaffolds based on mussel-inspired 3D centrifugal spun. Mater. Sci. Eng. C 2021, 121, 111849. [Google Scholar] [CrossRef]
- Garner, J.; Park, K. Chemically Modified Natural Polysaccharides to Form Gels. In Polysaccharides; Ramawat, K., Mérillon, J.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Moussa, D.G.; Aparicio, C. Present and future of tissue engineering scaffolds for dentin-pulp complex regeneration. J. Tissue Eng. Regen. Med. 2018, 13, 58–75. [Google Scholar] [CrossRef] [Green Version]
- Dalheim, M.Ø.; Vanacker, J.; Najmi, M.A.; Aachmann, F.L.; Strand, B.L.; Christensen, B.E. Efficient functionalization of alginate biomaterials. Biomaterials 2016, 80, 146–156. [Google Scholar] [CrossRef]
- Shaheen, T.; Montaser, A.; Li, S. Effect of cellulose nanocrystals on scaffolds comprising chitosan, alginate and hydroxyapatite for bone tissue engineering. Int. J. Biol. Macromol. 2019, 121, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Drury, J.L.; Mooney, D.J. Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Ganguly, S.; Das, P.; Itzhaki, E.; Hadad, E.; Gedanken, A.; Margel, S. Microwave-Synthesized Polysaccharide-Derived Carbon Dots as Therapeutic Cargoes and Toughening Agents for Elastomeric Gels. ACS Appl. Mater. Interfaces 2020, 12, 51940–51951. [Google Scholar] [CrossRef]
- Sharma, S.; Srivastava, D.; Grover, S.; Sharma, V. Biomaterials in tooth tissue engineering: A review. J. Clin. Diagn. Res. 2014, 8, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Dubey, N.; Ferreira, J.A.; Daghrery, A.; Aytac, Z.; Malda, J.; Bhaduri, S.B.; Bottino, M.C. Highly tunable bioactive fiber-reinforced hydrogel for guided bone regeneration. Acta Biomater. 2020, 113, 164–176. [Google Scholar] [CrossRef]
- Yan, J.; Miao, Y.; Tan, H.; Zhou, T.; Ling, Z.; Chen, Y.; Xing, X.; Hu, X. Injectable alginate/hydroxyapatite gel scaffold combined with gelatin microspheres for drug delivery and bone tissue engineering. Mater. Sci. Eng. C 2016, 63, 274–284. [Google Scholar] [CrossRef]
- Liu, P.; Shen, H.; Zhi, Y.; Si, J.; Shi, J.; Guo, L.; Shen, S.G. 3D bioprinting and in vitro study of bilayered membranous construct with human cells-laden alginate/gelatin composite hydrogels. Colloids Surf. B Biointerf. 2019, 181, 1026–1034. [Google Scholar] [CrossRef]
- Lopa, S.; Madry, H. Bioinspired Scaffolds for Osteochondral Regeneration. Tissue Eng. Part A 2014, 20, 2052–2076. [Google Scholar] [CrossRef]
- Joddar, B.; Garcia, E.; Casas, A.; Stewart, C. Development of functionalized multi-walled carbon-nanotube-based alginate hydrogels for enabling biomimetic technologies. Sci. Rep. 2016, 6, 32456. [Google Scholar] [CrossRef] [PubMed]
- Prati, C.; Gandolfi, M.G. Calcium silicate bioactive cements: Biological perspectives and clinical applications. Dent. Mater. 2015, 31, 351–370. [Google Scholar] [CrossRef]
- Gandolfi, M.G.; Taddei, P.; Siboni, F.; Modena, E.; Ciapetti, G.; Prati, C. Development of the foremost light-curable calcium-silicate MTA cement as root-end in oral surgery. Chemical–physical properties, bioactivity and biological behavior. Dent. Mater. 2011, 27, e134–e157. [Google Scholar] [CrossRef]
