
Interpretation of Single-Molecule Force Experiments on Proteins Using Normal Mode Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Considerations
2.2. Procedure
- Examine the structure to identify the expected effects of the force on its general form, taking into consideration the size and location of the packed core. Decide whether or not additional truncated forms should be studied.
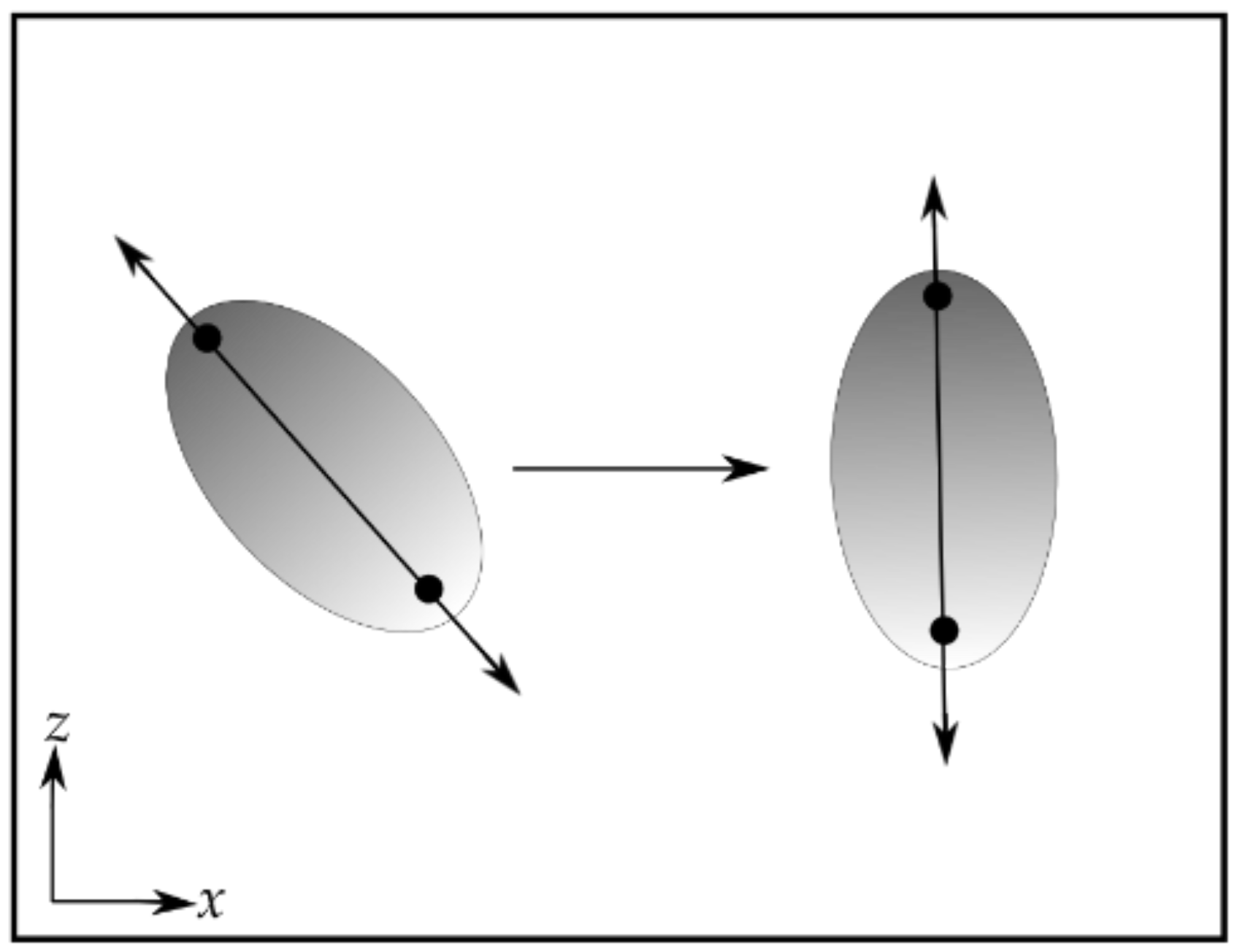
- Rotate the protein so that the direction of applied force in the experiments will be along one of the principal coordinate axes. This will allow those modes that separate the domains or subdomains to be more easily identified. In the case of all four of the proteins described below, it was most convenient to align the protein to the z-axis, though any other axis might have been used.
- Calculate the normal modes. This can be achieved using either conventional all-atom NMA or one of the coarse-grained ENMs. The first 50 non-zero normal modes will be sufficient for most purposes, but all modes will be needed to calculate the mechanical stiffness.
- Examine the results. For single-domain proteins, plots of the mean-squared fluctuations and the mechanical stiffness should be examined. In addition to these, for multi-domain proteins, the z-components of the principal components should be examined in order to identify those modes that move the domains in opposite directions along the direction of the applied force. These modes are likely to be amplified by the application of the force in the given direction. The z-components of the principal components should be examined. To aid in selecting these modes, it may be helpful to calculate the mean of the z-components of the individual domains.
