
Polycaprolactone Composite Micro/Nanofibrous Material as an Alternative to Restricted Access Media for Direct Extraction and Separation of Non-Steroidal Anti-Inflammatory Drugs from Human Serum Using Column-Switching Chromatography

 , , and
, , and
Abstract
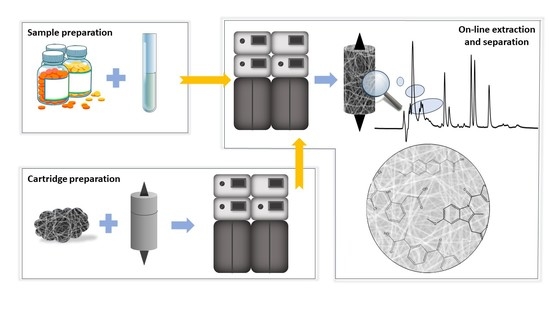
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Instrumentation
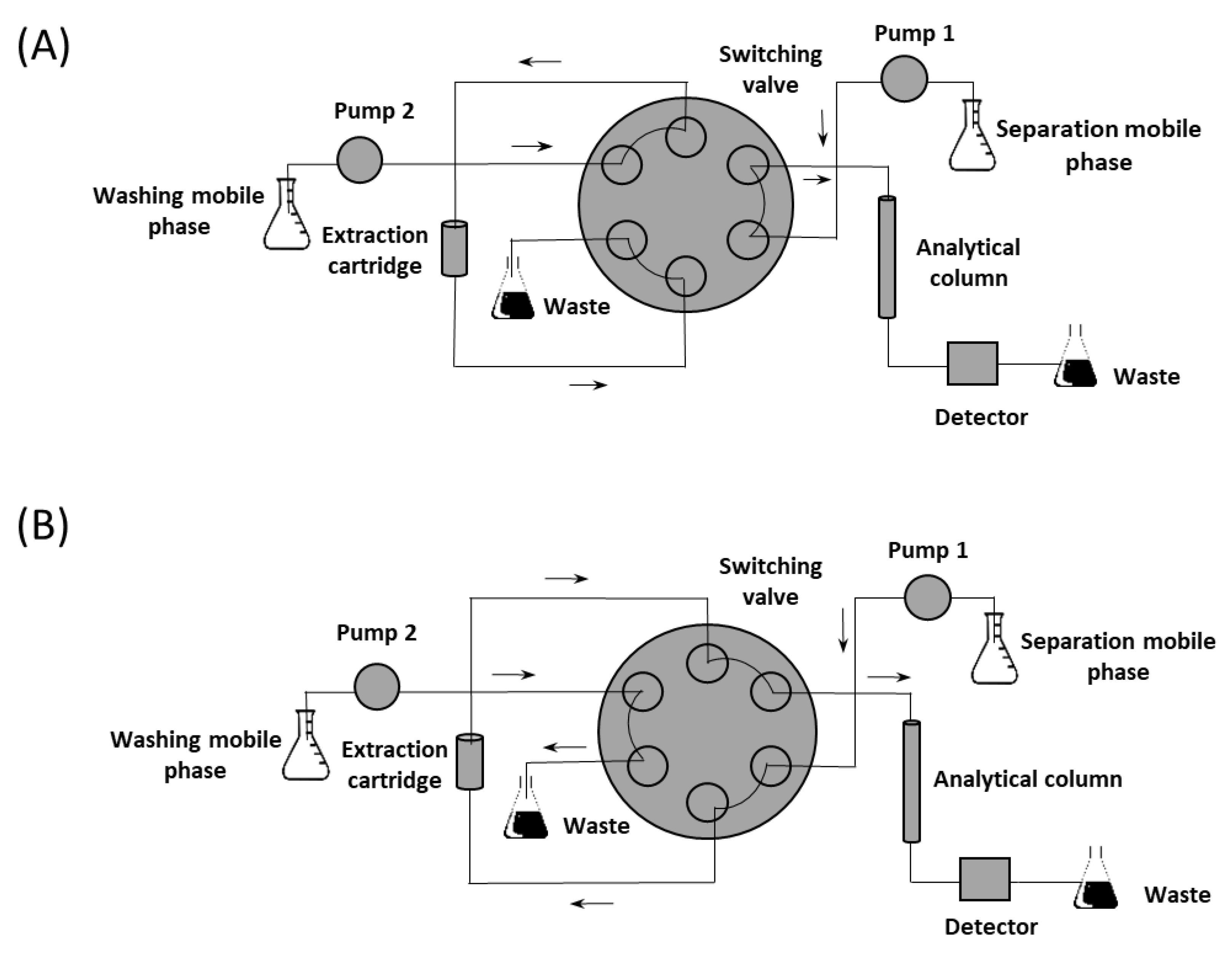
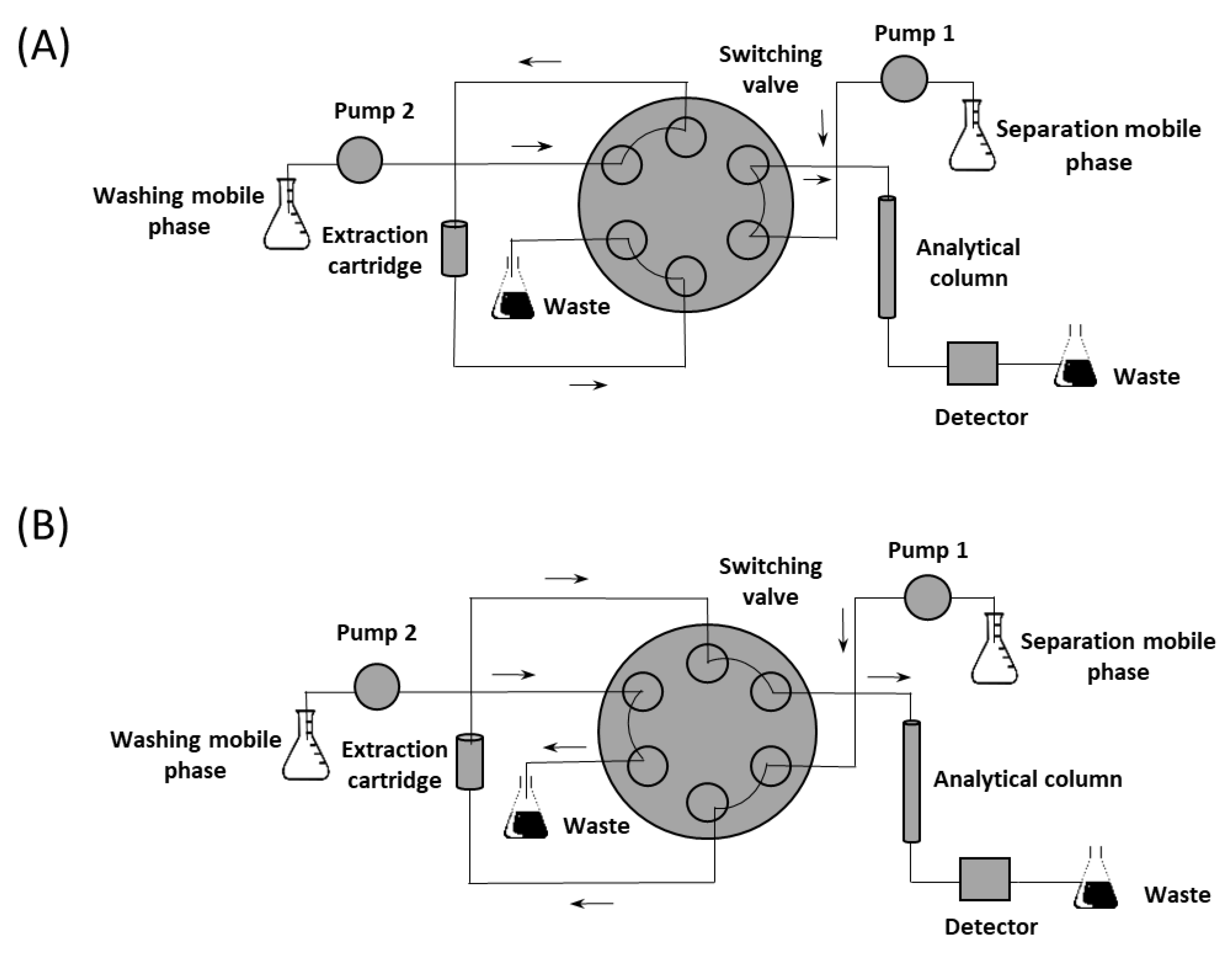
2.2.1. Column-Switching Chromatography System
2.2.2. Meltblown and Electrospun Fibers
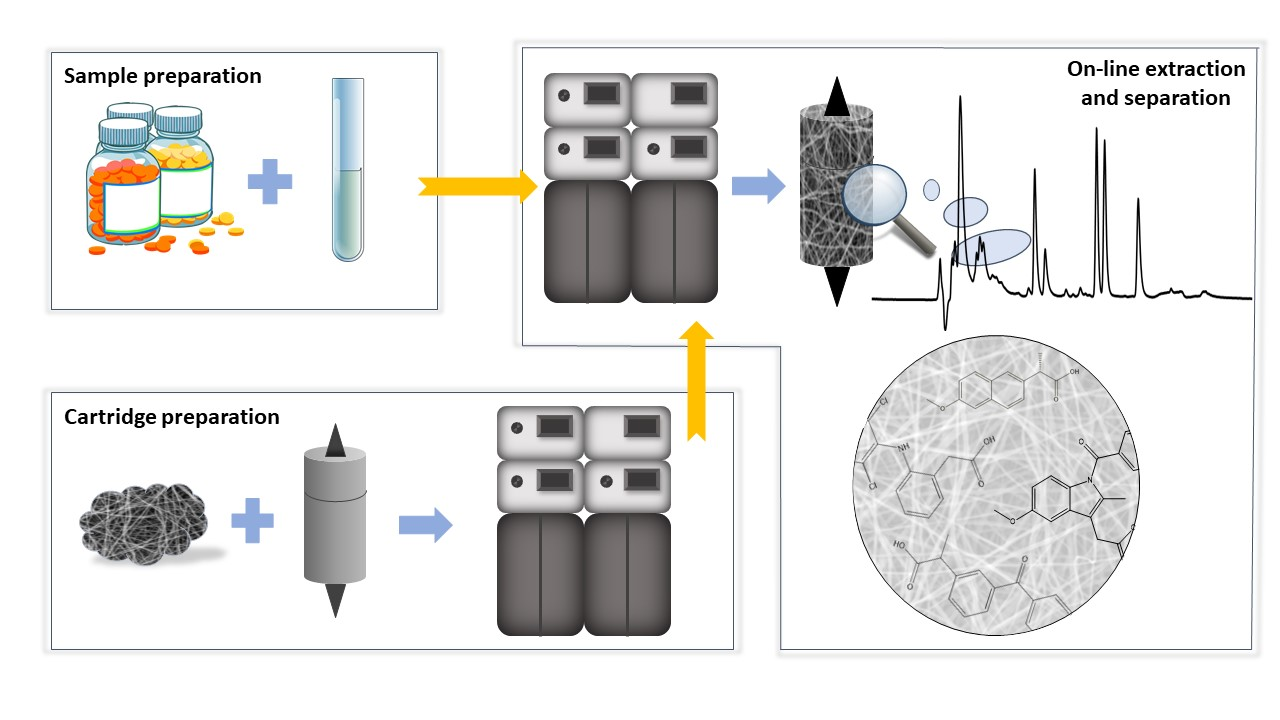
2.3. Preparation of Extraction Cartridge
2.4. Preparation of Standard and Matrix Solutions
2.5. Real Sample
2.6. Analytical Method
3. Results and Discussion
3.1. Optimization of Chromatographic Separation
3.2. Optimization of On-Line Extraction
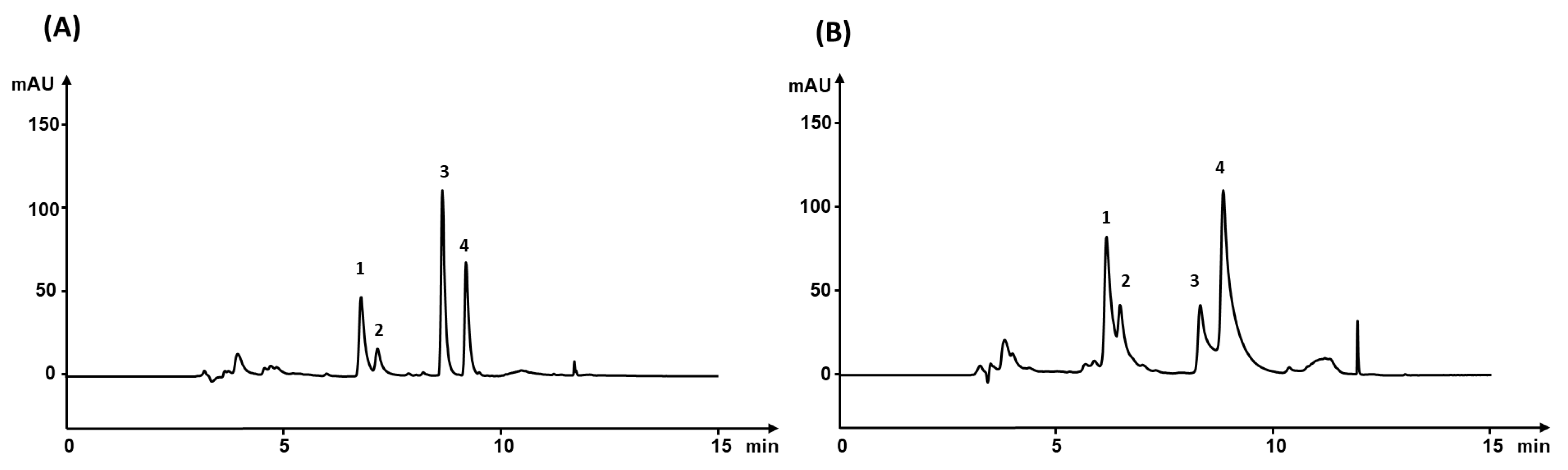
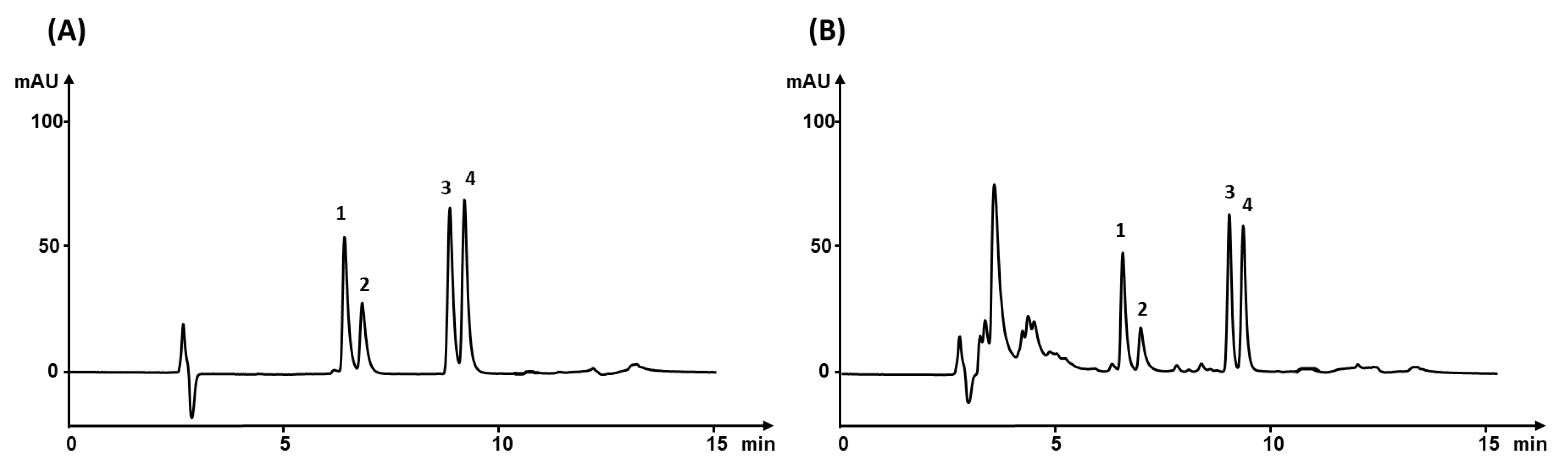
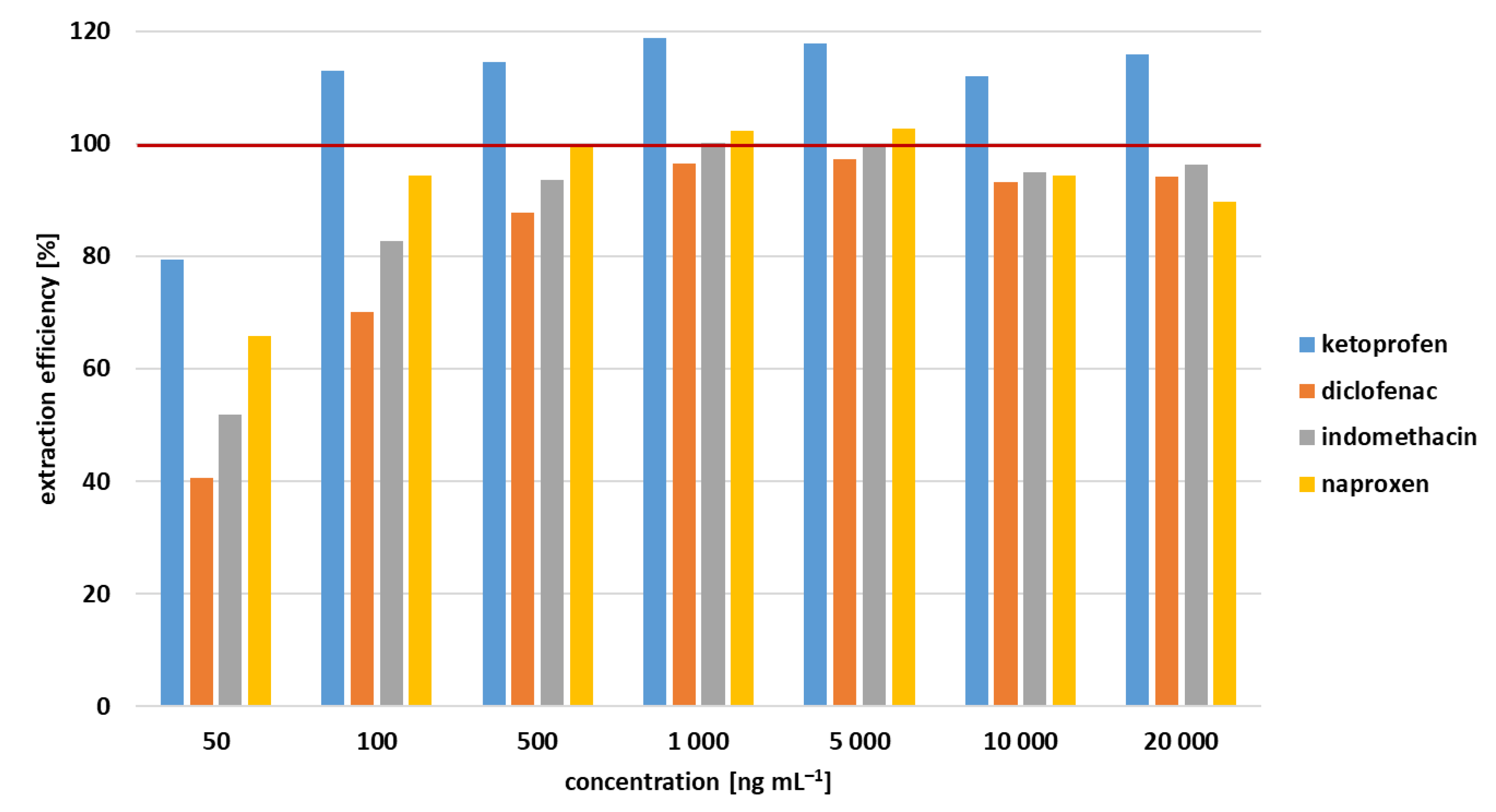
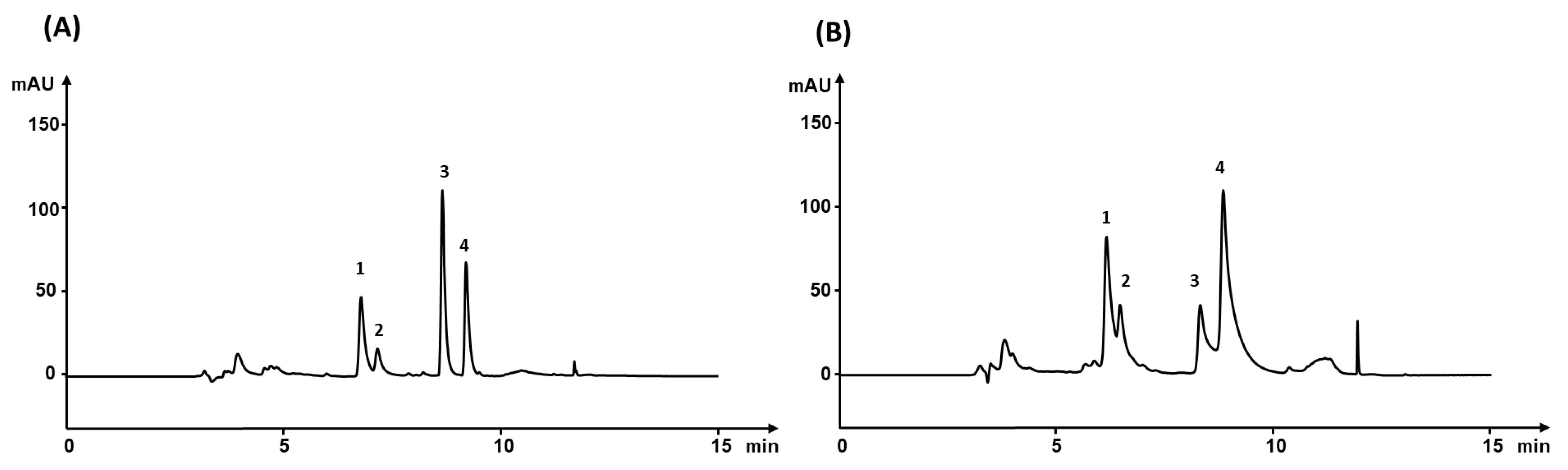
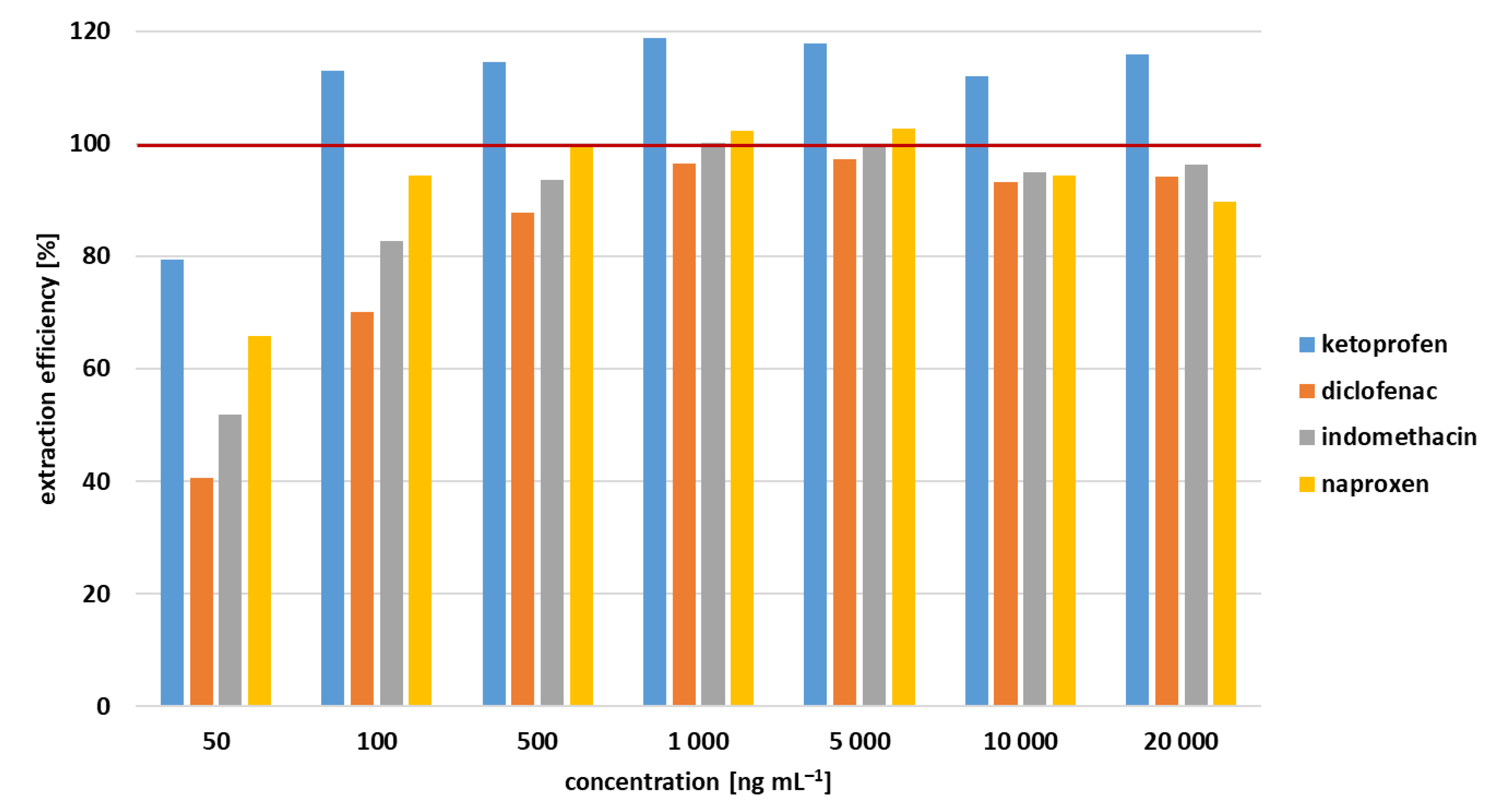
3.3. Extraction Efficiency
3.4. Optimization of Injected Sample Volume
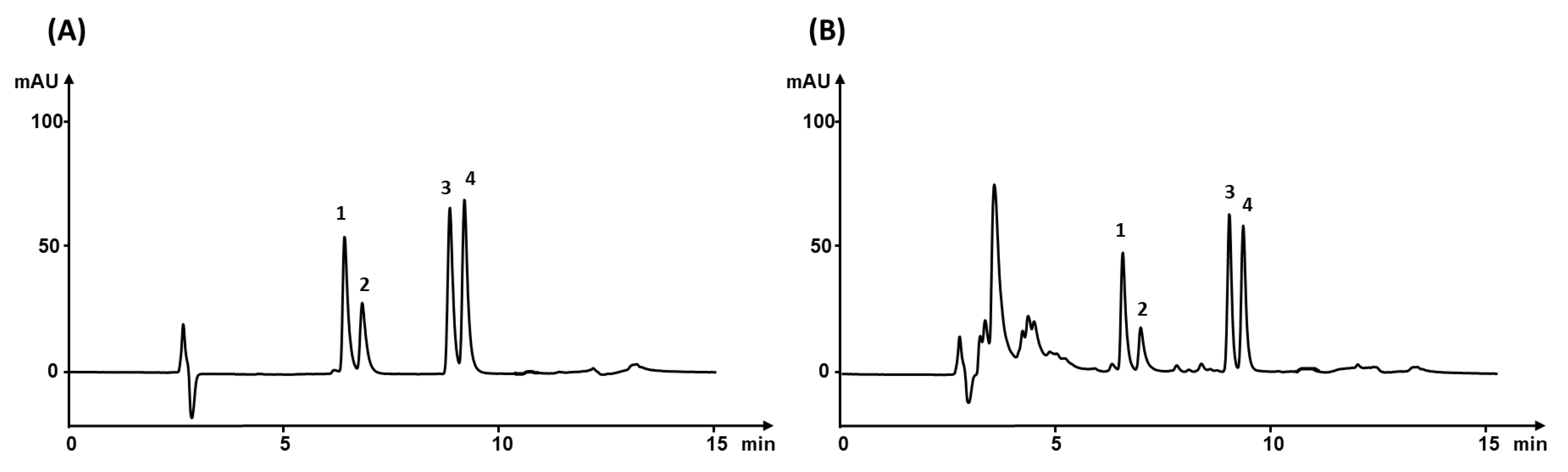
3.5. Validation
Linearity, Accuracy, Precision, and Limits of Detection and Quantification
3.6. Reusability of the Extraction Cartridge
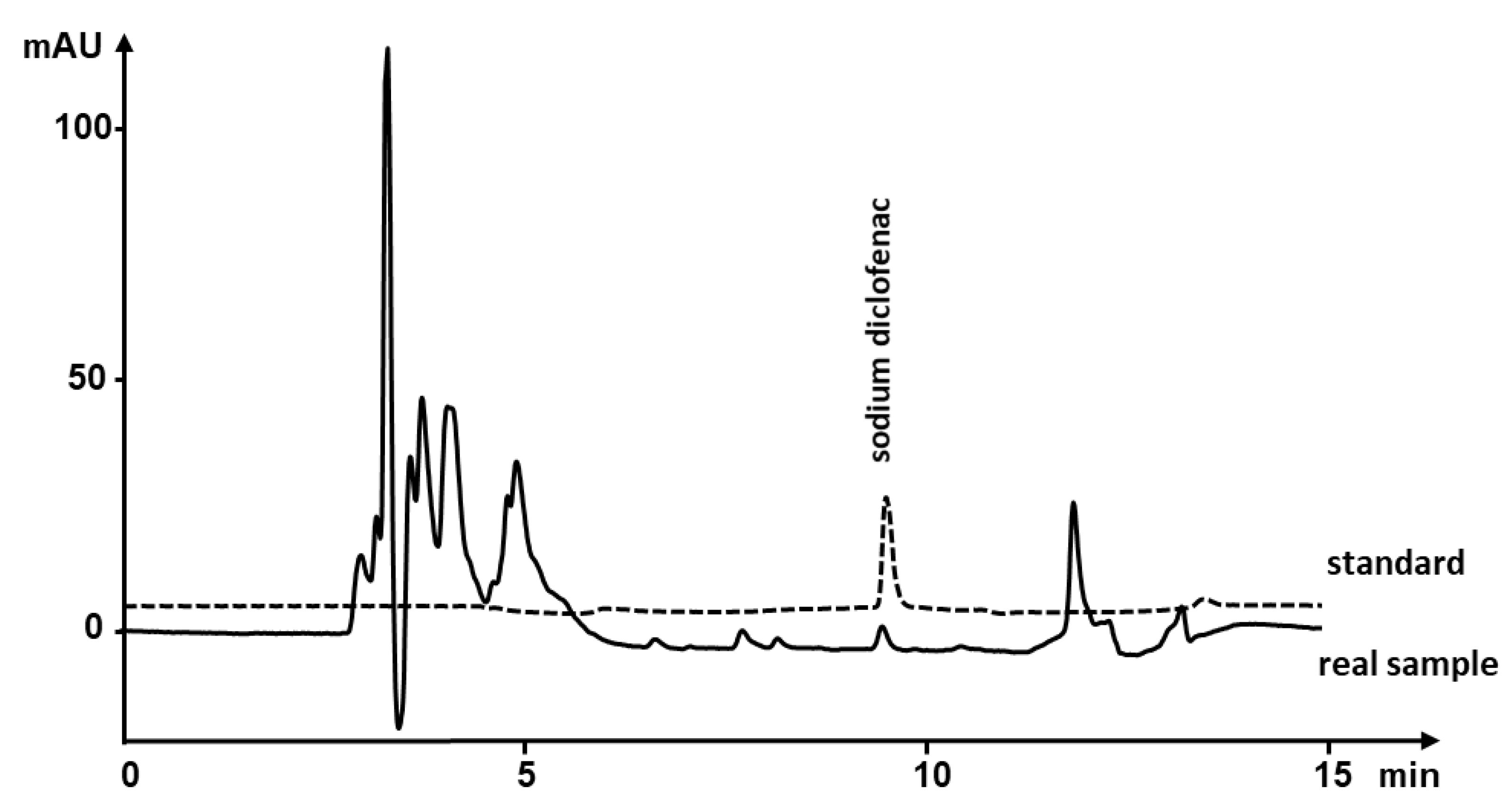
3.7. Analysis of Real Sample
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| DCF | Sodium diclofenac |
| H3PO4 | Phosphoric acid |
| IND | Indomethacin |
| KET | Ketoprofen |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| MeOH | Methanol |
| NAP | Naproxen |
