A Legendre–Fenchel Transform for Molecular Stretching Energies

 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Method
2.1. Simulation Details
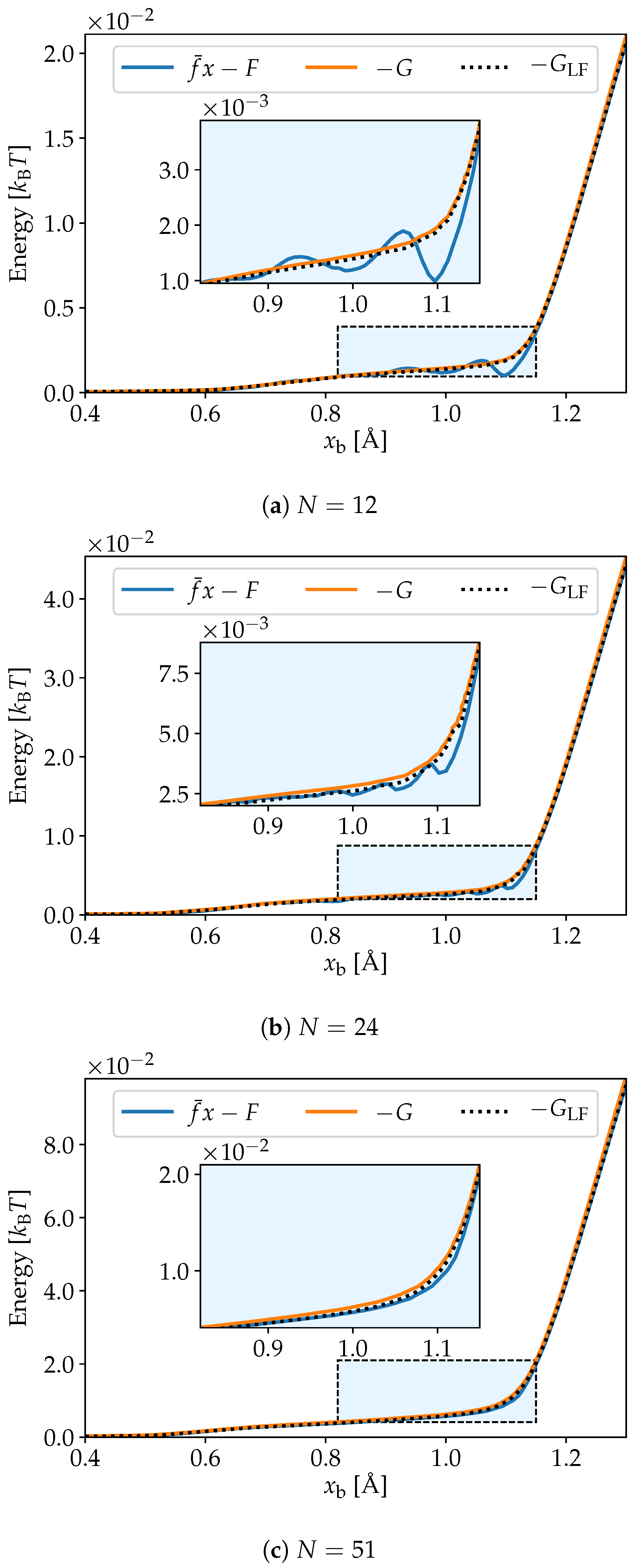
2.2. Energy Transforms
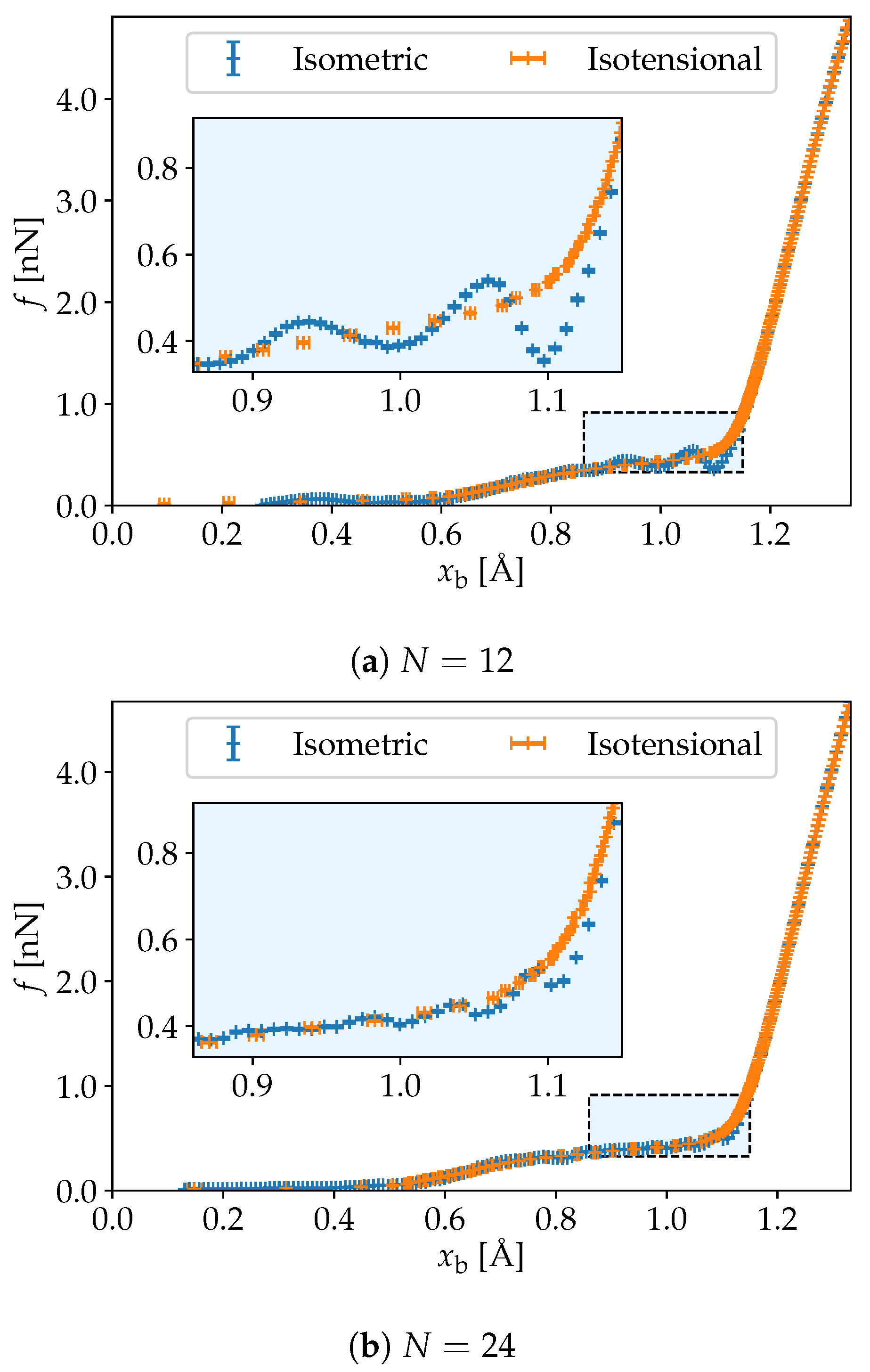
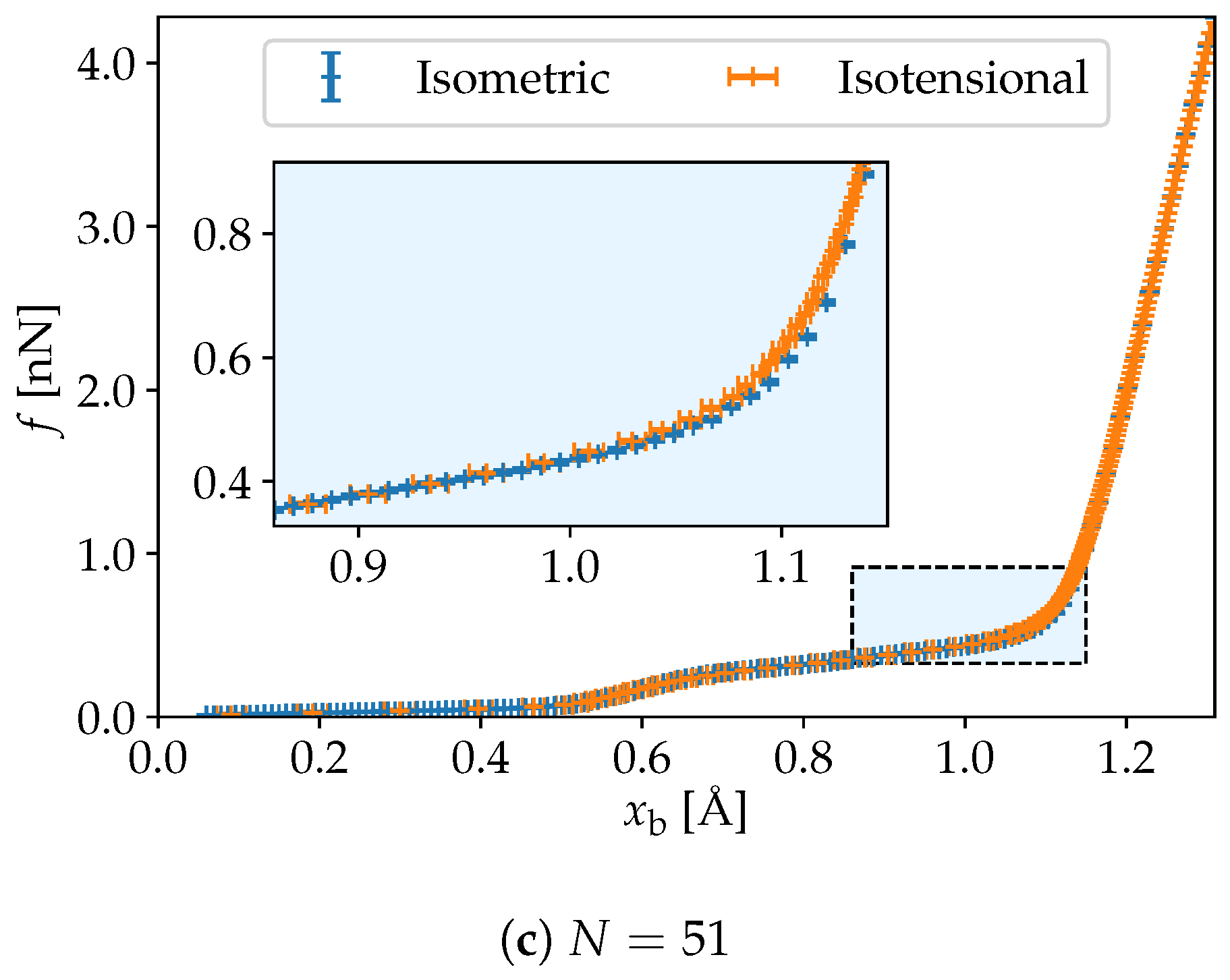
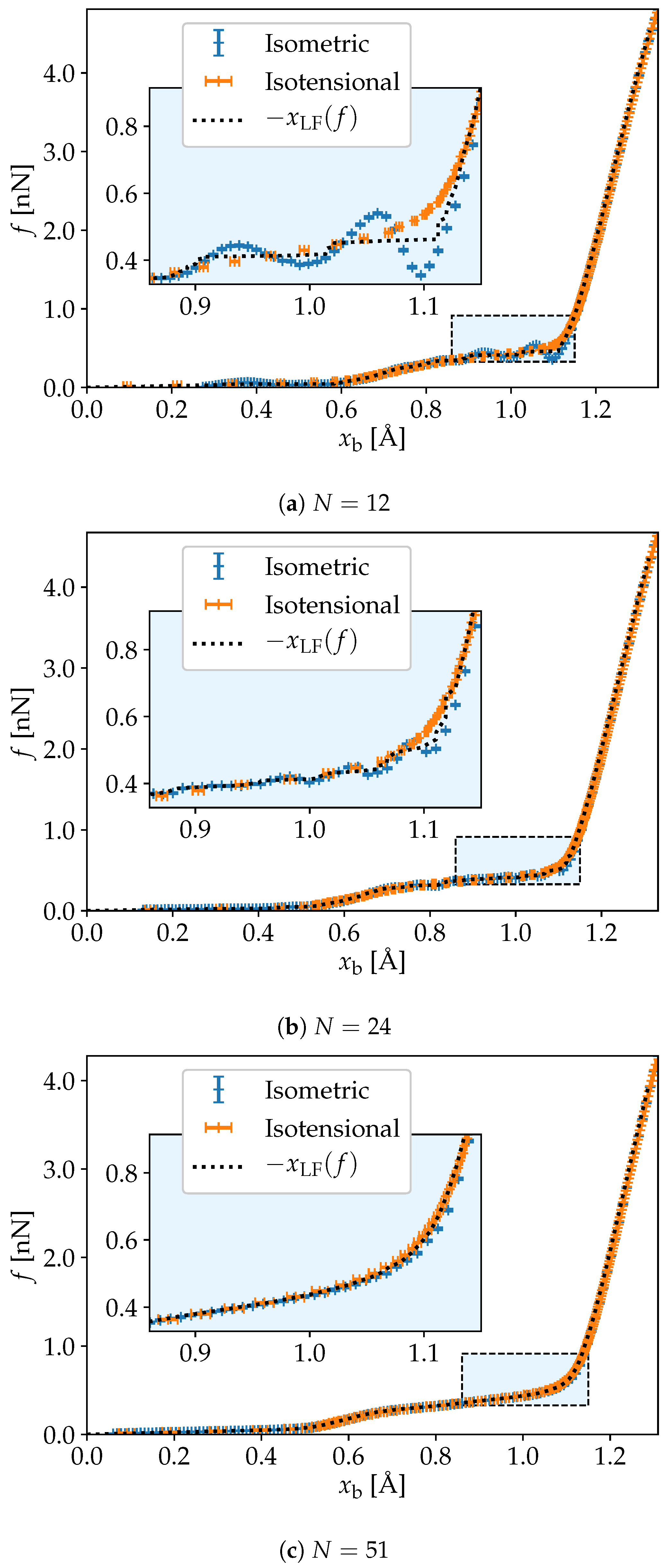
3. Simulation Results
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hill, T.L. Thermodynamics of Small Systems; W.A. Benjamin Inc.: New York, NY, USA, 1963; p. 210. [Google Scholar]
- Bedeaux, D.; Kjelstrup, S.; Schnell, S.K. Nanothermodynamics—General Theory, 1st ed.; PoreLab: Trondheim, Norway, 2020. [Google Scholar]
- Campa, A.; Dauxois, T.; Ruffo, S. Statistical mechanics and dynamics of solvable models with long-range interactions. Phys. Rep. 2009, 480, 57–159. [Google Scholar] [CrossRef]
- Bouchet, F.; Gupta, S.; Mukamel, D. Thermodynamics and dynamics of systems with long-range interactions. Phys. A Stat. Mech. Its Appl. 2010, 389, 4389–4405. [Google Scholar] [CrossRef]