- Gandolfi, M.G.; Taddei, P.; Modena, E.; Siboni, F.; Prati, C. Biointeractivity-related versus chemi/physisorption-related apatite precursor-forming ability of current root end filling materials. J. Biomed. Mater. Res. Part B Appl. Biomater. 2013, 101, 1107–1123. [Google Scholar] [CrossRef]
- Gandolfi, M.G.; Siboni, F.; Botero, T.; Bossù, M.; Riccitiello, F.; Prati, C. Calcium Silicate and Calcium Hydroxide Materials for Pulp Capping: Biointeractivity, Porosity, Solubility and Bioactivity of Current Formulations. J. Appl. Biomater. Funct. Mater. 2015, 13, 43–60. [Google Scholar] [CrossRef]
- Taddei, P.; Tinti, A.; Gandolfi, M.G.; Rossi, P.; Prati, C. Vibrational study on the bioactivity of Portland cement-based materials for endodontic use. J. Mol. Struct. 2009, 924–926, 548–554. [Google Scholar] [CrossRef]
- Gandolfi, M.G.; Taddei, P.; Siboni, F.; Modena, E.; Ginebra, M.P.; Prati, C. Fluoride-containing nanoporous calcium-silicate MTA cements for endodontics and oral surgery: Early fluorapatite formation in a phosphate-containing solution. Int. Endod. J. 2011, 44, 938–949. [Google Scholar] [CrossRef]
- Zamparini, F.; Siboni, F.; Prati, C.; Taddei, P.; Gandolfi, M.G. Properties of calcium silicate-monobasic calcium phosphate materials for endodontics containing tantalum pentoxide and zirconium oxide. Clin. Oral Investig. 2019, 23, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, M.G.; Ciapetti, G.; Perut, F.; Taddei, P.; Modena, E.; Rossi, P.L.; Prati, C. Biomimetic calcium-silicate cements aged in simulated body solutions. Osteoblast response and analyses of apatite coating. J. Appl. Biomater. Biomech. 2010, 7, 160–170. [Google Scholar]
- Gandolfi, M.G.; Ciapetti, G.; Taddei, P.; Perut, F.; Tinti, A.; Cardoso, M.V.; Van Meerbeek, B.; Prati, C. Apatite formation on bioactive calcium-silicate cements for dentistry affects surface topography and human marrow stromal cells proliferation. Dent. Mater. 2010, 26, 974–992. [Google Scholar] [CrossRef]
- Gandolfi, M.G.; Shah, S.N.; Feng, R.; Prati, C.; Akintoye, S.O. Biomimetic Calcium-Silicate Cements Support Differentiation of Human Orofacial Mesenchymal Stem Cells. J. Endod. 2011, 37, 1102–1108. [Google Scholar] [CrossRef] [Green Version]
- Hakki, S.S.; Bozkurt, B.S.; Ozcopur, B.; Gandolfi, M.G.; Prati, C.; Belli, S. The response of cementoblasts to calcium phosphate resin-based and calcium silicate-based commercial sealers. Int. Endod. J. 2012, 46, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Tatullo, M.; Spagnuolo, G.; Codispoti, B.; Zamparini, F.; Zhang, A.; Degli Esposti, M.; Aparicio, C.; Rengo, C.; Nuzzolese, M.; Manzoli, L.; et al. PLA-Based Mineral-Doped Scaffolds Seeded with Human Periapical Cyst-Derived MSCs: A Promising Tool for Regenerative Healing in Dentistry. Materials 2019, 12, 597. [Google Scholar] [CrossRef] [Green Version]