| NSAID | Non-steroidal anti-inflammatory drugs |
| micro/nano PCL | Composite sorbent containing poly-ε-caprolactone micro- and nanofibers |
| RAM | Restricted access media |
References
- Sun, Y.; Takaba, K.; Kido, H.; Nakashima, M.N.; Nakashima, K. Simultaneous determination of arylpropionic acidic nonsteroidal anti-inflammatory drugs in pharmaceutical formulations and human plasma by HPLC with UV detection. J. Pharm. Biomed. Anal. 2003, 30, 1611–1619. [Google Scholar] [CrossRef]
- Castilhos, T.S.; Barreto, F.; Meneghini, L.; Bergold, A.M. Development of high-throughput multi-residue method for non-steroidal anti-inflammatory drugs monitoring in swine muscle by LC-MS/MS. Food Addit. Contam. Part A 2016, 33, 1166–1174. [Google Scholar] [CrossRef]
- Martin, M.J.; Pablos, F.; Gonzalez, A.G. Simultaneous determination of caffeine and non-steroidal anti-inflammatory drugs in pharmaceutical formulations and blood plasma by reversed-phase HPLC from linear gradient elution. Talanta 1999, 49, 453–459. [Google Scholar] [CrossRef]
- El Haj, B.M.; Al Ainri, A.M.; Hassan, M.H.; Bin Khadem, R.K.; Marzouq, M.S. The GC/MS analysis of some commonly used non-steriodal anti-inflammatory drugs (NSAIDs) in pharmaceutical dosage forms and in urine. Forensic Sci. Int. 1999, 105, 141–153. [Google Scholar] [CrossRef]
- Kim, K.R.; Yoon, H.R. Rapid screening for acidic non-steroidal anti-inflammatory drugs in urine by gas chromatography mass spectrometry in the selected-ion monitoring mode. J. Chromatogr. B Biomed. Appl. 1996, 682, 55–66. [Google Scholar] [CrossRef]
