Combined In Vitro and In Vivo Approaches to Propose a Putative Adverse Outcome Pathway for Acute Lung Inflammation Induced by Nanoparticles: A Study on Carbon Dots


Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation and Characterization of CDs
2.2. Animal Experimentation
2.3. Lung Inflammation Measurement
2.4. Flow Cytometry Analysis of Cells Recovered in BALFs
2.5. Cell Culture
2.6. Assessment of CD Cell Uptake and Internalization Mechanisms by Cultured Cells
2.7. Cell Viability Assay
2.8. Lysosomal Membrane Integrity Assay
2.9. Oxidative Stress Assessment
2.10. Mitochondrial Membrane Potential Assay
2.11. Cytokine Assay
2.12. Statistical Analysis
3. Results and Discussion
3.1. Characterization of CDs
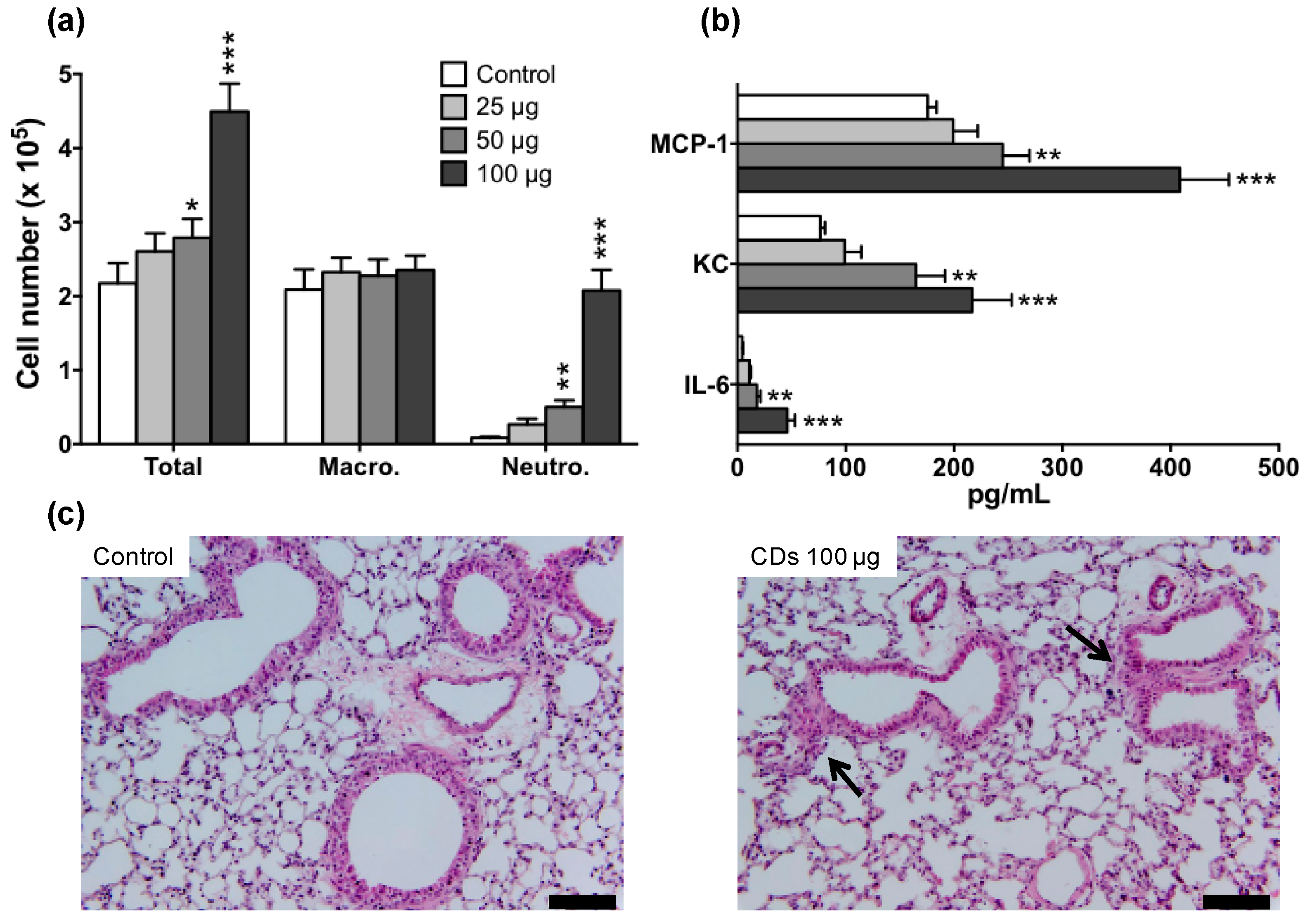
3.2. Lung Inflammation in Response to CDs
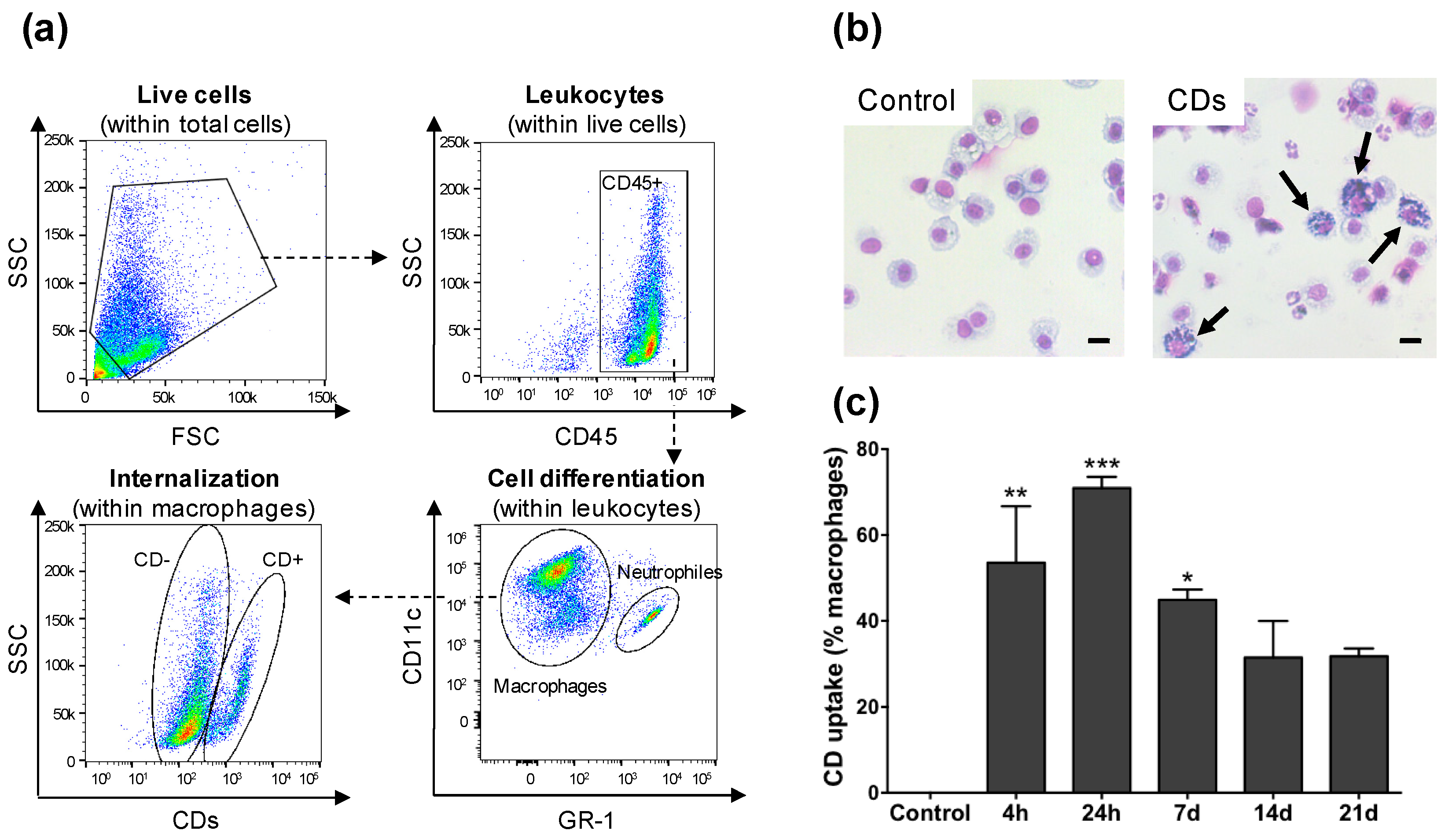
3.3. CD Cell Target in the Airway Lumen
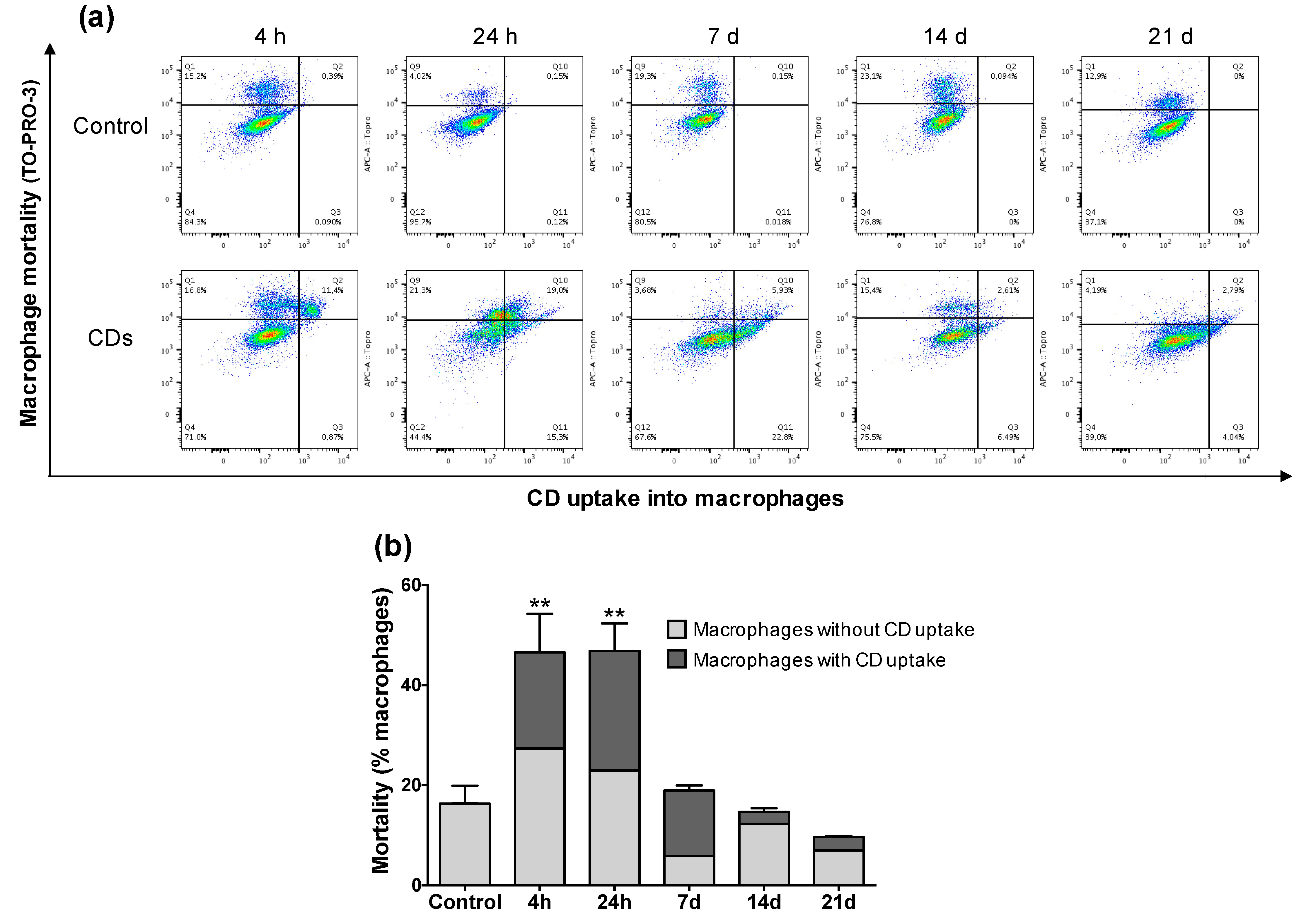
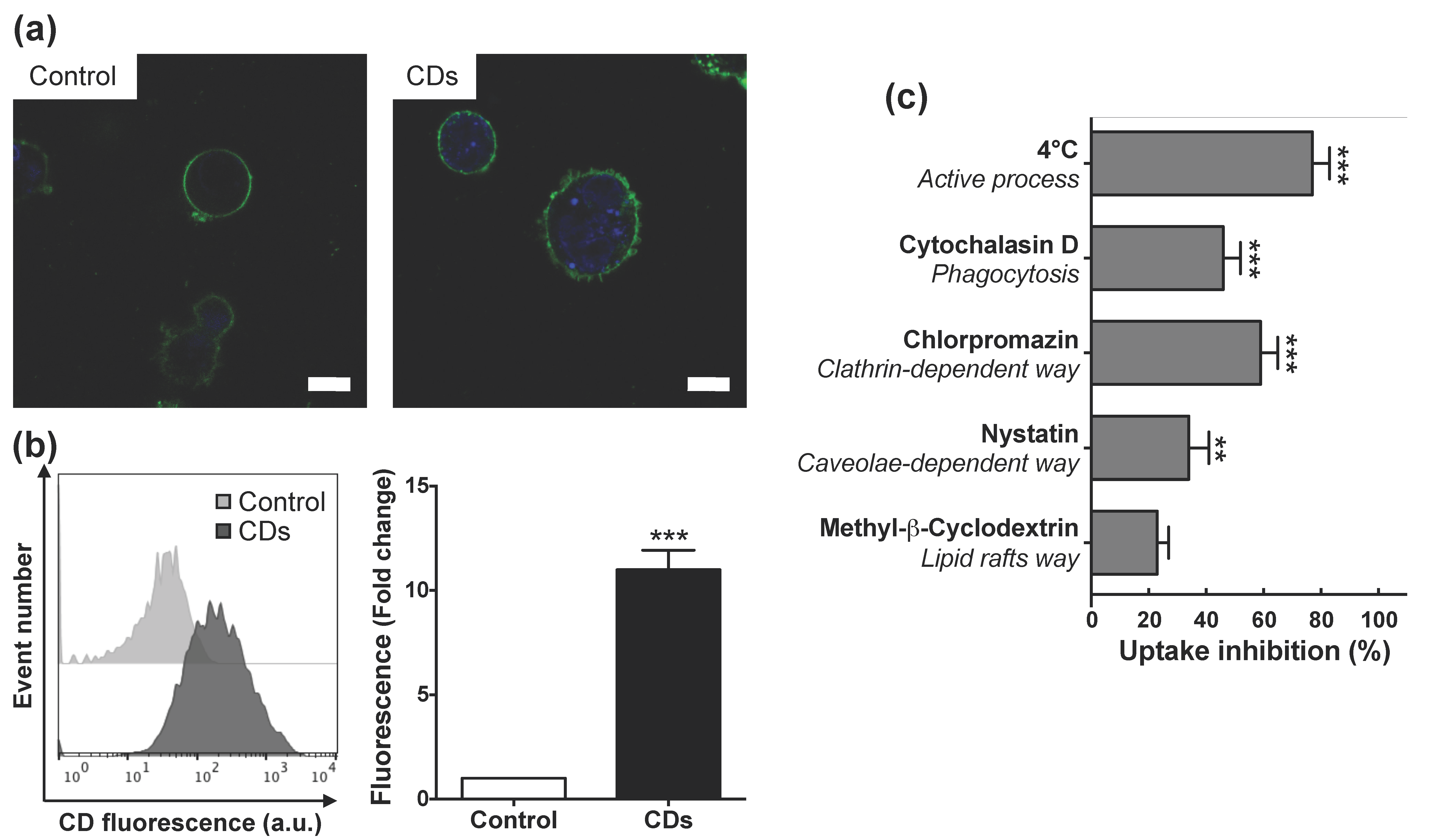
3.4. Internalization of CDs by Cultured Macrophages and Its Mechanisms
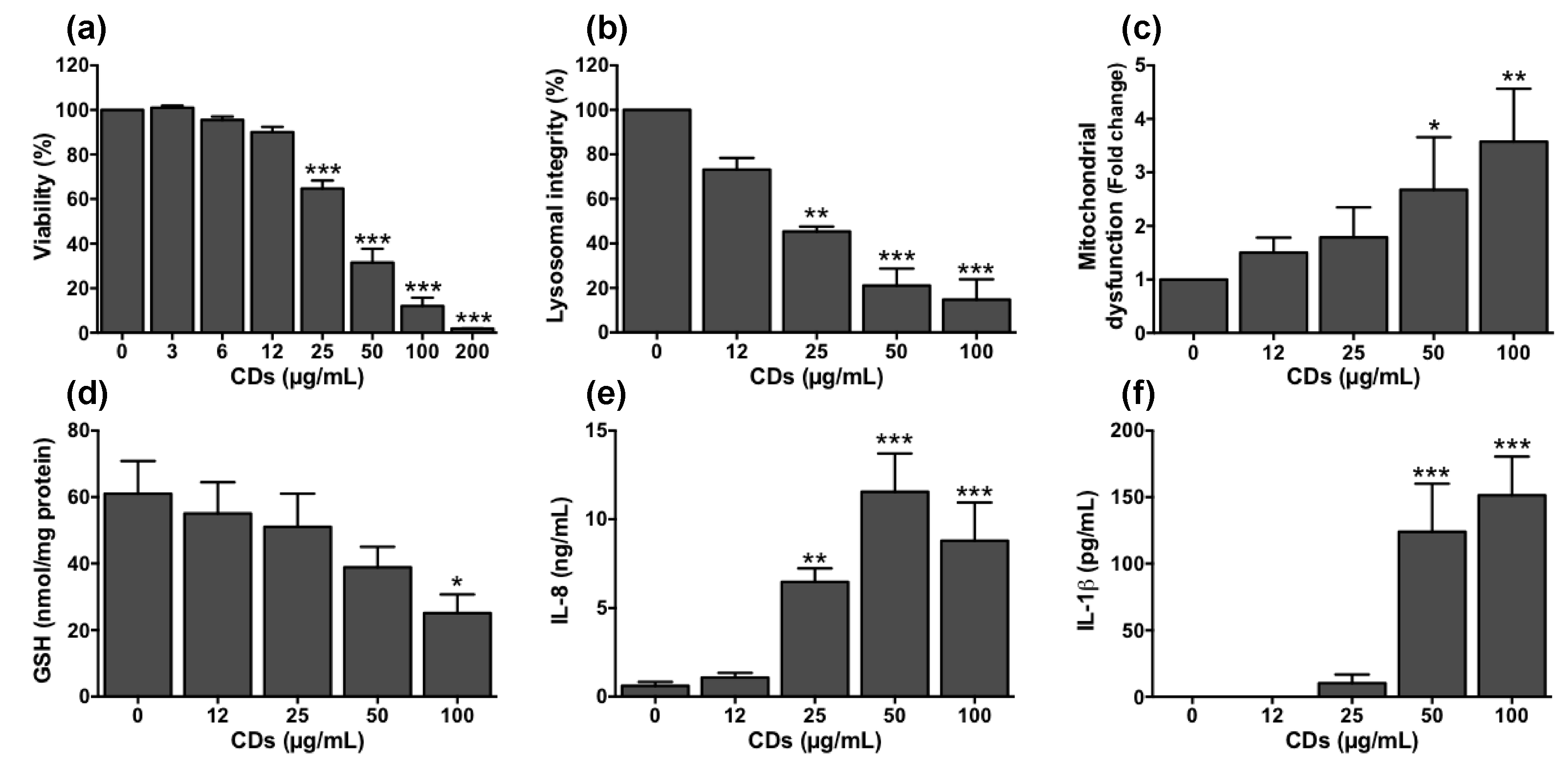
3.5. Cellular Mechanisms Involved in the Toxicity of CDs towards Macrophages
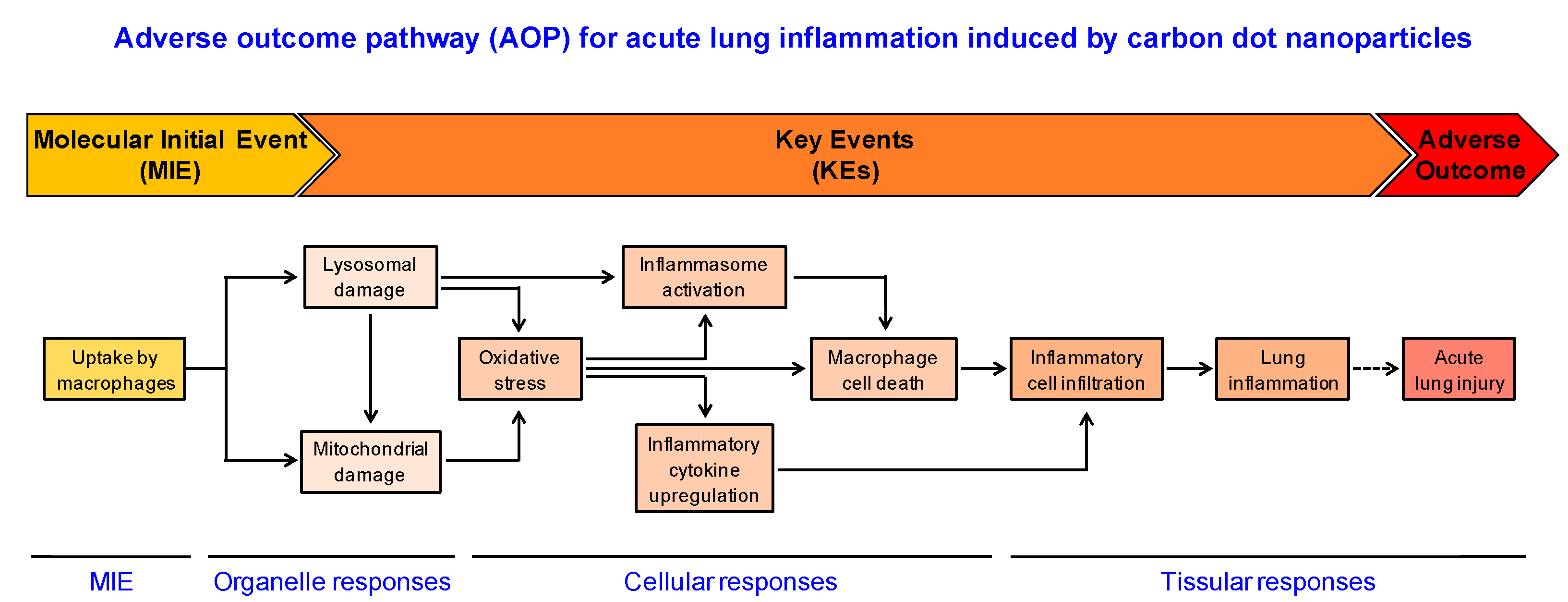
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vance, M.E.; Kuiken, T.; Vejerano, E.P.; McGinnis, S.P.; Hochella, M.F., Jr.; Rejeski, D.; Hull, M.S. Nanotechnology in the real world: Redeveloping the nanomaterial consumer products inventory. Beilstein J. Nanotechnol. 2015, 6, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Y.; Ray, R.; Gu, Y.L.; Ploehn, H.J.; Gearheart, L.; Raker, K.; Scrivens, W.A. Electrophoretic analysis and purification of fluorescent single-walled carbon nanotube fragments. J. Am. Chem. Soc. 2004, 126, 12736–12737. [Google Scholar] [CrossRef] [PubMed]
- Himaja, A.L.; Karthik, P.S.; Singh, S.P. Carbon Dots: The newest member of the carbon nanomaterials family. Chem. Rec. 2015, 15, 595–615. [Google Scholar] [CrossRef]
- Kang, C.; Huang, Y.; Yang, H.; Yan, X.F.; Chen, Z.P. A review of carbon dots produced from biomass wastes. Nanomaterials 2020, 10, 2316. [Google Scholar] [CrossRef] [PubMed]
- Claudel, M.; Fan, J.H.; Rapp, M.; Pons, F.; Lebeau, L. Influence of carbonization conditions on luminescence and gene delivery properties of nitrogen-doped carbon dots. RSC Adv. 2019, 9, 3493–3502. [Google Scholar] [CrossRef]
