Molecular imprinted polymers (MIPs) are crosslinked polymers with specific binding moieties for selectively binding target molecules. The interaction between the MIP and the corresponding target molecule is mimicking nature via various complementary binding processes (lock and key model), e.g., antigen-antibody, DNA-protein, ligand-receptor, and peptide–protein interaction. Therefore, imprinted polymers are expected to show a high affinity and selectivity towards the target molecule and should be able to discriminate and isolate them from closely related structures. MIPs can be prepared for a wide variety of targets, e.g., proteins, viruses, drugs or pesticides [
1,
2,
3,
4]. In contrast to natural receptors, MIPs can withstand much harsher conditions, such as high temperature, pressure, pH and organic solvents. Additionally, synthetic polymeric receptors are also less expensive to synthesize and their preparation can be easily scaled up for commercial and industrial applications [
4]. The technology of molecular imprinting enables the creation of artificial binding sites within a polymer matrix. These sites are tailored in situ via co-polymerization of functional monomers and crosslinkers around the template [
5,
6]. Afterwards, the template molecule is extracted from the obtained polymer, leaving complementary cavities (regarding shape, size, and distribution of functional groups) within the polymeric network [
1,
2,
3,
4]. For this reason, MIPs show notable potential for applications in many analytical areas, such as clinical analysis, medical diagnostics, environmental monitoring, but more recently also for assisting drug delivery [
4,
7]. For example, they can be employed in high-performance liquid chromatography [
8], solid phase extraction [
9], sensors [
10], separation [
11,
12], and catalysis [
13,
14]. However, the rebinding process depends on the diffusion of the template on the recognition sites. Therefore, small molecules are easier to imprint than macromolecules or entire biological species (i.e., peptides, proteins, DNA, viruses and bacteria), which can be limited by size, mass transfer, conformational instability, etc. [
15] Specifically, mass transfer to and through the crosslinked polymer matrix is limited due to the molecular weight, which limits template removal and fast rebinding, which also results in a low binding capacity [
15,
16,
17]. Because of this issue, MIPs prepared using the common bulky polymerization technique are not useful for proteins and other macromolecules [
5]. Moreover, the protein molecules can be trapped deeply in the polymer matrix, which is resulting in a deteriorated elution [
18,
19]. Surface imprinting via nanoscale carrier materials is well suited for proteins as a template, considering the mentioned problems [
15]. Moreover, the conditions here can be adapted more to the properties of the proteins. In the present study, core-shell imprinting was selected, which is a special application of the surface imprinting technique. Commonly used support materials for surface imprinting technique are TiO
2, [
20] Fe
2O
3, [
21] and carbonaceous materials [
22]. However, SiO
2 was the most frequently used solid support material reported by numerous studies [
5]. However, protein imprinting is still in the early stage, mainly because of the poor stability of the template protein during the polymerization, their limited solubility and stability (i.e., pH, temperature, etc.), as well as structural flexibility in solution [
23].
The protein used as a template in our study was the Hen egg white (HEW) lysozyme. HEW lysozyme consists of 129 amino acids, with a molar mass of 14.3 kDa. It is a single peptide chain protein and contains 4 pairs of cysteines on the molecule forming 4 S-S bonds. The lysozyme is elliptical and has a size of 4.5 nm × 3.0 nm × 3.0 nm. The isoelectric point is at pI 11.35. Lysozyme exhibits a globular structure composed of four α-helices and five β-sheets, with four disulfide bridges [
24]. The enzyme is active over a wide pH range (6.0 to 9.0). Maximum activity is observed at pH 6.2 [
25,
26,
27,
28]. The internal structure of lysozyme is almost non-polar, and hydrophobic interactions play an important role in the folded conformation of lysozyme [
25,
27]. Lysozymes preferentially hydrolyze the β-1,4-glycosidic bond between N-acetyl-D-muramic acid and 2-acetylamino-2-deoxy-D-glucose (=N-acetyl-D-glucosamine) in the sugar chains of the peptidoglucan scaffold of the bacterial cell wall. As a result, these enzymes have antibacterial properties. Lysozyme is abundant in a number of biological secretions, such as saliva, tears, sweat, human milk, earwax, and nasal and intestinal mucosa, as well as in blood plasma. Large amounts of lysozyme are found in hen egg white. In food industries, HEW lysozyme is approved within the EU as a food additive (i.e., number E 1105). For example, it is used at concentrations of up to 50 g/hl in winemaking to control acid biodegradation (i.e., bacterial breakdown of malic acid into lactic acid). Furthermore, it is used as a conservation agent for aged cheese and to preserve beer that has not been pasteurized or sterile-filtered [
25,
26,
27]. For these reasons, it is necessary to be able to isolate and purify lysozyme. A common method for this so far is pH precipitation (salting out) and via chromatography on a cation exchange column. The salting-out process, based on the addition of a neutral salt, compresses the solvation layer and increases protein–protein interactions. When the salt concentration of a solution is enhanced, the charges on the surface of the protein interact with the salt, not with the water, exposing hydrophobic regions on the protein surface and causing the protein to precipitate out of solution [
29]. The isoelectric point (pI) also plays an important role in this process. If the pH conforms to the pI, the protein has no netto charge. The negative and positive charges balance each other out, which reduces the repulsive electrostatic forces so that the attractive forces prevail. The attractive forces lead to aggregation and precipitation [
29]. Thus, the solubility is the lowest at or close to this point. Lysozyme has a minimum solubility at around pH 10 [
29]. This state of precipitation can be partially irreversible, so that the protein denatures. Moreover, the high pH can also lead to denaturation. This point is a disadvantage of this method [
29]. Furthermore, the cation exchange chromatography works with the pI of the proteins. It can separate molecules based on their netto surface charge [
30]. For this device, it is important that the target molecule, in this case lysozyme, has a positive surface charge. Thus, a pH of below 9 is required. This method involves several steps: the buffer preparation, the column equilibration, the sample loading, the column washing and the elution of the protein [
30,
31]. This involves a certain amount of effort. Furthermore, the effectiveness of this method is also highly pH dependent, if the pH is too close to the pI the amount of positive charges decreases. Moreover, to separate several proteins from each other using this method a salt gradient is then used to separate the protein of interest from other bound proteins, so they will be eluted in an order depending on their net surface charge [
30,
31]. This requires knowledge about their pIs. These points make this method more sophisticated in its usage. MIPs have the potential to represent an alternative, inexpensive method for lysozyme purification.