- Arroyo, D.; Ortiz, M.C.; Sarabia, L.A. Optimization of the derivatization reaction and the solid-phase microextraction conditions using a D-optimal design and three-way calibration in the determination of non-steroidal anti-inflammatory drugs in bovine milk by gas chromatography-mass spectrometry. J. Chromatogr. A 2011, 1218, 4487–4497. [Google Scholar] [CrossRef]
- Heitmeier, S.; Blaschke, G. Direct assay of nonopioid analgesics and their metabolites in human urine by capillary electrophoresis and capillary electrophoresis-mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1999, 721, 109–125. [Google Scholar] [CrossRef]
- Bechet, I.; Fillet, M.; Hubert, P.; Crommen, J. Quantitative-analysis of nonsteroidal antiinflammatory drugs by capillary zone electrophoresis. J. Pharm. Biomed. Anal. 1995, 13, 497–503. [Google Scholar] [CrossRef]
- Maboundou, C.W.; Paintaud, G.; Berard, M.; Bechtel, P.R. Separation of 15 nonsteroidal antiinflammatory drugs using micellar electrokinetic capillary chromatography. J. Chromatogr. B Biomed. Appl. 1994, 657, 173–183. [Google Scholar] [CrossRef]
- Navas, N.; Urena, R.; Capitan-Vallvey, L.-F. Determination of celecoxib, rofecoxib, sodium diclofenac and niflumic acid in human serum samples by HPLC with DAD detection. Chromatographia 2008, 67, 55–61. [Google Scholar] [CrossRef]
- Dadashzadeh, S.; Vali, A.M.; Rezagholi, N. LC determination of piroxicam in human plasma. J. Pharm. Biomed. Anal. 2002, 28, 1201–1204. [Google Scholar] [CrossRef]
- Kataoka, H.; Saito, K. Recent advances in column switching sample preparation in bioanalysis. Bioanalysis 2012, 4, 809–832. [Google Scholar] [CrossRef]