- Huang, D.P.; Zhou, H.F.; Wu, Y.Q.; Wang, T.; Sun, L.L.; Gao, P.; Sun, Y.Z.; Huang, H.N.; Zhou, G.J.; Hu, J.F. Bottom-up synthesis and structural design strategy for graphene quantum dots with tunable emission to the near infrared region. Carbon 2019, 142, 673–684. [Google Scholar] [CrossRef]
- Kang, Z.H.; Lee, S.T. Carbon dots: Advances in nanocarbon applications. Nanoscale 2019, 11, 19214–19224. [Google Scholar] [CrossRef]
- Ghosal, K.; Ghosh, A. Carbon dots: The next generation platform for biomedical applications. Mater. Sci. Eng. C 2019, 96, 887–903. [Google Scholar] [CrossRef]
- Du, J.J.; Xu, N.; Fan, J.L.; Sun, W.; Peng, X.J. Carbon dots for in vivo bioimaging and theranostics. Small 2019, 15, e1805087. [Google Scholar] [CrossRef]
- Gomez, I.J.; Arnaiz, B.; Cacioppo, M.; Arcudi, F.; Prato, M. Nitrogen-doped carbon nanodots for bioimaging and delivery of paclitaxel. J. Mater. Chem. B. 2018, 6, 5540–5548. [Google Scholar] [CrossRef]
- Yuan, Y.; Guo, B.; Hao, L.; Liu, N.; Lin, Y.; Guo, W.; Li, X.; Gu, B. Doxorubicin-loaded environmentally friendly carbon dots as a novel drug delivery system for nucleus targeted cancer therapy. Colloid Surf. B Biointerfaces 2017, 159, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Choi, Y.; Park, G.; Won, C.; Park, Y.J.; Lee, Y.; Kim, B.S.; Min, D.H. Highly efficient gene silencing and bioimaging based on fluorescent carbon dots in vitro and in vivo. Nano Res. 2017, 10, 503–519. [Google Scholar] [CrossRef]
- Pierrat, P.; Wang, R.; Kereselidze, D.; Lux, M.; Didier, P.; Kichler, A.; Pons, F.; Lebeau, L. Efficient in vitro and in vivo pulmonary delivery of nucleic acid by carbon dot-based nanocarriers. Biomaterials 2015, 51, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Gomes, V.G.; Dehghani, A.; Ardekani, S.M. Engineering carbon quantum dots for photomediated theranostics. Nano Res. 2018, 11, 1–41. [Google Scholar]
- Nel, A.; Xia, T.; Madler, L.; Li, N. Toxic potential of materials at the nanolevel. Science 2006, 311, 622–627. [Google Scholar] [CrossRef]
- Oberdorster, G.; Oberdorster, E.; Oberdorster, J. Nanotoxicology: An emerging discipline evolving from studies of ultrafine particles. Environ. Health Perspect. 2005, 113, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Kreyling, W.G.; Semmler-Behnke, M.; Takenaka, S.; Moller, W. Differences in the biokinetics of inhaled nano- versus micrometer-sized particles. Acc. Chem. Res. 2013, 46, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Ronzani, C.; Spiegelhalter, C.; Vonesch, J.L.; Lebeau, L.; Pons, F. Lung deposition and toxicological responses evoked by multi-walled carbon nanotubes dispersed in a synthetic lung surfactant in the mouse. Arch. Toxicol. 