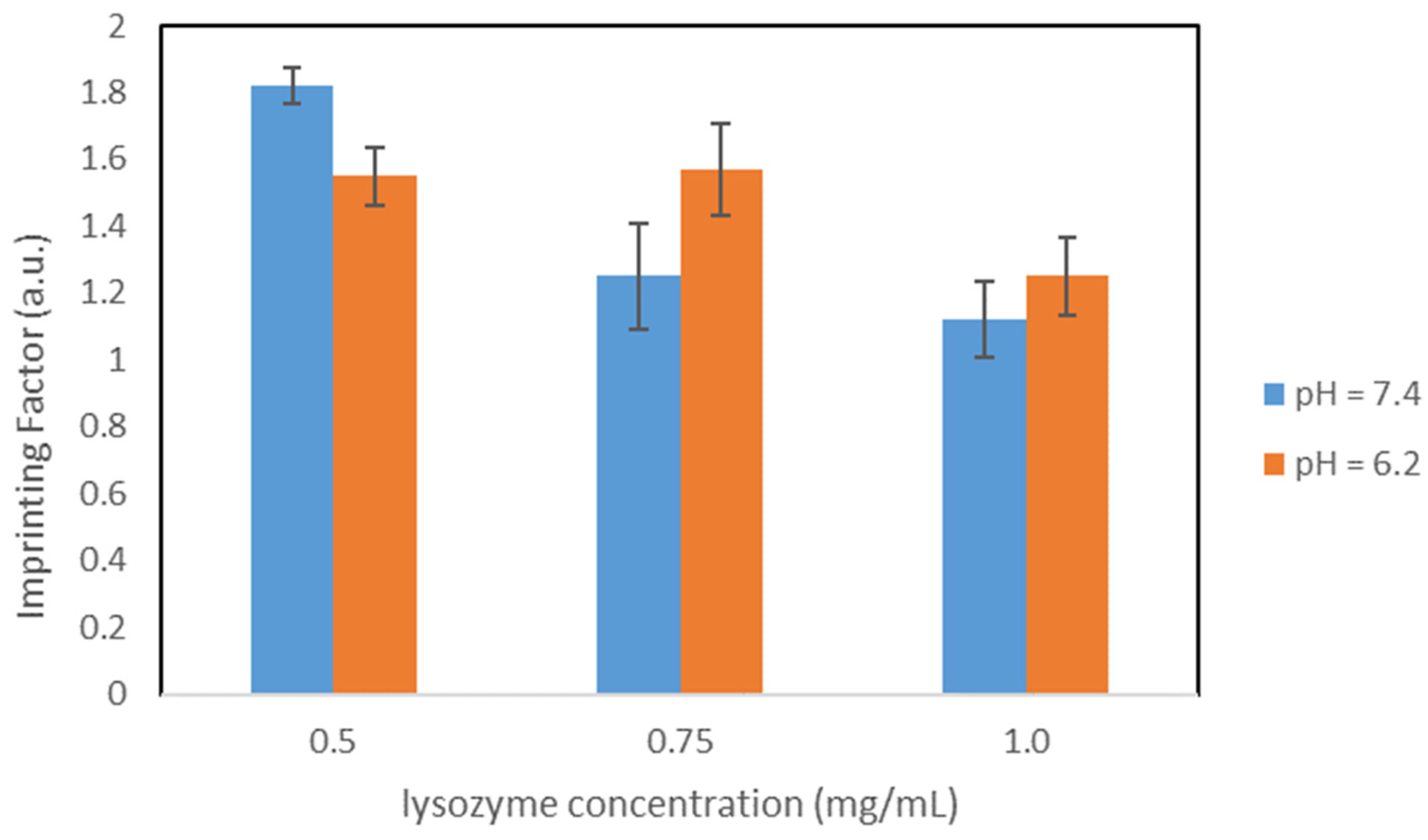