- Kataoka, H. New trends in sample preparation for clinical and pharmaceutical analysis. Trends Anal. Chem. 2003, 22, 232–244. [Google Scholar] [CrossRef]
- Nazario, C.E.D.; Fumes, B.H.; da Silva, M.R.; Lancas, F.M. New materials for sample preparation techniques in bioanalysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1043, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Cassiano, N.M.; Barreiro, J.C.; Moraes, M.C.; Oliveira, R.V.; Cass, Q.B. Restricted access media supports for direct high-throughput analysis of biological fluid samples: Review of recent applications. Bioanalysis 2009, 1, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Rudolphi, A.; Boos, K.S.; Grimm, C.H. Column switching and restricted access materials for sample preparation. Abstr. Pap. Am. Chem. Soc. 1997, 214, 186. [Google Scholar]
- Yang, S.H.; Fan, H.; Classon, R.J.; Schug, K.A. Restricted access media as a streamlined approach toward on-line sample preparation: Recent advancements and applications. J. Sep. Sci. 2013, 36, 2922–2938. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Jeong, C.K.; Choi, S.J.; Kim, S.B.; Lee, M.H.; Ko, G.I.; Sohn, D.H. Simultaneous determination of aceclofenac and diclofenac in human plasma by narrowbore HPLC using column-switching. J. Pharm. Biomed. Anal. 2000, 23, 775–781. [Google Scholar] [CrossRef]
- Suenami, K.; Lim, L.W.; Takeuchi, T.; Sasajima, Y.; Sato, K.; Takekoshi, Y.; Kanno, S. Direct determination of non-steroidal anti-inflammatory drugs by column-switching LC-MS. J. Sep. Sci. 2006, 29, 2725–2732. [Google Scholar] [CrossRef]