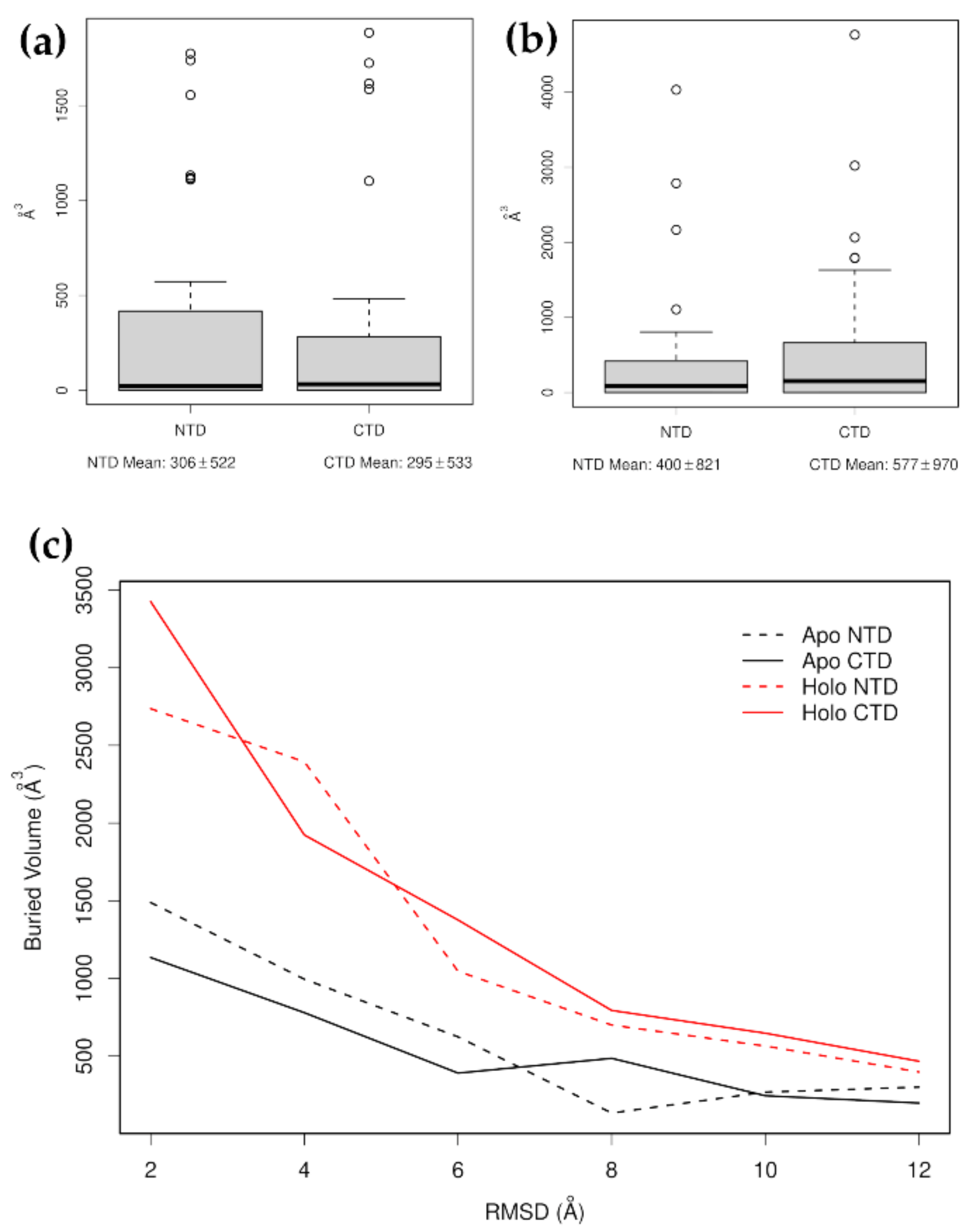
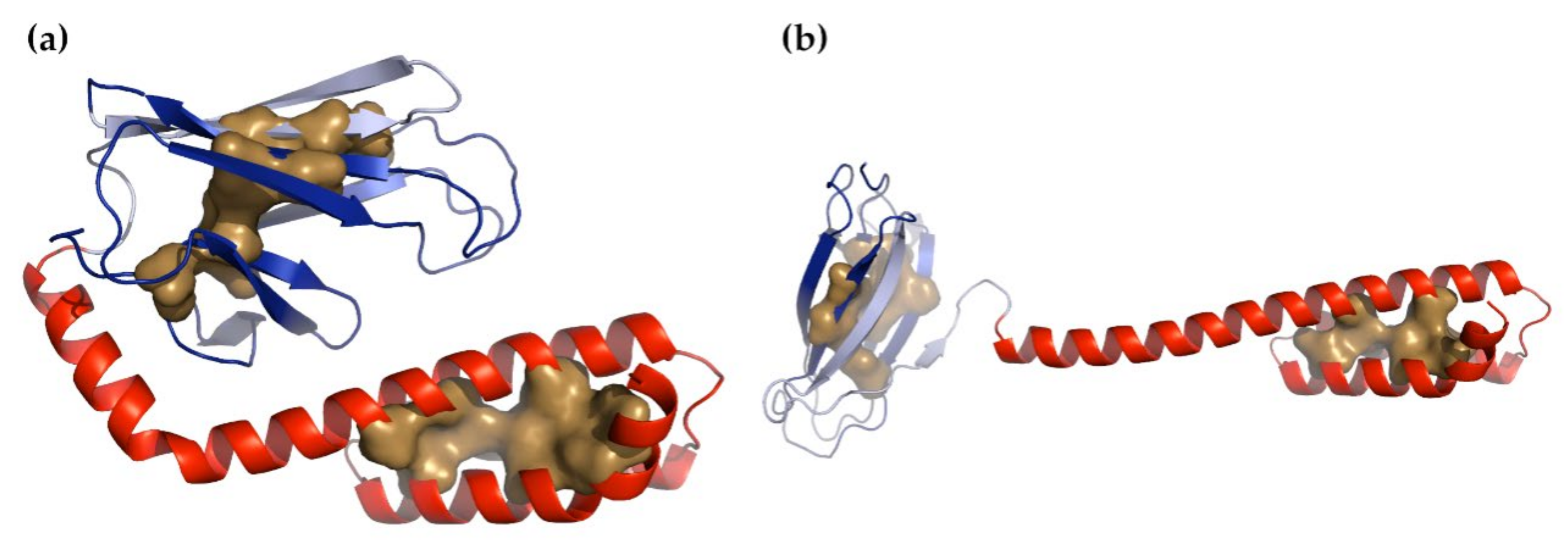
- Examine the characteristic motions of each selected mode. Create pseudo-trajectories to view the effects of these modes on the protein’s overall motion. Often, only a subset of the selected modes will actually produce motions that are relevant for studying the effect of the applied forces. For example, in several cases, the z-components may show clear shifts in opposite directions, but these may arise from rotational motions rather than lateral shifts. Those modes that shift the positions of the domains primarily through lateral shifts should be selected and used to create distorted structures. How these distorted structures are generated will depend on the software package being used. For example, ProDy [33,34] calculates an ensemble of structures using a target RMSD, but with the direction and amplitudes of the individual modes chosen randomly. In this case, the resulting ensembles have to be evaluated statistically. ElNémo [35], on the other hand, allows individual conformations to be calculated by specifying the amplitude and direction of individual modes; in this case, it is sufficient to calculate only one or a few distorted structures. These distorted structures can then be examined to determine which domain or other part of the structure is likely to come apart first. This can be approximately quantized by following the volume of the buried residues of each distorted domain: the domain with the lowest overall volume of the buried core is likely to have the lowest structural integrity.
3. Results
3.1. T4 Lysozyme
3.2. Hsp70
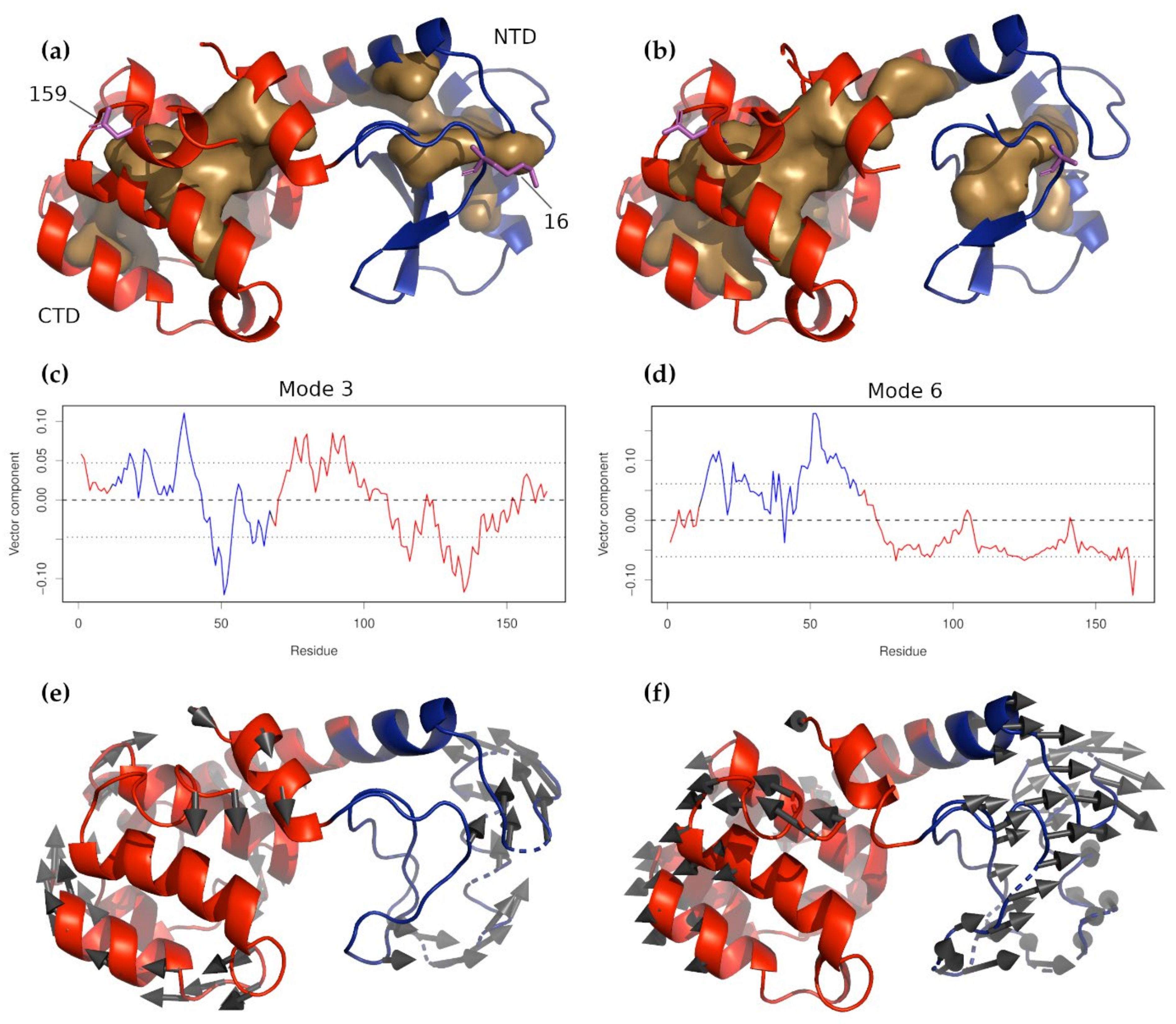
3.2.1. Nucleotide-Binding Domain
3.2.2. Substrate-Binding Domain
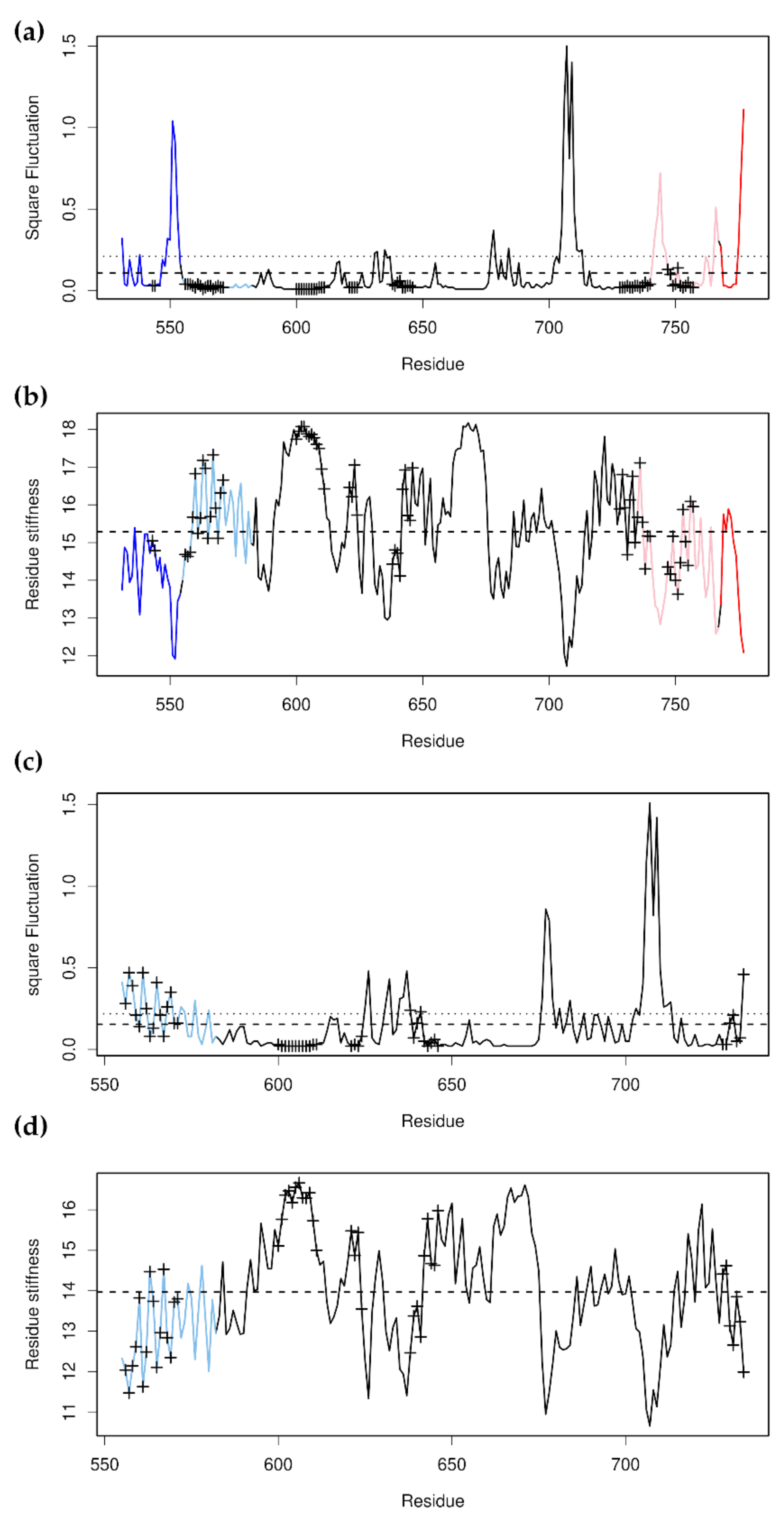
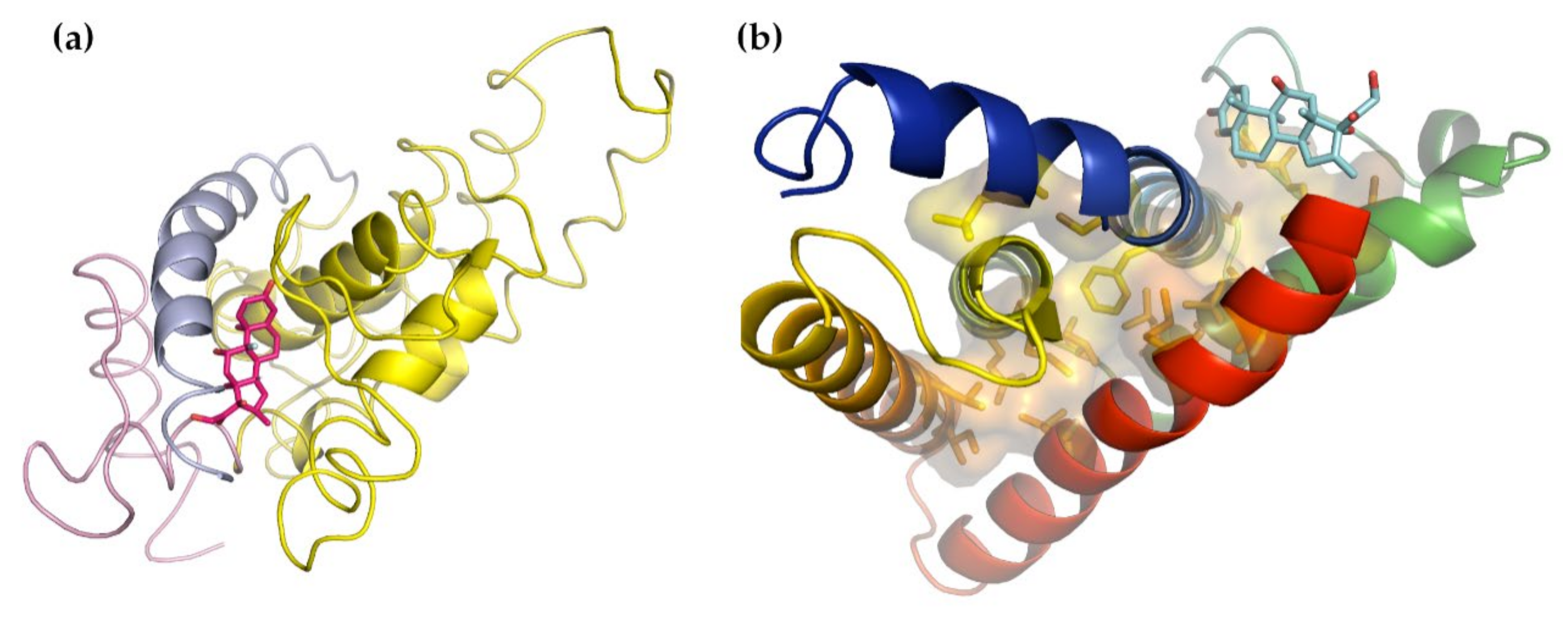
3.3. Glucocorticoid Receptor
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kellermayer, M.S.; Smith, S.B.; Granzier, H.L.; Bustamante, C. Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science 1997, 276, 1112–1116. [Google Scholar] [CrossRef] [Green Version]
- Rief, M.; Gautel, M.; Oesterhelt, F.; Fernandez, J.M.; Gaub, H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science 1997, 276, 1109–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, B.; Lipmanm, E.A.; Eaton, W.A. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature 2002, 419, 743–747. [Google Scholar]
- Rhoades, E.; Gussakovsky, E.; Haran, G. Watching proteins fold one molecule at a time. Proc. Natl. Acad. Sci. USA 2003, 100, 3197–3202. [Google Scholar] [PubMed] [Green Version]