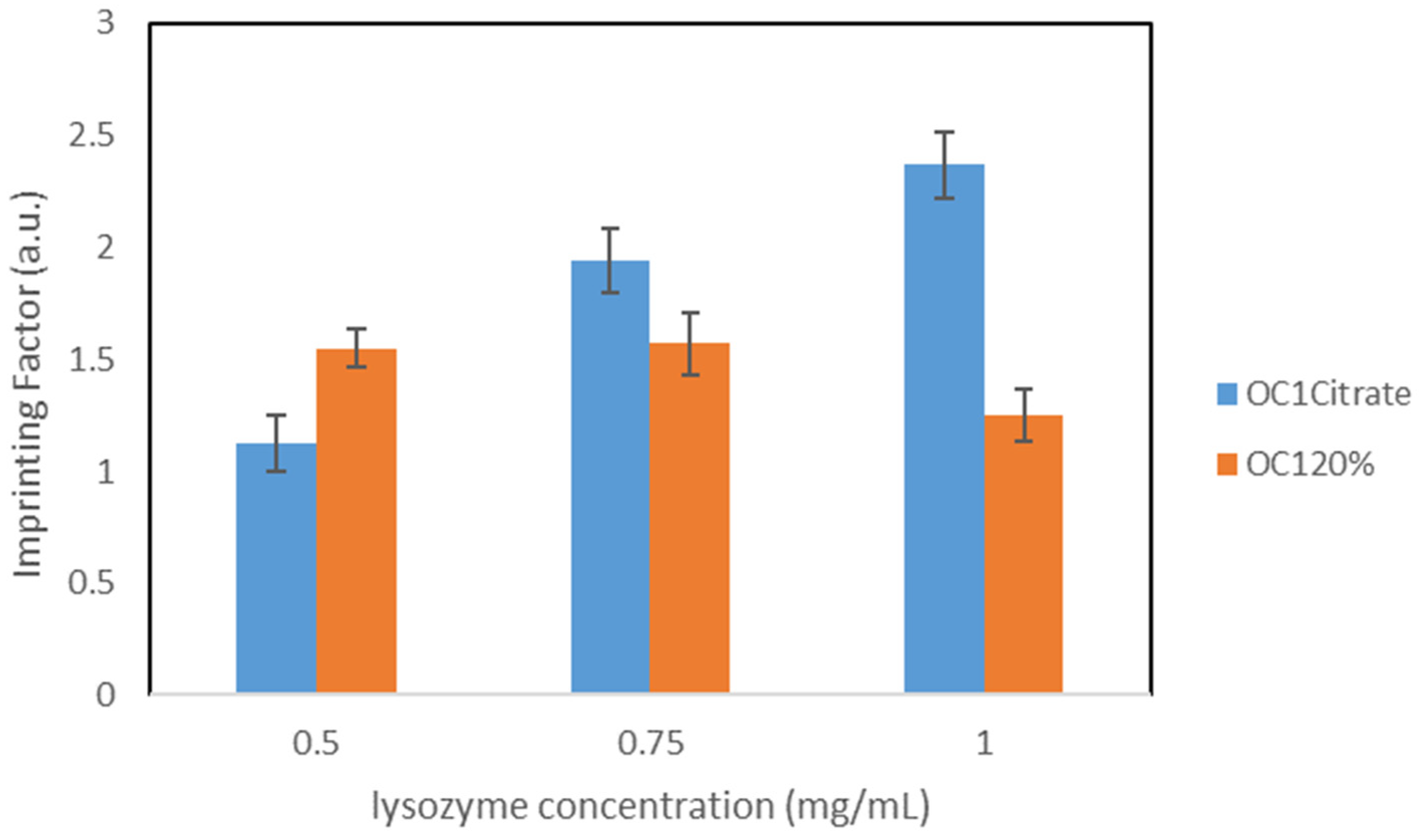
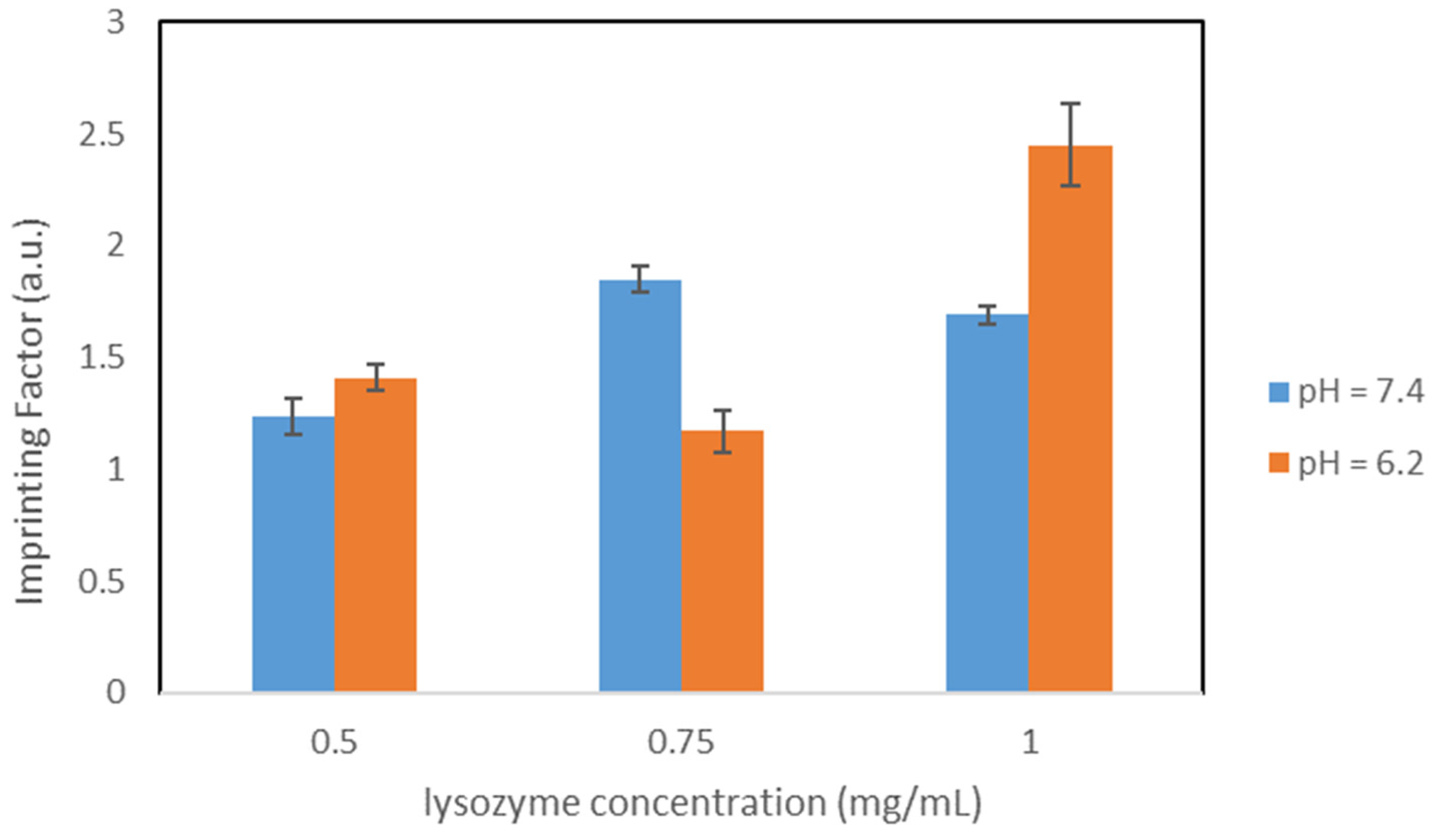