- Brabcová, I.; Šatínský, D.; Solich, P. A fast on-line micro-column sample pretreatment with a HPLC column switching technique and large volume sample injection for determination of non-steroidal anti-inflammatory drugs in human serum. Anal. Methods 2013, 5, 6153–6159. [Google Scholar] [CrossRef]
- Puoci, F.; Iemma, F.; Cirillo, G.; Curcio, M.; Parisi, O.I.; Spizzirri, U.G.; Picci, N. New restricted access materials combined to molecularly imprinted polymers for selective recognition/release in water media. Eur. Polym. J. 2009, 45, 1634–1640. [Google Scholar] [CrossRef]
- Xiao, D.L.; Liu, S.B.; Liang, L.Y.; Bi, Y.P. Magnetic restricted-access microspheres for extraction of adrenaline, dopamine and noradrenaline from biological samples. Mikrochim. Acta 2016, 183, 1417–1423. [Google Scholar] [CrossRef]
- de Faria, H.D.; Abrao, L.C.D.; Santos, M.G.; Barbosa, A.F.; Figueiredo, E.C. New advances in restricted access materials for sample preparation: A review. Anal. Chim. Acta 2017, 959, 43–65. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.F.; Barbosa, V.M.P.; Bettini, J.; Luccas, P.O.; Figueiredo, E.C. Restricted access carbon nanotubes for direct extraction of cadmium from human serum samples followed by atomic absorption spectrometry analysis. Talanta 2015, 131, 213–220. [Google Scholar] [CrossRef]
- Raabová, H.; Háková, M.; Chocholoušová-Havliková, L.; Erben, J.; Chvojka, J.; Solich, P.; Švec, F.; Šatínský, D. Poly-epsilon-caprolactone Nanofibrous Polymers: A Simple Alternative to Restricted Access Media for Extraction of Small Molecules from Biological Matrixes. Anal. Chem. 2020, 92, 6801–6805. [Google Scholar] [CrossRef]