- Campa, A.; Dauxois, T.; Fanelli, D.; Ruffo, S. Physics of Long-Range Interacting Systems; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Latella, I.; Pérez-Madrid, A. Local thermodynamics and the generalized Gibbs-Duhem equation in systems with long-range interactions. Phys. Rev. E 2013, 88, 42135. [Google Scholar] [CrossRef]
- Latella, I.; Pérez-Madrid, A.; Campa, A.; Casetti, L.; Ruffo, S. Thermodynamics of Nonadditive Systems. Phys. Rev. Lett. 2015, 114, 230601. [Google Scholar] [CrossRef]
- Gross, D.H.E. Microcanonical Thermodynamics; World Scientific: Singapore, 2001. [Google Scholar]
- Chomaz, P.; Gulminelli, F. Phase Transitions in Finite Systems. In Dynamics and Thermodynamics of Systems with Long Range Interactions; Dauxois, T., Ruffo, S., Arimondo, E., Wilkens, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; Volume 602, pp. 68–129. [Google Scholar]
- Süzen, M.; Sega, M.; Holm, C. Ensemble inequivalence in single-molecule experiments. Phys. Rev. E 2009, 79, 051118. [Google Scholar] [CrossRef]
- Bering, E.; Kjelstrup, S.; Bedeaux, D.; Miguel Rubi, J.; de Wijn, A.S. Entropy Production beyond the Thermodynamic Limit from Single-Molecule Stretching Simulations. J. Phys. Chem. B 2020, 124, 8909–8917. [Google Scholar] [CrossRef] [PubMed]
- Keller, D.; Swigon, D.; Bustamante, C. Relating Single-Molecule Measurements to Thermodynamics. Biophys. J. 2003, 84, 733–738. [Google Scholar] [CrossRef]
- Monge, A.M.; Manosas, M.; Ritort, F. Experimental test of ensemble inequivalence and the fluctuation theorem in the force ensemble in DNA pulling experiments. Phys. Rev. E 2018, 98, 032146. [Google Scholar] [CrossRef]
- Rockafellar, T. Convex Analysis; Princeton University Press: Princeton, NJ, USA, 1972. [Google Scholar]