- Gandolfi, M.G.; Spagnuolo, G.; Siboni, F.; Procino, A.; Rivieccio, V.; Pelliccioni, G.A.; Prati, C.; Rengo, S. Calcium silicate/calcium phosphate biphasic cements for vital pulp therapy: Chemical-physical properties and human pulp cells response. Clin. Oral Investig. 2015, 19, 2075–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandolfi, M.; Iezzi, G.; Piattelli, A.; Prati, C.; Scarano, A. Osteoinductive potential and bone-bonding ability of ProRoot MTA, MTA Plus and Biodentine in rabbit intramedullary model: Microchemical characterization and histological analysis. Dent. Mater. 2017, 33, e221–e238. [Google Scholar] [CrossRef]
- Gandolfi, M.G.; Taddei, P.; Tinti, A.; Dorigo, E.D.S.; Prati, C. Alpha-TCP improves the apatite-formation ability of calcium-silicate hydraulic cement soaked in phosphate solutions. Mater. Sci. Eng. C 2011, 31, 1412–1422. [Google Scholar] [CrossRef]
- Gandolfi, M.G.; Taddei, P.; Tinti, A.; Dorigo, E.D.S.; Rossi, P.L.; Prati, C. Kinetics of apatite formation on a calcium-silicate cement for root-end filling during ageing in physiological-like phosphate solutions. Clin. Oral Investig. 2009, 14, 659–668. [Google Scholar] [CrossRef] [PubMed]
- David, L.C. The constitution and specification of Portland cements. In Leas’s Chemistry of Cement and Concrete, 4th ed.; Hewlett, P.C., Ed.; Butterworth Heinemann: Oxford, UK, 1998; pp. 131–193. [Google Scholar]
- Siboni, F.; Taddei, P.; Prati, C.; Gandolfi, M.G. Properties of NeoMTA Plus and MTA Plus cements for endodontics. Int. Endod. J. 2017, 50, e83–e94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camilleri, J.; Gandolfi, M.G. Evaluation of the radiopacity of calcium silicate cements containing different radiopacifiers. Int. Endod. J. 2010, 43, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, M.G.; Zamparini, F.; Degli Esposti, M.; Chiellini, F.; Aparicio, C.; Fava, F.; Fabbri, P.; Taddei, P.; Prati, C. Polylactic acid-based porous scaffolds doped with calcium silicate and dicalcium phosphate dihydrate designed for biomedical application. Mater. Sci. Eng. C 2018, 82, 163–181. [Google Scholar] [CrossRef]
- Gandolfi, M.G.; Zamparini, F.; Degli Esposti, M.; Chiellini, F.; Fava, F.; Fabbri, P.; Taddei, P.; Prati, C. Highly porous polycaprolactone scaffolds doped with calcium silicate and dicalcium phosphate dihydrate designed for bone regeneration. Mater. Sci. Eng. C 2019, 102, 341–361. [Google Scholar] [CrossRef]
- Stoppel, W.L.; White, J.C.; Horava, S.D.; Henry, A.C.; Roberts, S.C.; Bhatia, S.R. Terminal sterilization of alginate hydrogels: Efficacy and impact on mechanical properties. J. Biomed. Mater. Res. Part B Appl. Biomater. 2014, 102, 877–884. [Google Scholar] [CrossRef]
- Valente, S.; Alviano, F.; Ciavarella, C.; Buzzi, M.; Ricci, F.; Tazzari, P.L.; Pagliaro, P.; Pasquinelli, G. Human cadaver multipotent stromal/stem cells isolated from arteries stored in liquid nitrogen for 5 years. Stem Cell Res. Ther. 2014, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, T.; Messmer, A.; Yeo, B.S.; Zhang, W.; Zenobi, R. Towards chemical analysis of nanostructures in biofilms II: Tip-enhanced Raman spectroscopy of alginates. Anal. Bioanal. Chem. 2008, 391, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Andersen, F.A.; Brecevic, L.; Beuter, G.; Dell’Amico, D.B.; Calderazzo, F.; Bjerrum, N.J.; Underhill, A.E. Infrared Spectra of Amorphous and Crystalline Calcium Carbonate. Acta Chem. Scand. 1991, 45, 1018–1024. [Google Scholar] [CrossRef]