- Háková, M.; Chocholoušová-Havliková, L.; Chvojka, J.; Erben, J.; Solich, P.; Švec, F.; Šatínský, D. A comparison study of nanofiber, microfiber, and new composite nano/microfiber polymers used as sorbents for on-line solid phase extraction in chromatography system. Anal. Chim. Acta 2018, 1023, 44–52. [Google Scholar] [CrossRef]
- Erben, J.; Pilařová, K.; Sanetrnik, F.; Chvojka, J.; Jenčová, V.; Blažková, L.; Havlíčeka, J.; Novák, O.; Mikeš, P.; Prosecká, E.; et al. The combination of meltblown and electrospinning for bone tissue engineering. Mater. Lett. 2015, 143, 172–176. [Google Scholar] [CrossRef]
- European Medicines Association. Note for Guidance on Validation of Analytical Procedures: Text and Methodology; European Medical Agency: Amsterdam, The Netherlands, 1995. [Google Scholar]
- Emara, L.H.; Taha, N.F.; El-Ashmawy, A.A.; Raslan, H.M.; Mursi, N.M. A rapid and sensitive bioanalytical hplc method for determining diclofenac sodium in human plasma for bioequivalence studies. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 2203–2216. [Google Scholar] [CrossRef]
- Sahoo, N.K.; Sahu, M.; Rao, P.S.; Ghosh, G. Solid phase extraction and quantification of diclofenac sodium in human plasma by liquid chromatography-tandem mass spectrometry. J. Anal. Chem. 2015, 70, 424–430. [Google Scholar] [CrossRef]