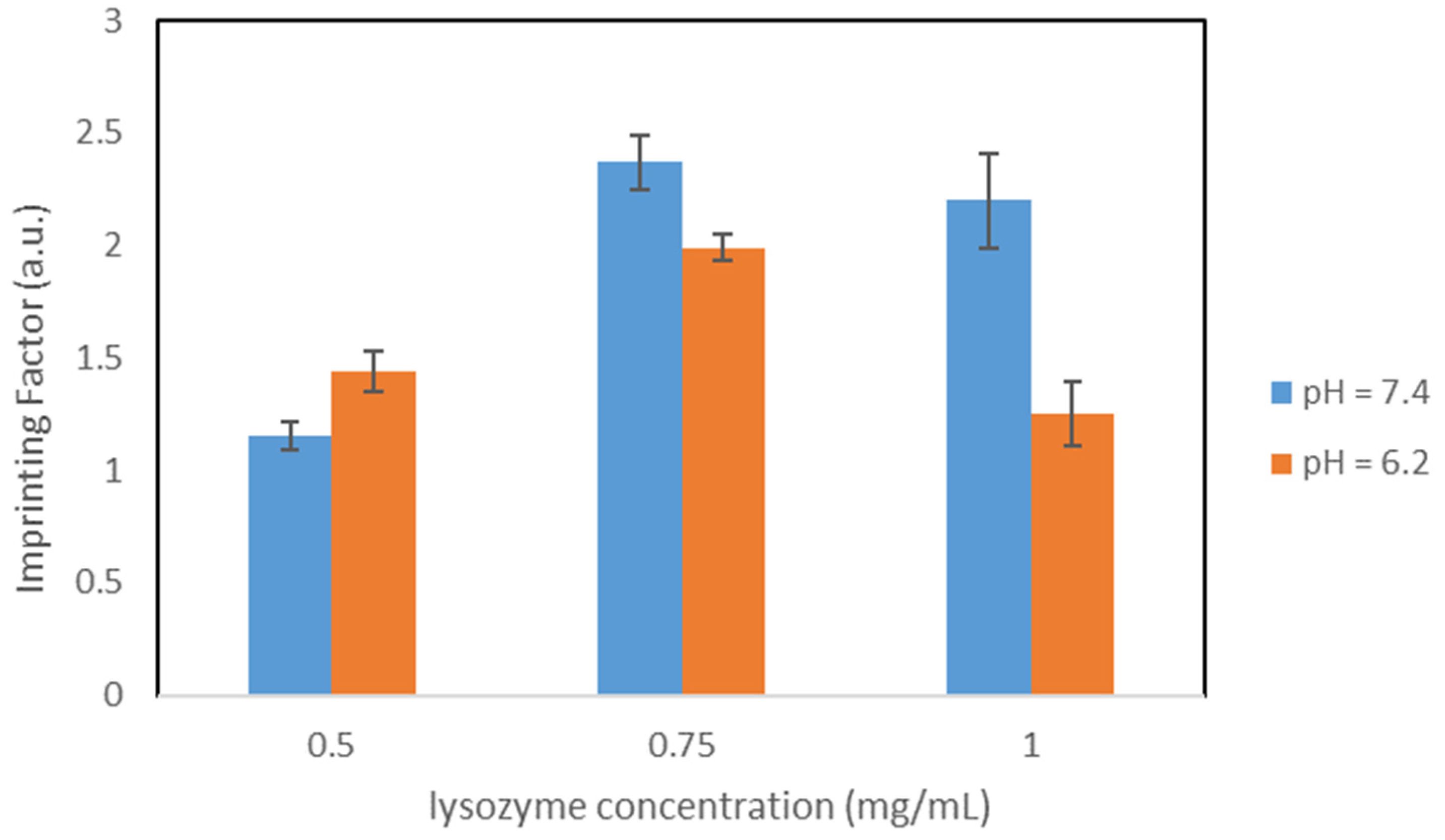
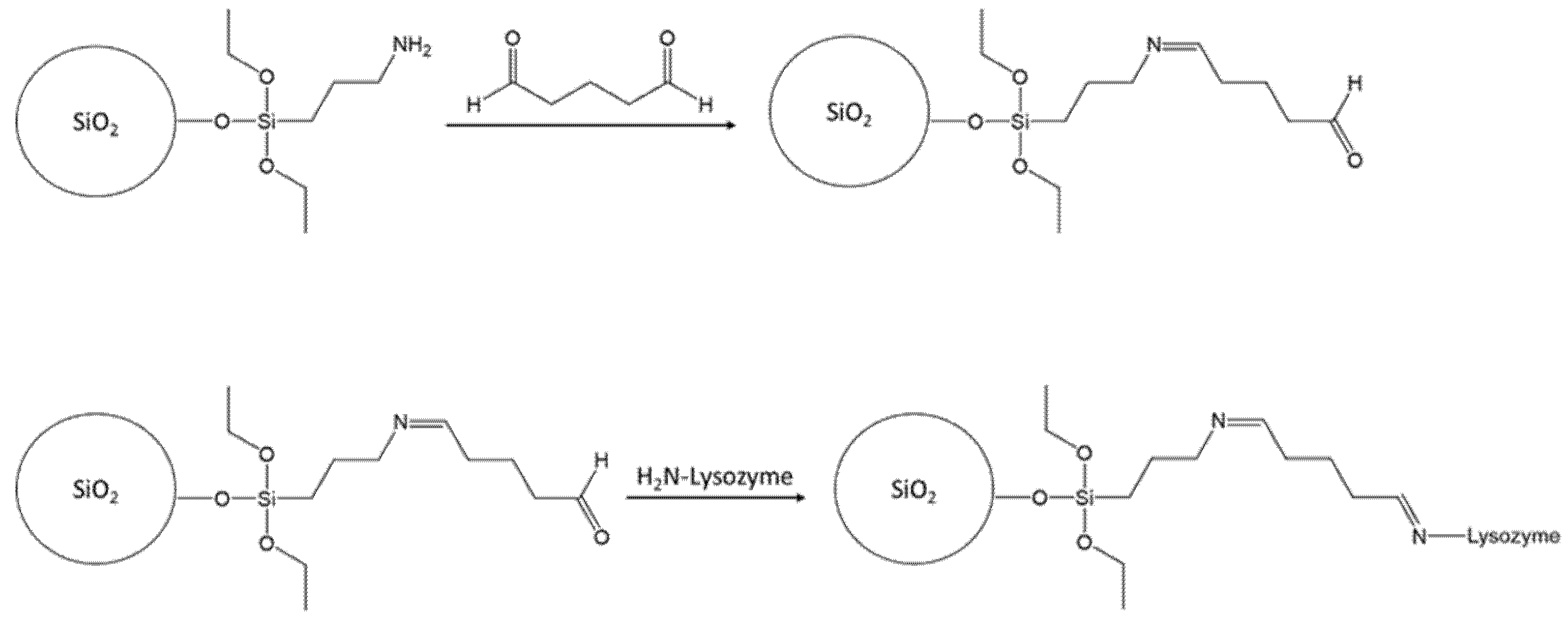