2012, 86, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Laskin, D.L.; Malaviya, R.; Laskin, J.D. Role of macrophages in acute lung injury and chronic fibrosis induced by pulmonary toxicants. Toxicol. Sci. 2019, 168, 287–301. [Google Scholar] [CrossRef]
- Manke, A.; Wang, L.Y.; Rojanasakul, Y. Mechanisms of nanoparticle-induced oxidative stress and toxicity. Biomed. Res. Int. 2013, 2013, 942916. [Google Scholar] [CrossRef]
- Nho, R. Pathological effects of nano-sized particles on the respiratory system. Nanomed. Nanotechnol. 2020, 29, 102242. [Google Scholar] [CrossRef]
- Krug, H.F.; Wick, P. Nanotoxicology: An interdisciplinary challenge. Angew. Chem. Int. Ed. 2011, 50, 1260–1278. [Google Scholar] [CrossRef] [PubMed]
- Gerloff, K.; Landesmann, B.; Worth, A.; Munn, S.; Palosaari, T.; Whelan, M. The adverse outcome pathway approach in nanotoxicology. Comput. Toxicol. 2017, 1, 3–11. [Google Scholar] [CrossRef]
- Ankley, G.T.; Edwards, S.W. The Adverse Outcome Pathway: A multifaceted framework supporting 21st century toxicology. Curr. Opin. Toxicol. 2018, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; LaLone, C.A. Adverse Outcome Pathways: Moving from a scientific concept to an internationally accepted framework. Environ. Toxicol. Chem. 2019, 38, 1152–1163. [Google Scholar] [CrossRef]
- Halappanavar, S.; van den Brule, S.; Nymark, P.; Gate, L.; Seidel, C.; Valentino, S.; Zhernovkov, V.; Danielsen, P.H.; De Vizcaya, A.; Wolff, H.; et al. Adverse outcome pathways as a tool for the design of testing strategies to support the safety assessment of emerging advanced materials at the nanoscale. Part. Fibre Toxicol. 2020, 17, 16. [Google Scholar] [CrossRef]
- Vietti, G.; Lison, D.; van den Brule, S. Mechanisms of lung fibrosis induced by carbon nanotubes: Towards an Adverse Outcome Pathway (AOP). Part. Fibre Toxicol. 2016, 13, 11. [Google Scholar] [CrossRef]
- Labib, S.; Williams, A.; Yauk, C.L.; Nikota, J.K.; Wallin, H.; Vogel, U.; Halappanavar, S. Nano-risk Science: Application of toxicogenomics in an adverse outcome pathway framework for risk assessment of multi-walled carbon nanotubes. Part. Fibre Toxicol. 2016, 13, 15. [Google Scholar] [CrossRef]
