
A Comparative Study of Three Approaches to Fibre’s Surface Functionalization



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Electrospinning
2.3. Optimization of Surface Activation Methods
2.3.1. Hydrolysis
2.3.2. Plasma
2.3.3. Aminolysis
2.4. Functionalization
2.5. Functionalization
2.5.1. Water Contact Angle
2.5.2. Nonwovens Morphology
2.5.3. Quantification of -COOH Groups
2.5.4. Fourier Transform Infrared Spectroscopy (FTIR)
2.5.5. Molecular Weight
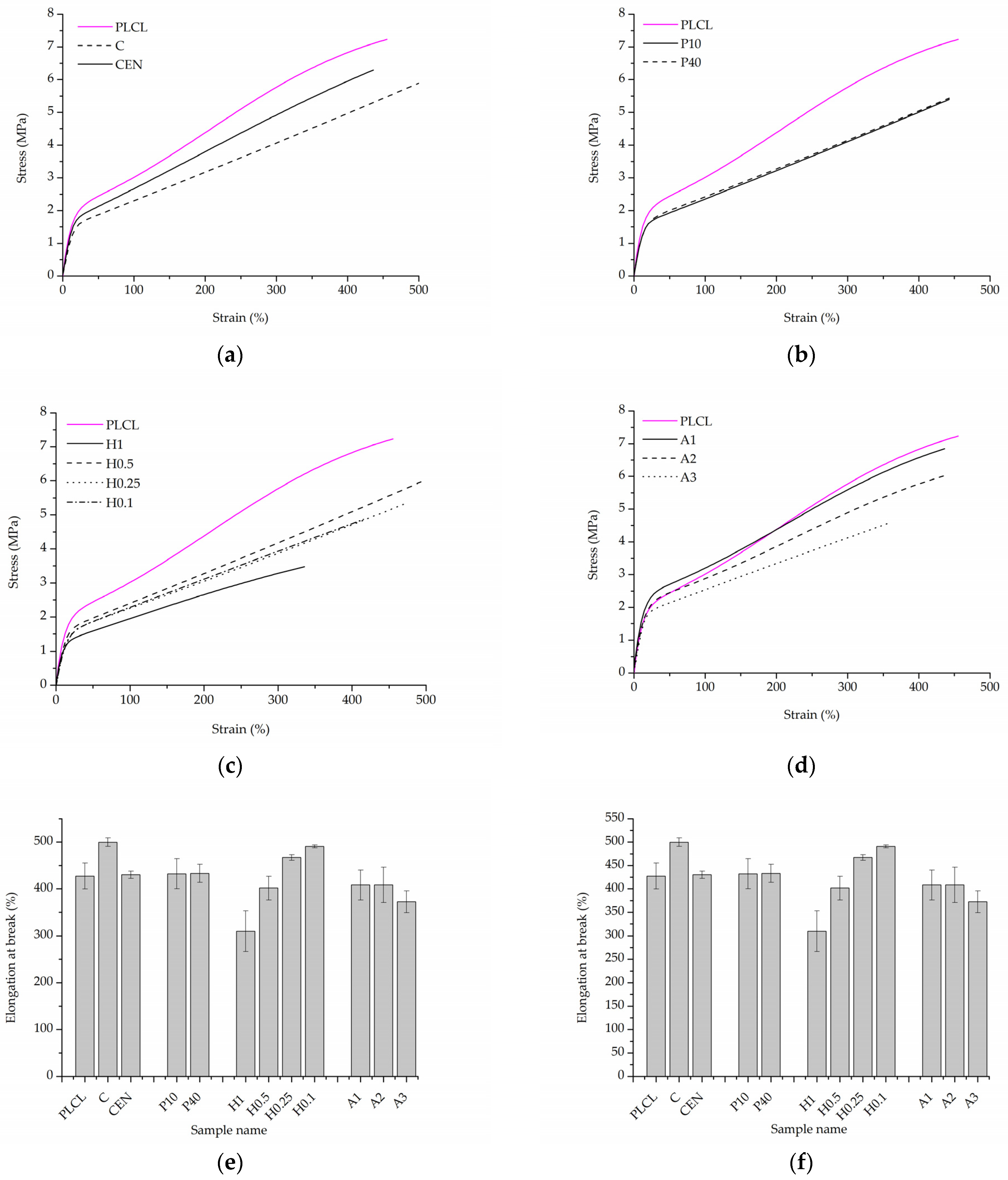
2.5.6. Mechanical Tests
2.5.7. Functionalization Stability
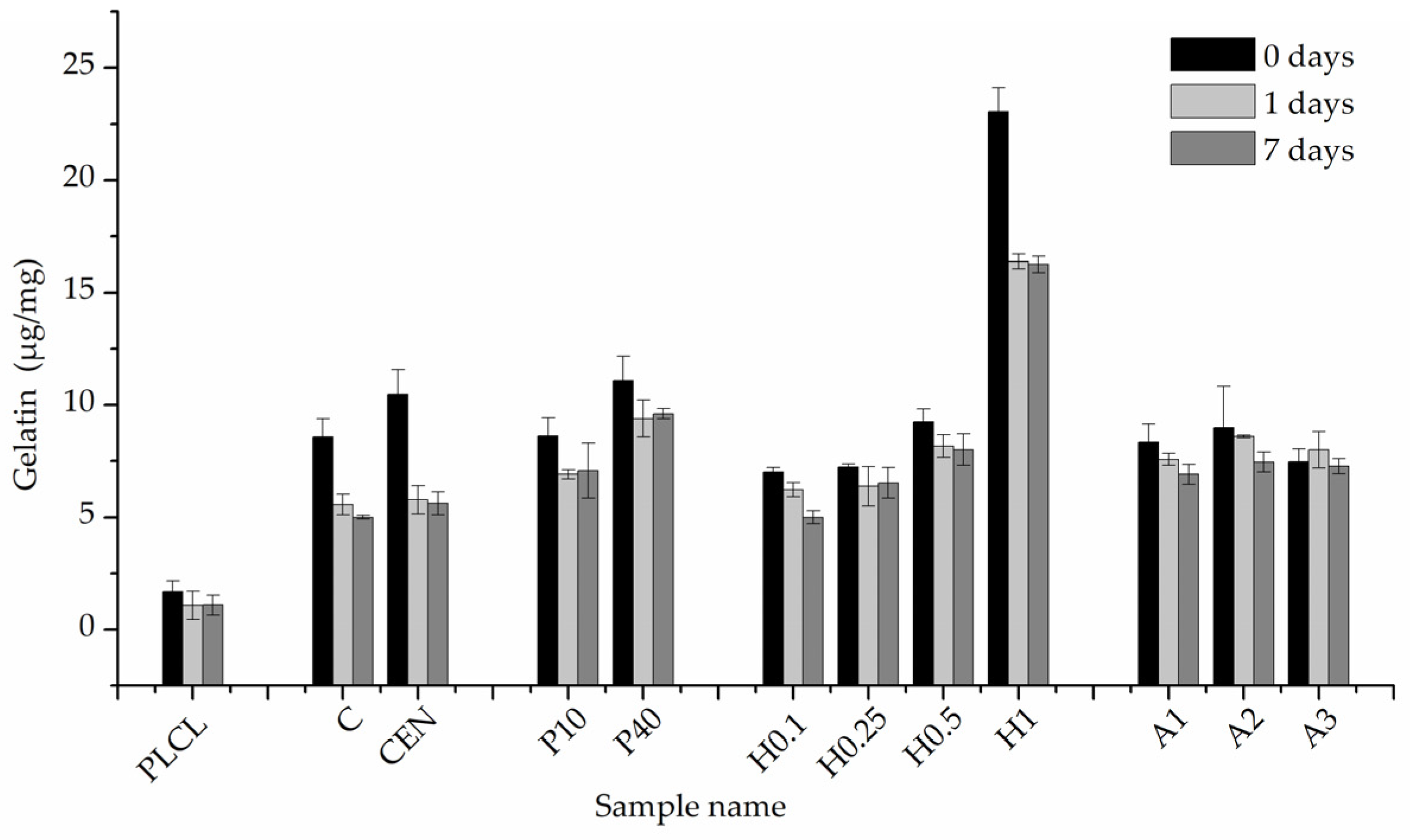