- Neupane, K.; Foster, D.A.; Dee, D.R.; Yu, H.; Wang, F.; Woodside, M.T. Direct observation of transition paths during the folding of proteins and nucleic acids. Science 2016, 352, 239–242. [Google Scholar] [PubMed]
- Greenleaf, W.J.; Woodside, M.T.; Block, S.M. High-resolution, single-molecule measurements of biomolecular motion. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 171–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashaghi, A.; Bezrukavnikov, S.; Minde, D.P.; Wentink, A.S.; Kityk, R.; Zachmann-Brand, B.; Mayer, M.P.; Kramer, G.; Bukau, B.; Tans, S.J. Alternative modes of client binding enable functional plasticity of Hsp70. Nature 2016, 539, 448–451. [Google Scholar] [PubMed]
- Del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching single talin rod molecules activates vinculin binding. Science. 2009, 323, 638–641. [Google Scholar]
- Jagannathan, B.; Marqusee, S. Protein Folding and Unfolding Under Force. Biopolymers 2013, 99, 860–869. [Google Scholar]
- Crooks, G.E. Entropy production fluctuation theorem and the nonequilibrium work relationfor free energy differences. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics 1999, 60, 2721–2726. [Google Scholar]
- Collin, D.; Ritort, F.; Jarzynski, C.; Smith, S.B.; Tinco, I., Jr.; Bustamante, C. Verification of the Crooks fluctuation theorem and recovery of RNA folding free energies. Nature 2005, 437, 231–234. [Google Scholar] [CrossRef]
- Dudko, O.K.; Hummer, G.; Szabo, A. Intrinsic Rates and Activation Free Energies from Single-Molecule Pulling Experiments. Phys. Rev. Lett. 2006, 96, 108101. [Google Scholar]
- Dudko, O.K.; Hummer, G.; Szabo, A. Theory, analysis, and interpretation of single-moleculeforce spectroscopy experiments. Proc. Natl. Acad. Sci. USA 2008, 105, 15755–15760. [Google Scholar] [PubMed] [Green Version]
- Puchner, E.M.; Franzen, G.; Gautel, M.; Gaub, H.E. Comparing Proteins by Their Unfolding Pattern. Biophys. J. 2008, 95, 426–434. [Google Scholar] [PubMed] [Green Version]
- Hyeon, C.; Dima, R.I.; Thirumalai, D. Pathways and Kinetic Barriers in MechanicalUnfolding and Refolding of RNA and Proteins. Structure 2006, 14, 1633–1645. [Google Scholar] [CrossRef] [Green Version]
- Zhmurov, A.; Dima, R.I.; Kholodov, Y.; Barsegov, V. SOP-GPU: Accelerating biomolecular simulations in the centisecond timescale using graphics processors. Proteins 2010, 78, 2984–2999. [Google Scholar] [PubMed]