- Nelson, D.G.; Featherstone, J.D. Preparation, analysis, and characterization of carbonated apatites. Calcif. Tissue Int. 1982, 34, 69–81. [Google Scholar]
- Pielesz, A.; Klimczak, M.; Bak, K. Raman spectroscopy and WAXS method as a tool for analyzing ion-exchange properties of alginate hydrogels. Int. J. Biol. Macromol. 2008, 43, 438–443. [Google Scholar] [CrossRef]
- Beata Łabowska, M.; Michalak, I.; Detyna, J. Methods of extraction, physicochemical properties of alginates and their applications in biomedical field—A review. Open Chem. 2019, 17, 738–762. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, R.; Farrell, P. Introduced brown algae in the North East Atlantic, with particular respect toUndaria pinnatifida (Harvey) suringar. Helgol. Meeresunters 1998, 52, 259–275. [Google Scholar] [CrossRef] [Green Version]
- Engelen, A.; Serebyakova, A.; Ang, P.; Britton-Simmon, K.; Mineur, F.; Pedersen, M.F.; Arenas, F.; Fernández, C.; Steen, S.; Svenson, R.; et al. Circumglobal Invasion by the Brown Seaweed Sargassum muticum. Ocean Mar. Biol. 2015, 53, 81–126. [Google Scholar]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [Green Version]
- Kohli, N.; Sharma, V.; Orera, A.; Sawadkar, P.; Owji, N.; Frost, O.G.; Bailey, R.J.; Snow, M.; Knowles, J.C.; Blunn, G.W.; et al. Pro-angiogenic and osteogenic composite scaffolds of fibrin, alginate and calcium phosphate for bone tissue engineering. J. Tissue Eng. 2021, 12. [Google Scholar] [CrossRef]
- Sathain, A.; Monvisade, P.; Siriphannon, P. Bioactive alginate/carrageenan/calcium silicate porous scaffolds for bone tissue engineering. Mater. Today Commun. 2021, 26, 102165. [Google Scholar] [CrossRef]
- Liu, D.; Liu, Z.; Zou, J.; Li, L.; Sui, X.; Wang, B.; Yang, N.; Wang, B. Synthesis and Characterization of a Hydroxyapatite-Sodium Alginate-Chitosan Scaffold for Bone Reg eneration. Front. Mater. 2021, 8, 69. [Google Scholar] [CrossRef]
- Solovieva, E.V.; Fedotov, A.Y.; E Mamonov, V.; Komlev, V.S.; A Panteleyev, A. Fibrinogen-modified sodium alginate as a scaffold material for skin tissue engineering. Biomed. Mater. 2017, 13, 025007. [Google Scholar] [CrossRef] [Green Version]
- Afjoul, H.; Shamloo, A.; Kamali, A. Freeze-gelled alginate/gelatin scaffolds for wound healing applications: An in vitro, in vivo study. Mater. Sci. Eng. C 2020, 113, 110957. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, G.O.; Hamilton, A.; Zhou, W.; Buser, D.; Chen, S. Implant placement and loading protocols in partially edentulous patients: A systematic review. Clin. Oral Implant. Res. 2018, 29, 106–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Yao, D.; Guo, R.; Deng, L.; Dong, A.; Zhang, J. Composites of Polymer Hydrogels and Nanoparticulate Systems for Biomedical and Pharmaceutical Applications. Nanomaterials 2015, 5, 2054–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utech, S.; Boccaccini, A.R. A review of hydrogel-based composites for biomedical applications: Enhancement of hydrogel properties by addition of rigid inorganic fillers. J. Mater. Sci. 2016, 51, 271–310. [Google Scholar] [CrossRef]