- Havrdova, M.; Hola, K.; Skopalik, J.; Tomankova, K.; Martin, P.A.; Cepe, K.; Polakova, K.; Tucek, J.; Bourlinos, A.B.; Zboril, R. Toxicity of carbon dots—Effect of surface functionalization on the cell viability, reactive oxygen species generation and cell cycle. Carbon 2016, 99, 238–248. [Google Scholar] [CrossRef]
- Liu, C.J.; Zhang, P.; Zhai, X.Y.; Tian, F.; Li, W.C.; Yang, J.H.; Liu, Y.; Wang, H.B.; Wang, W.; Liu, W.G. Nano-carrier for gene delivery and bioimaging based on carbon dots with PEI-passivation enhanced fluorescence. Biomaterials 2012, 33, 3604–3613. [Google Scholar] [CrossRef]
- Yang, Y.; Ren, X.L.; Sun, Z.N.; Fu, C.H.; Liu, T.L.; Meng, X.W.; Wang, Z.L. Toxicity and bio-distribution of carbon dots after single inhalation exposure in vivo. Chin. Chem. Lett. 2018, 29, 895–898. [Google Scholar] [CrossRef]
- Fan, J.H.; Claudel, M.; Ronzani, C.; Arezki, Y.; Lebeau, L.; Pons, F. Physicochemical characteristics that affect carbon dot safety: Lessons from a comprehensive study on a nanoparticle library. Int. J. Pharm. 2019, 569, 118521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Qu, J.; Yao, Y.; Zhang, Y.; Ma, Y.; Wu, D.; Cao, Y.; Yang, M.; Zhang, Y.; Tang, M.; et al. N-doped carbon dots triggered the induction of ROS-mediated cytoprotective autophagy in Hepa1-6 cells. Chemosphere 2020, 251, 126440. [Google Scholar] [CrossRef] [PubMed]
- Lategan, K.; Fowler, J.; Bayati, M.; de Cortalezzi, M.F.; Pool, E. The effects of carbon dots on immune system biomarkers, using the murine macrophage cell line RAW 264.7 and human whole blood cell cultures. Nanomaterials 2018, 8, 388. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.T.; Sun, H.Q.; Ruan, S.B.; Wang, Y.; Shen, S.; Xu, W.M.; He, Q.; Gao, H.L. In vitro and in vivo toxicology of bare and PEGylated fluorescent carbonaceous nanodots in mice and zebrafish: The potential relationship with autophagy. RSC Adv. 2015, 5, 38547–38557. [Google Scholar] [CrossRef]
- Ronzani, C.; Van Belle, C.; Didier, P.; Spiegelhalter, C.; Pierrat, P.; Lebeau, L.; Pons, F. Lysosome mediates toxicological effects of polyethyleneimine-based cationic carbon dots. J. Nanopart. Res. 2019, 21, 4. [Google Scholar] [CrossRef]
- Ronzani, C.; Casset, A.; Pons, F. Exposure to multi-walled carbon nanotubes results in aggravation of airway inflammation and remodeling and in increased production of epithelium-derived innate cytokines in a mouse model of asthma. Arch. Toxicol. 2014, 88, 489–499. [Google Scholar] [CrossRef]