- Lee, E.H.; Hsin, J.; Sotomayor, M.; Comellas, G.; Schulten, K. Discovery Through the Computational Microscope. Structure 2009, 17, 1295–1306. [Google Scholar] [CrossRef] [Green Version]
- Ackbarow, T.; Chen, X.; Keten, S.; Buehler, M.J. Hierarchies, multiple energy barriers, and robustness govern the fracture mechanics of α-helical and β-sheet protein domains. Proc. Natl. Acad. Sci. USA 2007, 104, 16410–16415. [Google Scholar]
- Lichter, S.; Rafferty, B.; Flohr, Z.; Martini, A. Protein High-Force Pulling Simulations Yield Low-Force Results. PLoS ONE 2012, 7, e34781. [Google Scholar]
- Kouza, M.; Hu, C.K.; Li, M.S.; Kolinski, A. A structure-based model fails to probe the mechanical unfolding pathways of the titin i27 domain. J. Chem. Phys. 2013, 139, 065103. [Google Scholar]
- Rico, F.; Gonzalez, L.; Casuso, I.; Puig-Vidal, M.; Scheuring, S. High-Speed Force Spectroscopy Unfolds Titin at the Velocity of Molecular Dynamics Simulations. Science 2013, 342, 741–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kmiecik, S.; Wabik, J.; Kolinski, M.; Kouza, M.; Kolinski, A. Coarse-Grained Modeling of Protein Dynamics. In Computational Methods to Study the Structure and Dynamics of Biomolecules and Biomolecular Processes; Liwo, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 55–79. [Google Scholar]
- Bauer, J.A.; Pavlović, J.; Bauerová-Hlinková, V. Normal Mode Analysis as a Routine Part of a Structural Investigation. Molecules 2019, 24, 2936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skjaerven, L.; Martinez, A.; Reuter, N. Principal component and normal mode analysis of proteins; a quantitative comparison using the GroEL subunit. Proteins 2011, 79, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Rueda, M.; Chacón, P.; Orozco, M. Thorough Validation of Protein Normal Mode Analysis: A Comparative Study with Essential Dynamics. Structure 2007, 15, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueda, M.; Ferrer-Costa, C.; Meyer, T.; Pérez, A.; Camps, J.; Hospital, A.; Gelpi, J.L.; Orozco, M. A consensus view of protein dynamics. Proc. Natl. Acad. Sci. USA 2007, 104, 796–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doruker, P.; Atilgan, A.R.; Bahar, I. Dynamics of Proteins Predicted by Molecular Dynamics Simulations and Analytical Approaches: Application to α-Amylase Inhibitor. Proteins 2000, 40, 512–524. [Google Scholar] [CrossRef]
- Knapp, E.W.; Fischer, S.F.; Parak, F. Protein Dynamics from Mössbauer Spectra. The Temperature Dependence. J. Phys. Chem. 1982, 86, 5042–5047. [Google Scholar] [CrossRef]