- Moini, N.; Jahandideh, A.; Anderson, G. Inorganic Nanocomposite Hydrogels: Present Knowledge and Future Challenge. In Sustainable Polymer Composites and Nanocomposites; Springer: Winsland House, Singapore, 2019; pp. 805–853. [Google Scholar]
- Saveleva, M.; Prikhozhdenko, E.; Gorin, D.; Skirtach, A.G.; Yashchenok, A.; Parakhonskiy, B. Polycaprolactone-Based, Porous CaCO3 and Ag Nanoparticle Modified Scaffolds as a SERS Platform With Molecule-Specific Adsorption. Front. Chem. 2020, 7, 888. [Google Scholar] [CrossRef] [Green Version]
- Lishchynskyi, O.; Stetsyshyn, Y.; Raczkowska, J.; Awsiuk, K.; Orzechowska, B.; Abalymov, A.; Skirtach, A.; Bernasik, A.; Nastyshyn, S.; Budkowski, A. Fabrication and Impact of Fouling-Reducing Temperature-Responsive POEGMA Coatings with Embedded CaCO3 Nanoparticles on Different Cell Lines. Materials 2021, 14, 1417. [Google Scholar] [CrossRef]
- Kaniewska, K.; Karbarz, M.; Katz, E. Nanocomposite hydrogel films and coatings—Features and applications. Appl. Mater. Today 2020, 20, 100776. [Google Scholar] [CrossRef]
- Nastyshyn, S.; Raczkowska, J.; Stetsyshyn, Y.; Orzechowska, B.; Bernasik, A.; Shymborska, Y.; Brzychczy-Włoch, M.; Gosiewski, T.; Lishchynskyi, O.; Ohar, H.; et al. Non-cytotoxic, temperature-responsive and antibacterial POEGMA based nanocomposite coatings with silver nanoparticles. RSC Adv. 2020, 10, 10155–10166. [Google Scholar] [CrossRef]
- Zamparini, F.; Pelliccioni, G.A.; Spinelli, A.; Gissi, D.B.; Gandolfi, M.G.; Prati, C. Root canal treatment of compromised teeth as alternative treatment for patients receiving bisphosphonates: 60-month results of a prospective clinical study. Int. Endod. J. 2021, 54, 156–171. [Google Scholar] [CrossRef]
- Chybowski, E.A.; Glickman, G.N.; Patel, Y.; Fleury, A.; Solomon, E.; He, J. Clinical Outcome of Non-Surgical Root Canal Treatment Using a Single-cone Technique with Endosequence Bioceramic Sealer: A Retrospective Analysis. J. Endod. 2018, 44, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Rashid, F.; Shiba, H.; Mizuno, N. The effect of extracellular calcium ion on gene expression of bone-related proteins in human pulp cells. J. Endod. 2003, 29, 104–107. [Google Scholar] [CrossRef]
- Matsumoto, S.; Hayashi, M.; Suzuki, Y.; Suzuki, N.; Maeno, M.; Ogiso, B. Calcium Ions Released from Mineral Trioxide Aggregate Convert the Differentiation Pathway of C2C12 Cells into Osteoblast Lineage. J. Endod. 2013, 39, 68–75. [Google Scholar] [CrossRef]
- Day, R.M. Bioactive Glass Stimulates the Secretion of Angiogenic Growth Factors and Angiogenesis in Vitro. Tissue Eng. 2005, 11, 768–777. [Google Scholar] [CrossRef]
- Zhai, W.; Lu, H.; Chen, L.; Lin, X.; Huang, Y.; Dai, K.; Naoki, K.; Chen, G.; Chang, J. Silicate bioceramics induce angiogenesis during bone regeneration. Acta Biomater. 2012, 8, 341–349. [Google Scholar] [CrossRef]
- Takagi, S.; Chow, L.; Ishikawa, K. Formation of hydroxyapatite in new calcium phosphate cements. Biomaterials 1998, 19, 1593–1599. [Google Scholar] [CrossRef]