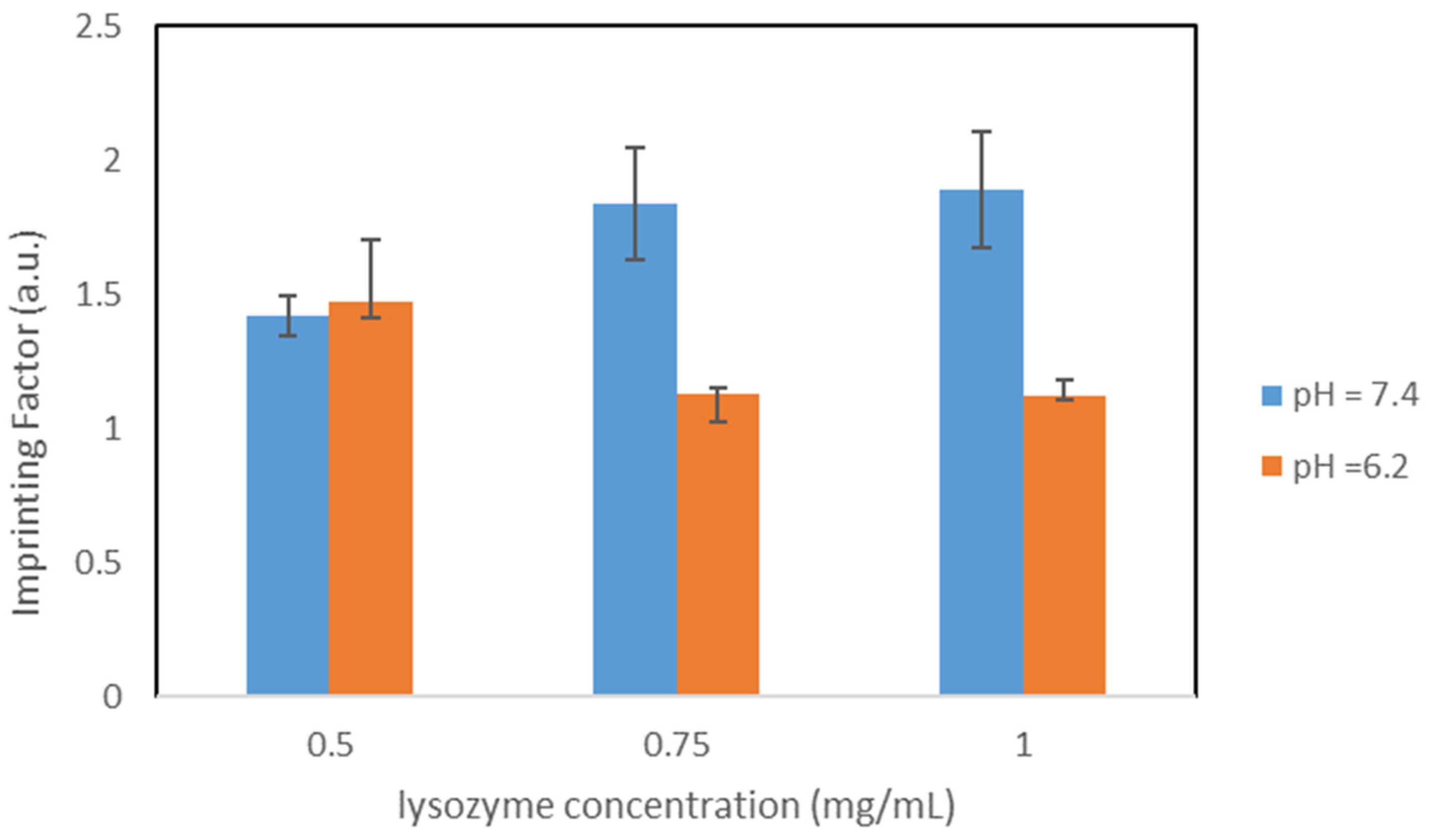