- Zaccai, G. How Soft Is a Protein? A Protein Dynamics Force Constant Measured by Neutron Scattering. Science 2000, 288, 1604–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, J.H.; Novikov, V.N.; Gregory, R.B.; Curtis, J.E.; Chowdhuri, Z.; Sokolov, A.P. Onsets of Anharmonicity in Protein Dynamics. Phys. Rev. Lett. 2005, 95, 038101. [Google Scholar] [CrossRef] [PubMed]
- Eyal, E.; Bahar, I. Toward a Molecular Understanding of the Anisotropic Response of Proteins to External Forces: Insights from Elastic Network Models. Biophys. J. 2008, 94, 3424–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikulska-Ruminska, K.; Kulik, A.J.; Benadiba, C.; Bahar, I.; Dietler, G.; Nowak, W. Nanomechanics of multidomain neuronal cell adhesion protein contactin revealed by single molecule AFM and SMD. Sci. Rep. 2017, 7, 8852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiment. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakan, A.; Dutta, A.; Mao, W.; Liu, Y.; Chennubhotla, C.; Lezon, T.R.; Bahar, I. Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 2014, 30, 2681–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suhre, K.; Sanejouand, Y.H. ElNémo: A normal mode web server for protein movements analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004, 32, W610–W614. [Google Scholar] [CrossRef] [PubMed]
- Baase, W.A.; Liu, L.; Tronrud, D.E.; Matthews, B.W. Lessons from the lysozyme of phage T4. Protein Sci. 2010, 19, 631–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Cecconi, C.; Baase, W.A.; Vetter, I.R.; Breyer, W.A.; Haack, J.A.; Matthews, B.W.; Dahlquist, F.W.; Bustamante, C. Solid-state synthesis and mechanical unfolding of polymers of T4 lysozyme. Proc. Natl. Acad. Sci. USA 2000, 97, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Li, H. Atomic force microscopy reveals parallel mechanicalunfolding pathways of T4 lysozyme: Evidence for a kinetic partitioning mechanism. Proc. Natl. Acad. Sci. USA 2008, 105, 1885–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shank, E.A.; Cecconi, C.; Dill, J.W.; Marqusee, S.; Bustamante, C. The folding cooperativity of a protein is controlled by its chain topology. Nature 2010, 465, 637–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Glenn, P. Probing the folded state and mechanical unfolding pathways of T4 lysozyme using all-atom and coarse-grained molecular simulation. J. Chem. Phys. 2015, 142, 035101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, L.H.; Matthews, B.W. Structure of bacteriophage T4 lysozyme refined at 1.7 Å resolution. J. Mol. Biol. 1987, 193, 189–199. [Google Scholar] [CrossRef]