- Okabe, T.; Sakamoto, M.; Takeuchi, H.; Matsushima, K. Effects of pH on Mineralization Ability of Human Dental Pulp Cells. J. Endod. 2006, 32, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wei, L.; Liu, X.; Li, J.; Li, B.; Wang, G.; Meng, F. Influences of ionic dissolution products of dicalcium silicate coating on osteoblastic proliferation, differentiation and gene expression. Acta Biomater. 2009, 5, 1284–1293. [Google Scholar] [CrossRef]
- Gandolfi, M.G.; Taddei, P.; Tinti, A.; Prati, C. Apatite forming ability of ProRoot MTA. Int. Endod. J. 2010, 43, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, R.T.; Iyer, B.S. Relationship between collagen synthesis and expression of the osteoblast phenotype in MC3T3-E1 cells. J. Bone Miner. Res. 2009, 7, 235–246. [Google Scholar] [CrossRef]
- Vanchinathan, V.; Mizramani, N.; Kantipudi, R.; Schwartz, E.J.; Sundram, U.N. The Vascular Marker CD31 Also Highlights Histiocytes and Histiocyte-Like Cells within Cutaneous Tumors. Am. J. Clin. Pathol. 2015, 143, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Bramfeld, H.; Sabra, G.; Centis, V.; Vermette, P. Scaffold Vascularization: A Challenge for Three-Dimensional Tissue Engineering. Curr. Med. Chem. 2010, 17, 3944–3967. [Google Scholar] [CrossRef]
- Gandolfi, M.G.; Gardin, C.; Zamparini, F.; Ferroni, L.; Degli Esposti, M.; Parchi, G.; Ercan, B.; Manzoli, L.; Fava, F.; Fabbri, P.; et al. Mineral-Doped Poly(L-lactide) Acid Scaffolds Enriched with Exosomes Improve Osteogenic Commitment of Human Adipose-Derived Mesenchymal Stem Cells. Nanomaterials 2020, 10, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forni, M.; Bernardini, C.; Zamparini, F.; Zannoni, A.; Salaroli, R.; Ventrella, D.; Parchi, G.; Degli Esposti, M.; Polimeni, A.; Fabbri, P.; et al. Vascular Wall–Mesenchymal Stem Cells Differentiation on 3D Biodegradable Highly Porous CaSi-DCPD Doped Poly (α-hydroxy) Acids Scaffolds for Bone Regeneration. Nanomaterials 2020, 10, 243. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Zhang, X.; Song, W.; Pan, T.; Wang, H.; Ning, T.; Wei, Q.; Xu, H.H.; Wu, B.; Ma, D. Effects of 3-dimensional Bioprinting Alginate/Gelatin Hydrogel Scaffold Extract on Proliferation and Differentiation of Human Dental Pulp Stem Cells. J. Endod. 2019, 45, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Karageorgiou, V.; Kaplan, D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials 2005, 26, 5474–5491. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.A.; Mestres, G. Role of pore size and morphology in musculo-skeletal tissue regeneration. Mater. Sci. Eng. C 2016, 61, 922–939. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence | Reverse Sequence |
|---|---|---|
| GAPDH | 5′–AATGGGCAGCCGTTAGGAAA–3′ | 5′–AGGAGAAATCGGGCCAGCTA–3′ |
| CD31 | 5′–CACAGATGAGAACCACGCCT–3′ | 5′–GGCCCCTCAGAACAACAT–3′ |
| ALP | 5′–GACCTCCTCGGAAGACACT–3′ | 5′–TGAAGGGCTTCTTGTCTGT–3′ |
| OCN | 5′–CACCGAGACACCATGAGAGC–3′ | 5′–CTGCTTGGACACAAAGGCT–3′ |
| Materials | 0–3 h | 3 h–1 Day | 1–3 Days | 3–7 Days | 7–14 Days | 14–28 Days | Cumulative |
|---|---|---|---|---|---|---|---|
| PP-16:16 | 43.73 ± 5.7 aA | 60.85 ± 10.89 aB | 29.52 ± 5.68 aC | 37.53 ± 10.31 aA | 31.95 ± 4.65 aC | 32.30 ± 13.18 cA | 192.17 ± 14.45 a |