- Daubeuf, F.; Becker, J.; Aguilar-Pimentel, J.A.; Ebel, C.; Hrabe de Angelis, M.; Herault, Y.; Frossard, N. A fast, easy, and customizable eight-color flow cytometric method for analysis of the cellular content of bronchoalveolar lavage fluid in the mouse. Curr. Protoc. Mouse Biol. 2017, 7, 88–99. [Google Scholar] [CrossRef]
- Jhingran, A.; Kasahara, S.; Hohl, T.M. Flow cytometry of lung and bronchoalveolar lavage fluid cells from mice challenged with fluorescent Aspergillus Reporter (FLARE) Conidia. Bio Protoc. 2016, 6, e1927. [Google Scholar] [CrossRef]
- Collot, M.; Kreder, R.; Tatarets, A.L.; Patsenker, L.D.; Mely, Y.; Klymchenko, A.S. Bright fluorogenic squaraines with tuned cell entry for selective imaging of plasma membrane vs. endoplasmic reticulum. Chem. Commun. 2015, 51, 17136–17139. [Google Scholar] [CrossRef]
- Safar, R.; Ronzani, C.; Diab, R.; Chevrier, J.; Bensoussan, D.; Grandemange, S.; Le Faou, A.; Rihn, B.H.; Joubert, O. Human monocyte response to S-nitrosoglutathione-loaded nanoparticles: Uptake, viability, and transcriptome. Mol. Pharm. 2015, 12, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Nie, W.; He, Z.; Yang, J.; Shao, B.; Ma, X.; Zhang, X.; Bi, Z.; Sun, L.; Liang, X.; et al. Carbon black nanoparticles induce cell necrosis through lysosomal membrane permeabilization and cause subsequent inflammatory response. Theranostics 2020, 10, 4589–4605. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.X.; Li, P.Y.; Zhang, M.Y.; Han, B.; Chu, C.; Su, X.; Li, B.H.; Kang, H.; Ning, J.; Zhang, B.Y.; et al. Carbon black nanoparticles induce pulmonary fibrosis through NLRP3 inflammasome pathway modulated by miR-96 targeted FOXO3a. Chemosphere 2020, 241, 125075. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, M.; Vennemann, A.; Sauer, U.G.; Wiench, K.; Ma-Hock, L.; Landsiedel, R. An in vitro alveolar macrophage assay for predicting the short-term inhalation toxicity of nanomaterials. J. Nanobiotechnol. 2016, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.; Walter, J.M.; Misharin, A.V. Alveolar macrophages. Cell Immunol. 2018, 330, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Elgrabli, D.; Dachraoui, W.; Menard-Moyon, C.; Liu, X.J.; Begin, D.; Begin-Colin, S.; Bianco, A.; Gazeau, F.; Alloyeau, D. Carbon nanotube degradation in macrophages: Live nanoscale monitoring and understanding of biological pathway. ACS Nano 2015, 9, 10113–10124. [Google Scholar] [CrossRef]