- Háková, M.; Chocholoušová-Havliková, L.; Chvojka, J.; Solich, P.; Šatínský, D. An on-line coupling of nanofibrous extraction with column-switching high performance liquid chromatography—A case study on the determination of bisphenol A in environmental water samples. Talanta 2018, 178, 141–146. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ketoprofen | Naproxen | Sodium Diclofenac | Indomethacin | |
|---|---|---|---|---|
| Retention time [min] | 6.41 | 6.82 | 9.18 | 10.49 |
| Retention time repeatability, RSD [%] | 0.03 | 0.03 | 0.02 | 0.02 |
| Repeatability of peak areas, RSD [%] | 0.85 | 0.49 | 0.21 | 0.56 |
| Peak resolution | 1.82 | 10.32 | 5.99 | |
| Peak symmetry | 1.80 | 1.63 | 2.00 | 1.60 |
| Peak capacity | 14.03 | 13.18 | 21.65 | 18.80 |
| Ketoprofen | Naproxen | Sodium Diclofenac | Indomethacin | ||
|---|---|---|---|---|---|
| Standard calibration range [ng mL−1] 1 | 50–20,000 | 50–10,000 | 50–20,000 | 50–20,000 | |
| Correlation coefficient | 0.9997 | 0.9996 | 0.9999 | 0.9998 | |
| Matrix calibration range [ng mL−1] 2 | 50–20,000 1 | 50–10,000 2 | 50–20,000 1 | 50–20,000 1 | |
| Correlation coefficient | 0.9999 | 0.9960 | 0.9999 | 0.9999 | |
| Intra-day precision [%] 3 | c1 | 8.54 | 7.89 | 10.24 | 8.37 |
| c2 | 0.60 | 0.90 | 0.38 | 0.17 | |
| c3 | 0.46 | 0.29 | 0.09 | 0.21 | |
| Inter-day precision [%] 4 | 6.79 | 3.62 | 7.44 | 7.88 | |
| Accuracy [%] 5 | c1 | 112.91 ± 4.50 | 94.31 ± 4.09 | 70.07 ± 6.20 | 82.76 ± 6.96 |
| c2 | 118.66 ± 0.84 | 102.28 ± 0.46 | 96.42 ± 0.15 | 100.06 ± 0.22 | |
| c3 | 111.87 ± 0.70 | 94.24 ± 0.23 | 93.12 ± 0.83 | 94.81 ± 0.20 | |
| LOD [ng mL−1] 6 | 2.49 | 24.40 | 3.58 | 26.57 | |