- Cellitti, J.; Llinas, M.; Echols, N.; Shank, E.A.; Gillespie, B.; Kwon, E.; Crowder, S.M.; Dahlquist, F.W.; Alber, T.; Marqusee, S. Exploring subdomain cooperativity in T4 lysozyme I: Structural and energetic studies of a circular permutant and protein fragment. Protein Sci. 2007, 16, 842–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlin, S.; Brocchieri, L. Heat Shock Protein 70 Family: Multiple Sequence Comparisons, Function, and Evolution. J. Mol. Evol. 1998, 47, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Fernáandez-Fernáandez, M.R.; Gragera, M.; Ochoa-Ibarrola, L.; Quintana-Gallardo, L.; Valpuesta, J.M. Hsp70—A master regulator in protein degradation. FEBS Lett. 2017, 591, 2648–2660. [Google Scholar]
- Havalová, H.; Ondrovičová, G.; Keresztesová, B.; Bauer, J.A.; Pevala, V.; Kutejová, E.; Kunová, N. Mitochondrial HSP70 Chaperone System—The Influence of Post-translational Modifications and Involvement in Human Disease. Int. J. Mol. Sci. 2021, 22, 8077. [Google Scholar] [CrossRef]
- Genevaux, P.; Georgopoulos, C.; Kelley, W.L. The Hsp70 chaperone machines of Escherichia coli: A paradigm for the repartition of chaperone functions. Mol. Microbiol. 2007, 66, 840–857. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.; Merz, D.R.; Pelz, B.; Theisen, K.E.; Yacyshyn, G.; Mokranjac, D.; Dima, R.I.; Rief, M.; Žoldák, G. Nucleotides regulate the mechanical hierarchy between subdomains of the nucleotide binding domain of the Hsp70 chaperone DnaK. Proc. Natl. Acad. Sci. USA 2015, 112, 10389–10394. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.S.; Merz, D.R.; Buchsteiner, M.; Dima, R.I.; Rief, M.; Žoldák, G. Nanomechanics of the substrate binding domain of Hsp70 determine its allosteric ATP-induced conformational change. Proc. Natl. Acad. Sci. USA 2017, 114, 6040–6045. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.; Meinhold, S.; Jakob, R.P.; Stigler, J.; Merkel, U.; Maier, T.; Rief, M.; Žoldák, G. A folding nucleus and minimal ATP binding domain of Hsp70 identified by single-molecule force spectroscopy. Proc. Natl. Acad. Sci. USA 2018, 115, 4666–4671. [Google Scholar] [CrossRef] [Green Version]
- Meinhold, S.; Bauer, D.; Huber, J.; Merkel, U.; Weißl, A.; Žoldák, G.; Rief, M. An Active, Ligand-Responsive Pulling Geometry Reports on Internal Signaling between Subdomains of the DnaK Nucleotide-Binding Domain in Single-Molecule Mechanical Experiments. Biochemistry 2019, 58, 4744–4750. [Google Scholar] [CrossRef]