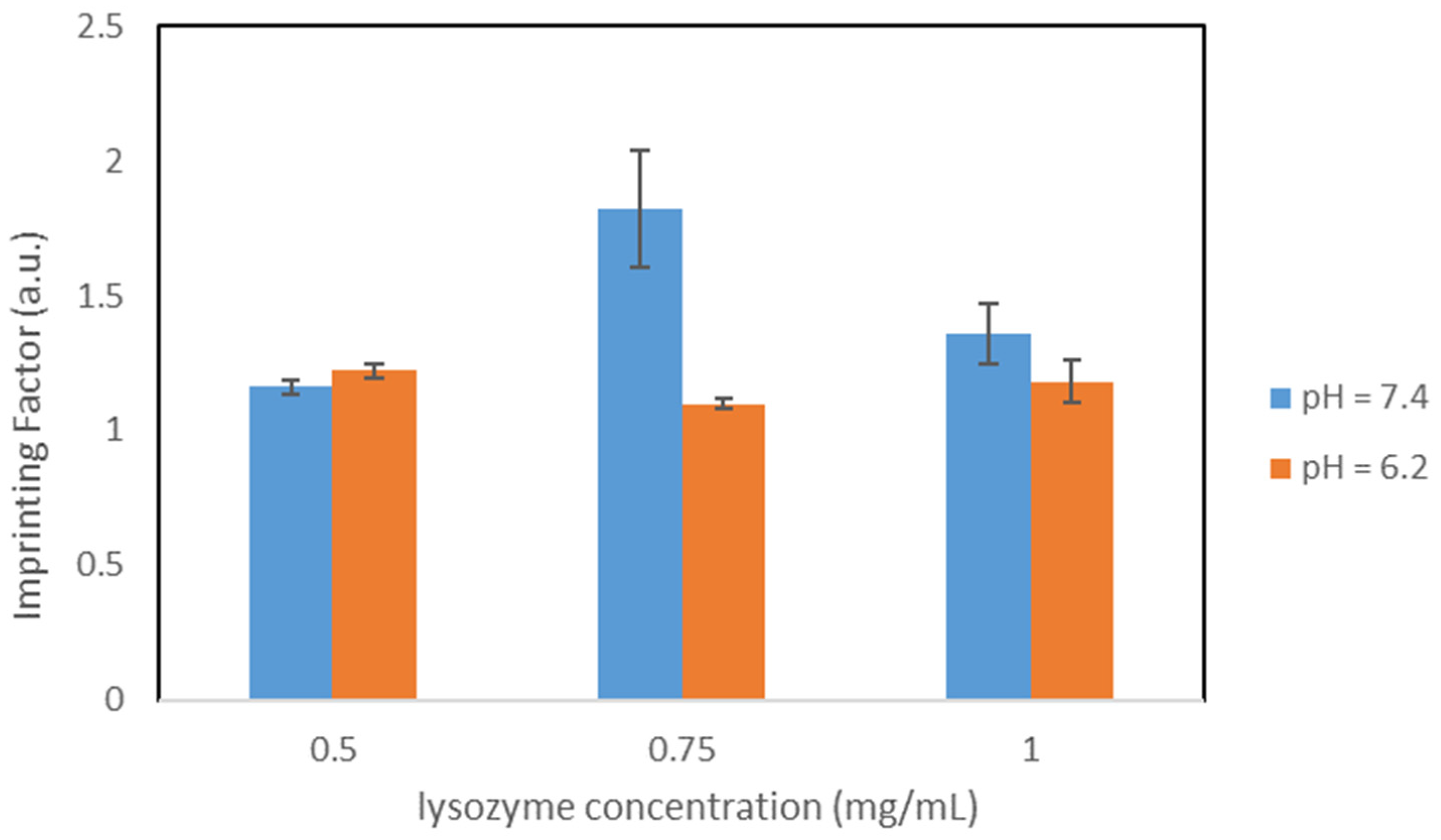

{kind=link}
{kind=link}