- Elgrabli, D.; Floriani, M.; Abella-Gallart, S.; Meunier, L.; Gamez, C.; Delalain, P.; Rogerieux, F.; Boczkowski, J.; Lacroix, G. Biodistribution and clearance of instilled carbon nanotubes in rat lung. Part. Fibre Toxicol. 2008, 5, 20. [Google Scholar] [CrossRef]
- Marano, F.; Hussain, S.; Rodrigues-Lima, F.; Baeza-Squiban, A.; Boland, S. Nanoparticles: Molecular targets and cell signalling. Arch. Toxicol. 2011, 85, 733–741. [Google Scholar] [CrossRef]
- Zhou, N.; Zhu, S.J.; Maharjan, S.; Hao, Z.Y.; Song, Y.B.; Zhao, X.H.; Jiang, Y.F.; Yang, B.; Lu, L.J. Elucidating the endocytosis, intracellular trafficking, and exocytosis of carbon dots in neural cells. RSC Adv. 2014, 4, 62086–62095. [Google Scholar] [CrossRef]
- Lund, T.; Callaghan, M.F.; Williams, P.; Turmaine, M.; Bachmann, C.; Rademacher, T.; Roitt, I.M.; Bayford, R. The influence of ligand organization on the rate of uptake of gold nanoparticles by colorectal cancer cells. Biomaterials 2011, 32, 9776–9784. [Google Scholar] [CrossRef]
- Donahue, N.D.; Acar, H.; Wilhelm, S. Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Adv. Drug Deliv. Rev. 2019, 143, 68–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Gao, H.J.; Bao, G. Physical principles of nanoparticle cellular endocytosis. ACS Nano 2015, 9, 8655–8671. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Lynch, I.; Ejtehadi, M.R.; Monopoli, M.P.; Bombelli, F.B.; Laurent, S. Protein-nanoparticle interactions: Opportunities and challenges. Chem. Rev. 2011, 111, 5610–5637. [Google Scholar] [CrossRef] [PubMed]
- Drasler, B.; Sayre, P.; Steinhauser, K.G.; Petri-Fink, A.; Rothen-Rutishauser, B. In vitro approaches to assess the hazard of nanomaterials. Nanoimpact 2017, 8, 99–116. [Google Scholar] [CrossRef]
- Loret, T.; Rogerieux, F.; Trouiller, B.; Braun, A.; Egles, C.; Lacroix, G. Predicting the in vivo pulmonary toxicity induced by acute exposure to poorly soluble nanomaterials by using advanced in vitro methods. Part. Fibre Toxicol. 2018, 15, 25. [Google Scholar] [CrossRef]
- Sima, M.; Vrbova, K.; Zavodna, T.; Honkova, K.; Chvojkova, I.; Ambroz, A.; Klema, J.; Rossnerova, A.; Polakova, K.; Malina, T.; et al. The differential effect of carbon dots on gene expression and DNA methylation of human embryonic lung fibroblasts as a function of surface charge and dose. Int. J. Mol. Sci. 2020, 21, 4763. [Google Scholar] [CrossRef]