| PP-33:22 | 44.64 ± 11.06 aA | 53.078 ± 14.8 aB | 54.95 ± 15.07 bB | 51.95 ± 9.1 bB | 49.01 ± 8.5 bAB | 44.15 ± 11.75 bA | 253.15 ± 40.64 b |
| PP-31:31 | 68.19 ± 19.01 bA | 71.65 ± 7.6 bA | 51.51 ± 5.3 bB | 46.72 ± 17.3 bB | 32.92 ± 9.01 aC | 27.73 ± 7.01 aC | 230.54 ± 33.05 b |
| PP-CTRL | 1.56 ± 0.28 cA | 6.47 ± 0.04 cB | 3.58 ± 0.68 cA | 2.31 ± 0.94 cA | 2.41 ± 1.43 cA | 2.46 ± 1.45 cA | 17.23 ± 3.9 c |
| Materials | 3 h | 1 Day | 3 Days | 7 Days | 14 Days | 28 Days |
|---|---|---|---|---|---|---|
| PP-16:16 | 8.82 ± 0.10 aA | 8.86 ± 0.28 aA | 8.614 ± 0.34 aA | 8.45 ± 0.13 aB | 8.33 ± 0.15 aB | 8.13 ± 0.16 aC |
| PP-33:22 | 8.79 ± 0.20 aA | 9.23 ± 0.23 bB | 8.86 ± 0.12 abA | 8.61 ± 0.18 bA | 8.49 ± 0.09 aC | 8.41 ± 0.23 aC |
| PP-31:31 | 9.07 ± 0.18 bA | 9.27 ± 0.17 bA | 8.98 ± 0.18 bA | 8.64 ± 0.25 bB | 8.46 ± 0.13 aBC | 8.32 ± 0.14 aC |
| PP-CTRL | 7.74 ± 0.08 cA | 7.83 ± 0.17 cA | 7.42 ± 0.16 cA | 7.75 ± 0.23 aA | 7.69 ± 0.16 bA | 7.68 ± 0.45 bA |
| WS (%) | S (%) | P (%) | VOP (cm3) | VIP (cm3) | |
|---|---|---|---|---|---|
| PP-16:16 | 74.45 ± 15.5 a | 5.14 ± 1.51 a | 46.65 ± 7.45 a | 1.554 ± 0.45 a | 1.764 ± 0.26 a |
| PP-33:22 | 147.15 ± 26.2 b | 10.12 ± 3.2 b | 63.15 ± 9.25 b | 2.123 ± 0.38 b | 1.265 ± 0.24 b |
| PP-31:31 | 224.29 ± 26.56 c | 21.31 ± 9.4 c | 76.23 ± 12.65 b | 2.514 ± 0.15 b | 0.912 ± 0.22 b |
| PP-CTRL | 54.45 ± 16.0 a | 5.26 ± 3.94 a | 36.47 ± 0.91 c | 1.21 ± 0.15 a | 2.17 ± 0.15 c |
| PP-16:16 | <1 |
| PP-33:22 | <1 |
| PP-31:31 | <1 |
| PP-CTRL | <1 |
| PP-16:16 | 96 |
| PP-33:22 | 96 |
| PP-31:31 | 96 |
| PP-CTRL | Unset after 144 h |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandolfi, M.G.; Zamparini, F.; Valente, S.; Parchi, G.; Pasquinelli, G.; Taddei, P.; Prati, C. Green Hydrogels Composed of Sodium Mannuronate/Guluronate, Gelatin and Biointeractive Calcium Silicates/Dicalcium Phosphate Dihydrate Designed for Oral Bone Defects Regeneration. Nanomaterials 2021, 11, 3439. https://doi.org/10.3390/nano11123439
Gandolfi MG, Zamparini F, Valente S, Parchi G, Pasquinelli G, Taddei P, Prati C. Green Hydrogels Composed of Sodium Mannuronate/Guluronate, Gelatin and Biointeractive Calcium Silicates/Dicalcium Phosphate Dihydrate Designed for Oral Bone Defects Regeneration. Nanomaterials. 2021; 11(12):3439. https://doi.org/10.3390/nano11123439
Chicago/Turabian StyleGandolfi, Maria Giovanna, Fausto Zamparini, Sabrina Valente, Greta Parchi, Gianandrea Pasquinelli, Paola Taddei, and Carlo Prati. 2021. "Green Hydrogels Composed of Sodium Mannuronate/Guluronate, Gelatin and Biointeractive Calcium Silicates/Dicalcium Phosphate Dihydrate Designed for Oral Bone Defects Regeneration" Nanomaterials 11, no. 12: 3439. https://doi.org/10.3390/nano11123439
APA StyleGandolfi, M. G., Zamparini, F., Valente, S., Parchi, G., Pasquinelli, G., Taddei, P., & Prati, C. (2021). Green Hydrogels Composed of Sodium Mannuronate/Guluronate, Gelatin and Biointeractive Calcium Silicates/Dicalcium Phosphate Dihydrate Designed for Oral Bone Defects Regeneration. Nanomaterials, 11(12), 3439. https://doi.org/10.3390/nano11123439