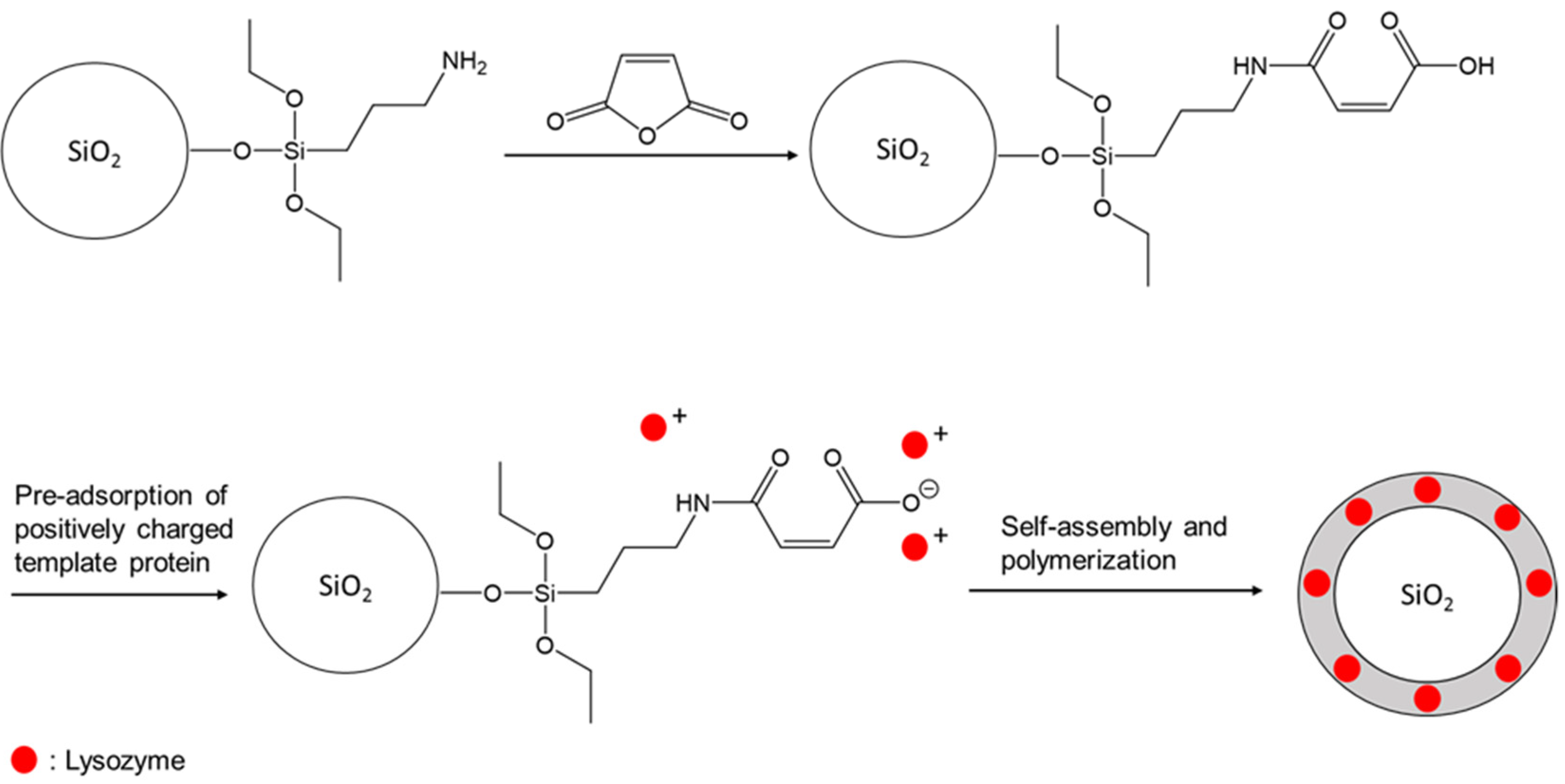{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