- Liu, N.; Tang, M. Toxic effects and involved molecular pathways of nanoparticles on cells and subcellular organelles. J. Appl. Toxicol. 2020, 40, 16–36. [Google Scholar] [CrossRef]
- Moore, M.N.; Allen, J.I.; McVeigh, A. Environmental prognostics: An integrated model supporting lysosomal stress responses as predictive biomarkers of animal health status. Mar. Environ. Res. 2006, 61, 278–304. [Google Scholar] [CrossRef]
- Stern, S.T.; Adiseshaiah, P.P.; Crist, R.M. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part. Fibre Toxicol. 2012, 9, 20. [Google Scholar] [CrossRef]
- Zhao, F.; Zhao, Y.; Liu, Y.; Chang, X.; Chen, C.; Zhao, Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small 2011, 7, 1322–1337. [Google Scholar] [CrossRef]
- Tahara, Y.; Nakamura, M.; Yang, M.; Zhang, M.; Iijima, S.; Yudasaka, M. Lysosomal membrane destabilization induced by high accumulation of single-walled carbon nanohorns in murine macrophage RAW 264.7. Biomaterials 2012, 33, 2762–2769. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; Wang, Z.X.; Lv, Q.Y.; Dong, P.X.; Zhao, L.X.; Yang, Y.; Guo, L.H. Single-walled carbon nanotubes and graphene oxides induce autophagosome accumulation and lysosome impairment in primarily cultured murine peritoneal macrophages. Toxicol. Lett. 2013, 221, 118–127. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, Y.J.; Teng, X.Y.; Yan, M.Q.; Bi, H.; Morais, P.C. Mitochondria-targeting nanoplatform with fluorescent carbon dots for long time imaging and magnetic field-enhanced cellular uptake. ACS Appl. Mater. Interfaces 2015, 7, 10201–10212. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xia, T.; Nel, A.E. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic. Biol. Med. 2008, 44, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Palic, D. Micro- and nano-plastics activation of oxidative and inflammatory adverse outcome pathways. Redox Biol. 2020, 37, 101620. [Google Scholar] [CrossRef]
- Shirasuna, K.; Karasawa, T.; Takahashi, M. Exogenous nanoparticles and endogenous crystalline molecules as danger signals for the NLRP3 inflammasomes. J. Cell Physiol. 2019, 234, 5436–5450. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Svadlakova, T.; Hubatka, F.; Knotigova, P.T.; Kulich, P.; Masek, J.; Kotoucek, J.; Macak, J.; Motola, M.; Kalbac, M.; Kolackova, M.; et al. Proinflammatory effect of carbon-based nanomaterials: In vitro study on stimulation of inflammasome NLRP3 via destabilisation of lysosomes. Nanomaterials 2020, 10, 418. [Google Scholar] [CrossRef]