2.5.8. Quantification of Gelatin Amount on Fibres’ Surfaces
2.6. Functionalization
2.6.1. Quantitative Tests: Cytotoxicity on Extracts and Viability Test in Direct Contact
2.6.2. Qualitative Morphology Tests
3. Results and Discussion
3.1. Optimisation of Surface Activation Methods
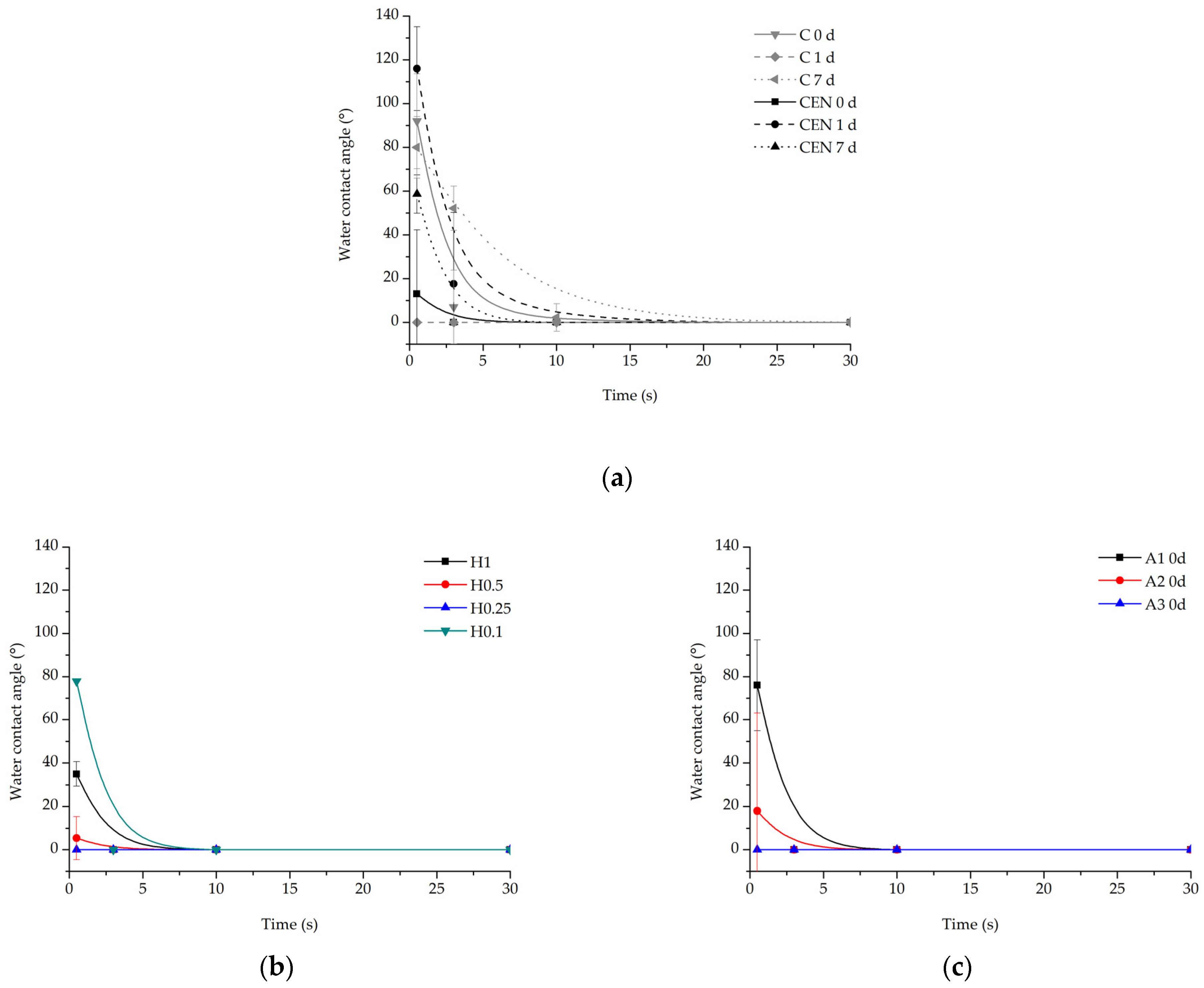
3.1.1. Water Contact Angle