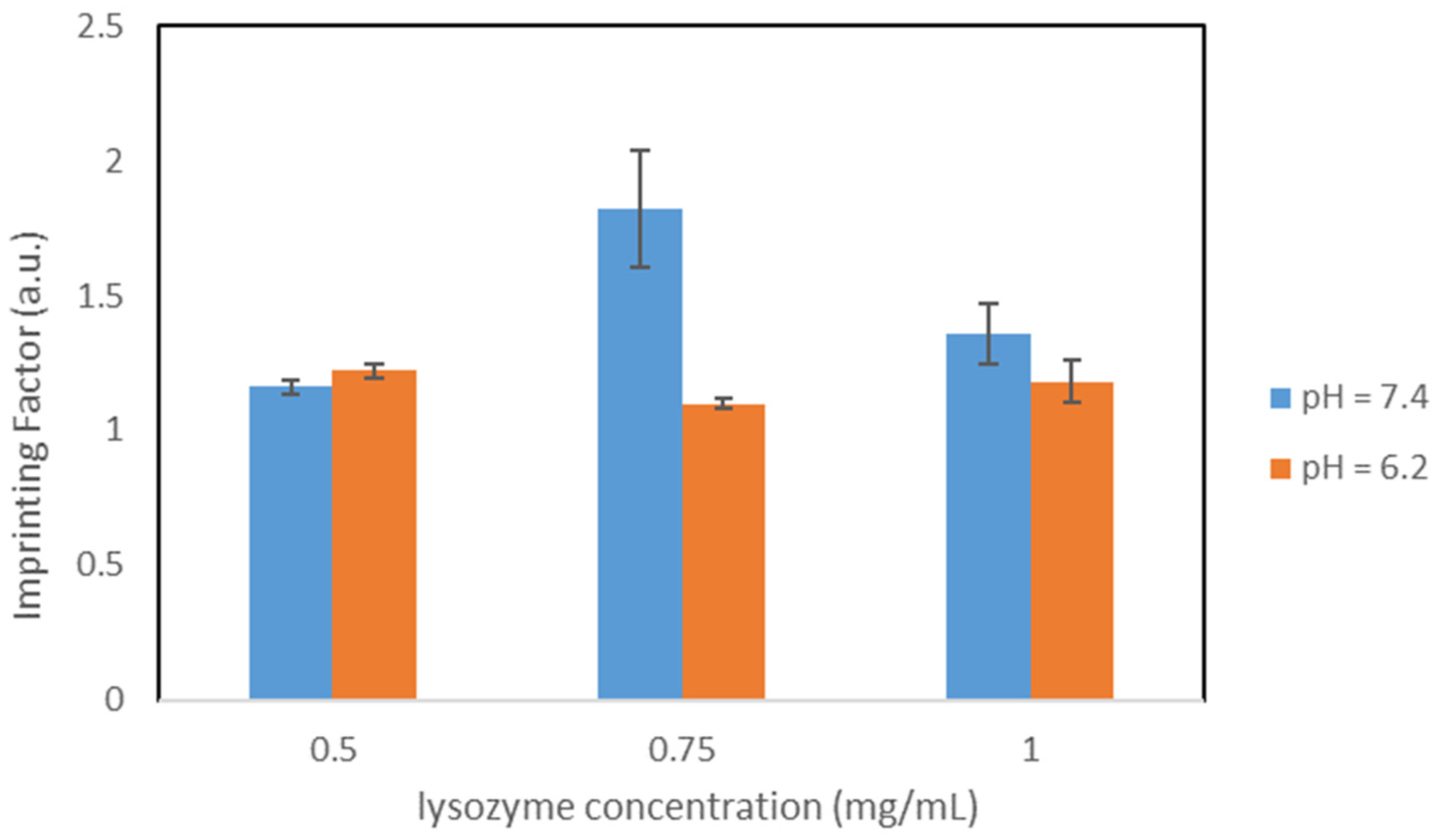{kind=link}