- Bertelsen, E.B.; Chang, L.; Gestwicki, J.E.; Zuiderweg, E.R.P. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar] [CrossRef] [Green Version]
- Kityk, R.; Kopp, J.; Sinning, I.; Mayer, M.P. Structure and Dynamics of the ATP-Bound Open Conformation of Hsp70 Chaperones. Mol. Cell 2012, 48, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [PubMed]
- Giguère, V.; Hollenberg, S.M.; Rosenfeld, M.G.; Evans, R.M. Functional domains of the human glucocorticoid receptor. Cell 1986, 46, 645–652. [Google Scholar] [CrossRef]
- Bledsoe, R.K.; Montana, V.G.; Stanley, T.B.; Delves, C.J.; Apolito, C.J.; McKee, D.D.; Consler, T.G.; Parks, D.J.; Stewart, E.L.; Willson, T.M.; et al. Crystal Structure of the Glucocorticoid Receptor Ligand Binding Domain Reveals a Novel Mode of Receptor Dimerization and Coactivator Recognition. Cell 2002, 110, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Schoch, G.A.; D’Arcy, B.; Stihle, M.; Burger, D.; Bär, D.; Benz, J.; Thoma, R.; Ruf, A. Molecular Switch in the Glucocorticoid Receptor: Active and Passive Antagonist Conformations. J. Mol. Biol. 2010, 395, 568–577. [Google Scholar] [CrossRef]
- Suren, T.; Rutz, D.; Mößmer, P.M.; Merkel, U.; Buchner, J.; Rief, M. Single-molecule force spectroscopy reveals folding steps associated with hormone binding and activation of the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 11688–11693. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, O.R.; Freiburger, L.; Rutz, D.A.; Krause, M.; Zierer, B.K.; Alvira, S.; Cuéllar, J.; Valpuesta, J.M.; Madl, T.; Sattler, M.; et al. Modulation of the Hsp90 Chaperone Cycle by a Stringent Client Protein. Mol. Cell 2014, 53, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandevyver, S.; Dejager, L.; Libert, C. On the Trail of the Glucocorticoid Receptor: Into the Nucleus and Back. Traffic 2012, 13, 364–374. [Google Scholar] [CrossRef]
- Dong, D.D.; Jewell, C.M.; Bienstock, R.J.; Cidlowski, J.A. Functional analysis of the LXXLL motifs of the human glucocorticoid receptor: Association with altered ligand affinity. J. Steroid Biochem. Mol. Biol. 2006, 101, 106–117. [Google Scholar] [CrossRef]
- He, C.; Genchev, G.Z.; Lu, H.; Li, H. Mechanically untying a protein slipknot: Multiple pathways revealed by force spectroscopy and steered molecular dynamics simulations. J. Am. Chem. Soc. 2012, 134, 10428–10435. [Google Scholar]