- Villeneuve, D.L.; Landesmann, B.; Allavena, P.; Ashley, N.; Bal-Price, A.; Corsini, E.; Halappanavar, S.; Hussell, T.; Laskin, D.; Lawrence, T.; et al. Representing the process of inflammation as Key Events in Adverse Outcome Pathways. Toxicol. Sci. 2018, 163, 346–352. [Google Scholar] [CrossRef]
- Braakhuis, H.M.; Park, M.V.D.Z.; Gosens, I.; De Jong, W.H.; Cassee, F.R. Physicochemical characteristics of nanomaterials that affect pulmonary inflammation. Part. Fibre Toxicol. 2014, 11, 18. [Google Scholar] [CrossRef]
- Clippinger, A.J.; Allen, D.; Behrsing, H.; BeruBe, K.A.; Bolger, M.B.; Casey, W.; DeLorme, M.; Gaca, M.; Gehen, S.C.; Glover, K.; et al. Pathway-based predictive approaches for non-animal assessment of acute inhalation toxicity. Toxicol. In Vitro 2018, 52, 131–145. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Data |
|---|---|
| Size | DLS: 10.1 ± 0.4 nm, PdI = 0.240 ± 0.061 TEM: 39.5 nm |
| Surface charge | Zeta potential: ζ = +23.9 ± 2.0 mV |
| Chemical composition | Nitrogen: 14.6% Carbon: 29.0% Hydrogen: 6.95% |
| Photoluminescence | λmax: 354 nm λex: 368 nm λem: 462 nm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, M.; Fan, J.; Claudel, M.; Lebeau, L.; Pons, F.; Ronzani, C. Combined In Vitro and In Vivo Approaches to Propose a Putative Adverse Outcome Pathway for Acute Lung Inflammation Induced by Nanoparticles: A Study on Carbon Dots. Nanomaterials 2021, 11, 180. https://doi.org/10.3390/nano11010180
Weiss M, Fan J, Claudel M, Lebeau L, Pons F, Ronzani C. Combined In Vitro and In Vivo Approaches to Propose a Putative Adverse Outcome Pathway for Acute Lung Inflammation Induced by Nanoparticles: A Study on Carbon Dots. Nanomaterials. 2021; 11(1):180. https://doi.org/10.3390/nano11010180
Chicago/Turabian StyleWeiss, Maud, Jiahui Fan, Mickaël Claudel, Luc Lebeau, Françoise Pons, and Carole Ronzani. 2021. "Combined In Vitro and In Vivo Approaches to Propose a Putative Adverse Outcome Pathway for Acute Lung Inflammation Induced by Nanoparticles: A Study on Carbon Dots" Nanomaterials 11, no. 1: 180. https://doi.org/10.3390/nano11010180
APA StyleWeiss, M., Fan, J., Claudel, M., Lebeau, L., Pons, F., & Ronzani, C. (2021). Combined In Vitro and In Vivo Approaches to Propose a Putative Adverse Outcome Pathway for Acute Lung Inflammation Induced by Nanoparticles: A Study on Carbon Dots. Nanomaterials, 11(1), 180. https://doi.org/10.3390/nano11010180