| LOQ [ng mL−1] 7 | 8.29 | 81.34 | 11.92 | 88.57 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raabová, H.; Havlíková, L.C.; Erben, J.; Chvojka, J.; Švec, F.; Šatínský, D. Polycaprolactone Composite Micro/Nanofibrous Material as an Alternative to Restricted Access Media for Direct Extraction and Separation of Non-Steroidal Anti-Inflammatory Drugs from Human Serum Using Column-Switching Chromatography. Nanomaterials 2021, 11, 2669. https://doi.org/10.3390/nano11102669
Raabová H, Havlíková LC, Erben J, Chvojka J, Švec F, Šatínský D. Polycaprolactone Composite Micro/Nanofibrous Material as an Alternative to Restricted Access Media for Direct Extraction and Separation of Non-Steroidal Anti-Inflammatory Drugs from Human Serum Using Column-Switching Chromatography. Nanomaterials. 2021; 11(10):2669. https://doi.org/10.3390/nano11102669
Chicago/Turabian StyleRaabová, Hedvika, Lucie Chocholoušová Havlíková, Jakub Erben, Jiří Chvojka, František Švec, and Dalibor Šatínský. 2021. "Polycaprolactone Composite Micro/Nanofibrous Material as an Alternative to Restricted Access Media for Direct Extraction and Separation of Non-Steroidal Anti-Inflammatory Drugs from Human Serum Using Column-Switching Chromatography" Nanomaterials 11, no. 10: 2669. https://doi.org/10.3390/nano11102669
APA StyleRaabová, H., Havlíková, L. C., Erben, J., Chvojka, J., Švec, F., & Šatínský, D. (2021). Polycaprolactone Composite Micro/Nanofibrous Material as an Alternative to Restricted Access Media for Direct Extraction and Separation of Non-Steroidal Anti-Inflammatory Drugs from Human Serum Using Column-Switching Chromatography. Nanomaterials, 11(10), 2669. https://doi.org/10.3390/nano11102669