- Ellis, R.; Haven, K.; Turkington, B. Large Deviation Principles and Complete Equivalence and Nonequivalence Results for Pure and Mixed Ensembles. J. Stat. Phys. 2000, 101, 999–1064. [Google Scholar] [CrossRef]
- Touchette, H. The large deviation approach to statistical mechanics. Phys. Rep. 2009, 478, 1–69. [Google Scholar] [CrossRef]
- Bering, E.; de Wijn, A.S. Stretching and breaking of PEO nanofibres. A classical force field and ab initio simulation study. Soft Matter 2020, 16, 2736–2752. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Depa, P.; Sakai, V.G.; Maranas, J.K.; Lynn, J.W.; Peral, I.; Copley, J.R.D. A comparison of united atom, explicit atom, and coarse-grained simulation models for poly(ethylene oxide). J. Chem. Phys. 2006, 124, 234901(11). [Google Scholar] [CrossRef] [PubMed]
- van Zon, A.; Mos, B.; Verkerk, P.; de Leeuw, S. On the dynamics of PEO-NaI polymer electrolytes. Electrochim. Acta 2001, 46, 1717–1721. [Google Scholar] [CrossRef]
- Neyertz, S.; Brown, D.; Thomas, J.O. Molecular dynamics simulation of crystalline poly(ethylene oxide). J. Chem. Phys. 1994, 101, 10064. [Google Scholar] [CrossRef]
- Beyer, M.K. The mechanical strength of a covalent bond calculated by density functional theory. J. Chem. Phys. 2000, 112, 7307–7312. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Steinbach, P.J.; Brooks, B.R. Protein simulation below the glass-transition temperature. Dependence on cooling protocol. Chem. Phys. Lett. 1994, 226, 447–452. [Google Scholar] [CrossRef]
- Hill, T.L. An Introduction to Statistical Thermodynamics; Dover Publications Inc.: New York, NY, USA, 1986. [Google Scholar]
- Campa, A.; Casetti, L.; Latella, I.; Pérez-Madrid, A.; Ruffo, S. Concavity, Response Functions and Replica Energy. Entropy 2018, 20, 907. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