- Peng, Q.; Zhuang, S.; Wang, M.; Cao, Y.; Khor, Y.; Li, H. Mechanical design of the third FnIII domain of tenascin-C. J. Mol. Biol. 2009, 386, 1327–1342. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.W. Structural and genetic analysis of the folding and function of T4 lysozyme. FASEB J. 1996, 10, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Žoldák, G.; Carstensen, L.; Scholz, C.; Schmid, F.X. Consequences of domain insertion on the stability and folding mechanism of a protein. J. Mol. Biol. 2009, 386, 1138–1152. [Google Scholar] [CrossRef] [PubMed]
- Llinás, M.; Marqusee, S. Subdomain interactions as a determinant in the folding and stability of T4 lysozyme. Protein Sci. 1998, 7, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Brooks, B.R.; Doniach, S.; Thirumalai, D. Network of Dynamically Important Residues in the Open/Closed Transition in Polymerases Is Strongly Conserved. Structure 2005, 13, 565–577. [Google Scholar] [PubMed]
- Zheng, W.; Brooks, B.R.; Thirumalai, D. Low-frequency normal modes that describe allosteric transitions in biological nanomachines are robust to sequence variations. Proc. Natl. Acad. Sci. USA 2006, 103, 7664–7669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Brooks, B.R.; Thirumalai, D. Allosteric Transitions in the Chaperonin GroEL are Captured by a Dominant Normal Mode that is Most Robust to Sequence Variations. Biophys. J. 2007, 93, 2289–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Thirumalai, D. Coupling between Normal Modes Drives Protein Conformational Dynamics: Illustrations Using Allosteric Transitions in Myosin II. Biophys. J. 2009, 96, 2128–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elms, P.J.; Chodera, J.D.; Bustamante, C.; Marqusee, S. The molten globule state is unusually deformable under mechanical force. Proc. Natl. Acad. Sci. USA 2012, 109, 3796–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, J.; Žoldák, G. Interpretation of Single-Molecule Force Experiments on Proteins Using Normal Mode Analysis. Nanomaterials 2021, 11, 2795. https://doi.org/10.3390/nano11112795
Bauer J, Žoldák G. Interpretation of Single-Molecule Force Experiments on Proteins Using Normal Mode Analysis. Nanomaterials. 2021; 11(11):2795. https://doi.org/10.3390/nano11112795
Chicago/Turabian StyleBauer, Jacob, and Gabriel Žoldák. 2021. "Interpretation of Single-Molecule Force Experiments on Proteins Using Normal Mode Analysis" Nanomaterials 11, no. 11: 2795. https://doi.org/10.3390/nano11112795
APA StyleBauer, J., & Žoldák, G. (2021). Interpretation of Single-Molecule Force Experiments on Proteins Using Normal Mode Analysis. Nanomaterials, 11(11), 2795. https://doi.org/10.3390/nano11112795