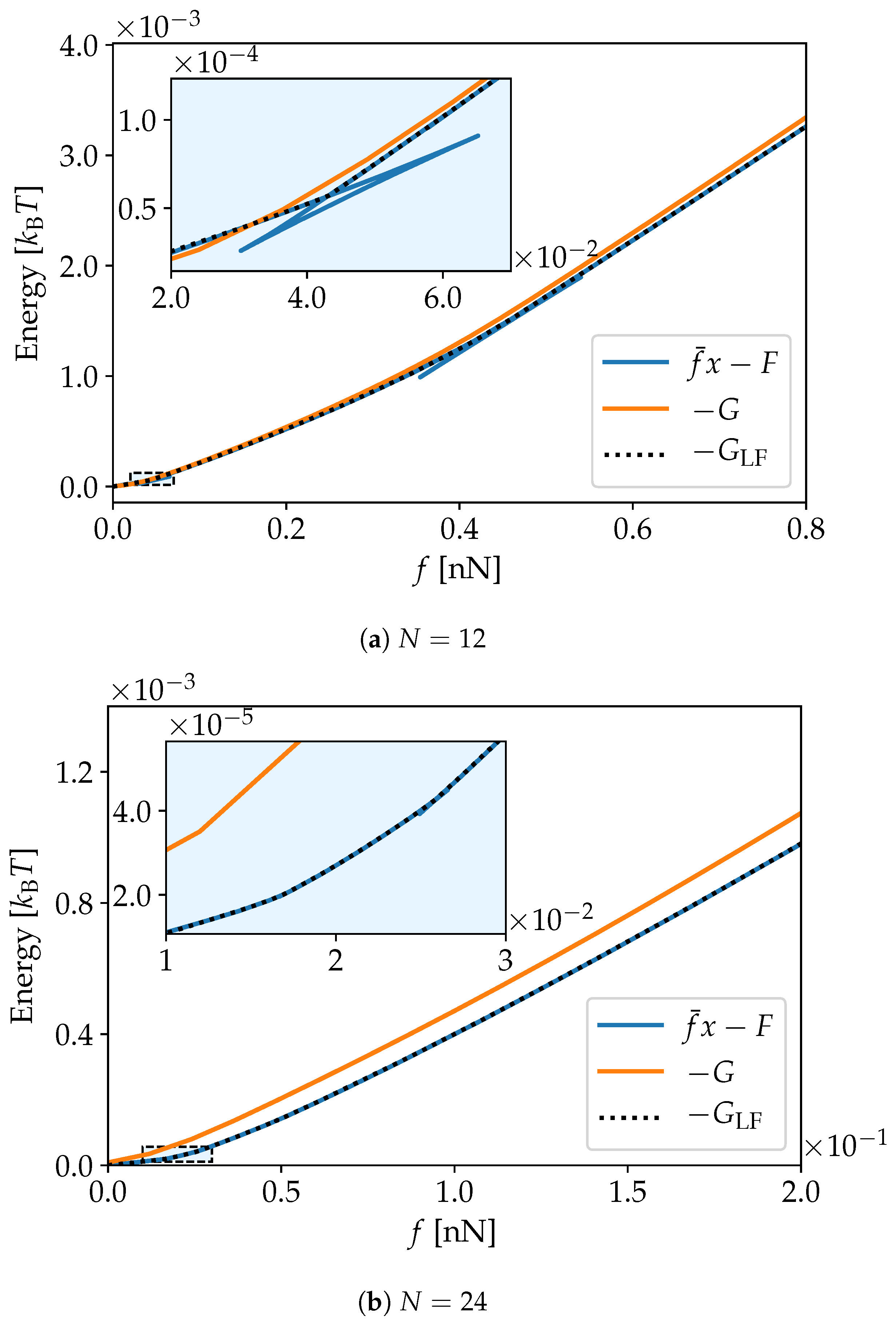{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bering, E.; Bedeaux, D.; Kjelstrup, S.; de Wijn, A.S.; Latella, I.; Rubi, J.M. A Legendre–Fenchel Transform for Molecular Stretching Energies. Nanomaterials 2020, 10, 2355. https://doi.org/10.3390/nano10122355
Bering E, Bedeaux D, Kjelstrup S, de Wijn AS, Latella I, Rubi JM. A Legendre–Fenchel Transform for Molecular Stretching Energies. Nanomaterials. 2020; 10(12):2355. https://doi.org/10.3390/nano10122355
Chicago/Turabian StyleBering, Eivind, Dick Bedeaux, Signe Kjelstrup, Astrid S. de Wijn, Ivan Latella, and J. Miguel Rubi. 2020. "A Legendre–Fenchel Transform for Molecular Stretching Energies" Nanomaterials 10, no. 12: 2355. https://doi.org/10.3390/nano10122355
APA StyleBering, E., Bedeaux, D., Kjelstrup, S., de Wijn, A. S., Latella, I., & Rubi, J. M. (2020). A Legendre–Fenchel Transform for Molecular Stretching Energies. Nanomaterials, 10(12), 2355. https://doi.org/10.3390/nano10122355