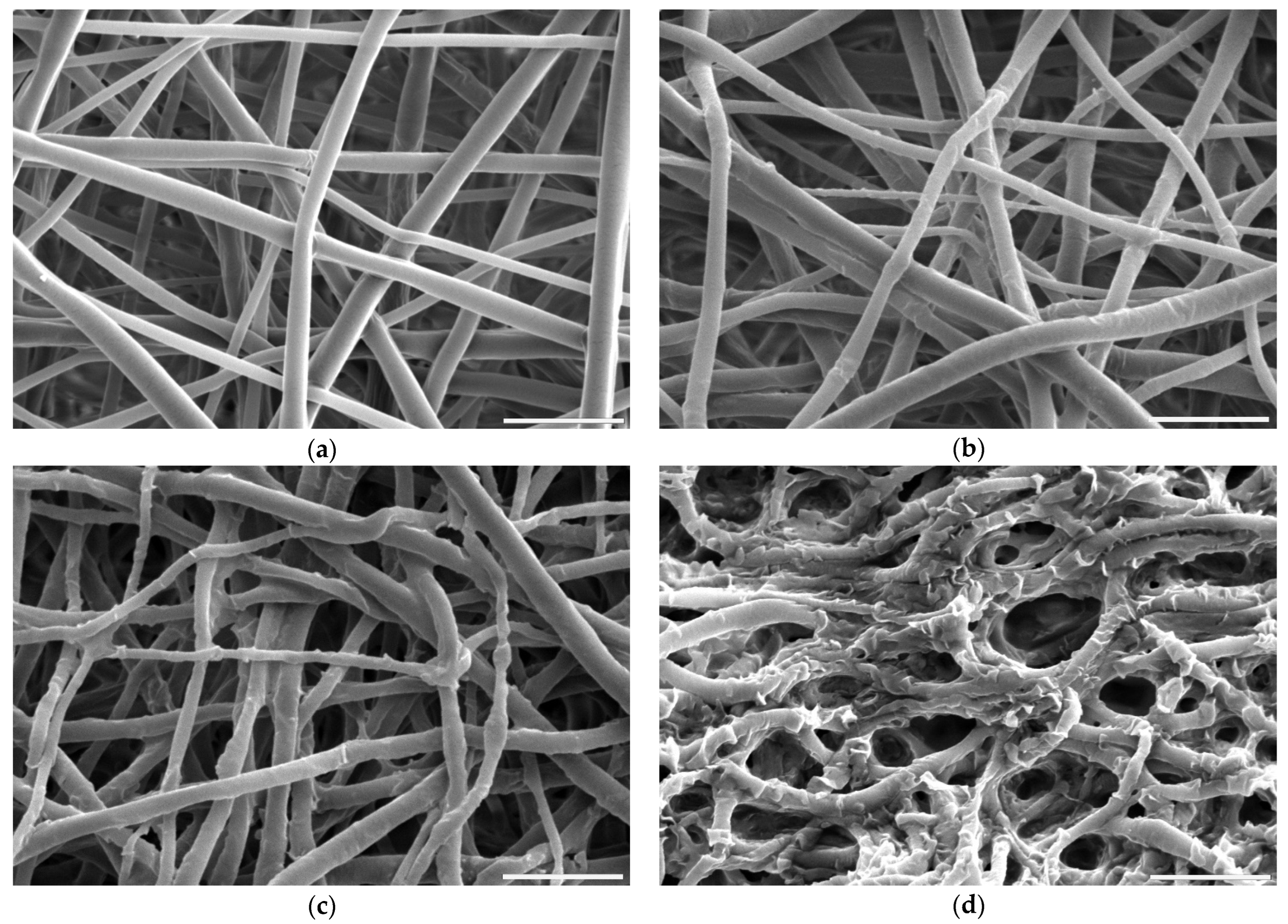
3.1.2. Morphology
3.1.3. The Efficacy of the -COOH Group Introduction to the Material’s Surface
3.1.4. Molecular Mass Change
3.2. Comparison of Functionalized Materials
3.2.1. Gelatin Attachment and Stability
3.2.2. Water Contact Angle
3.2.3. Mechanical Tests
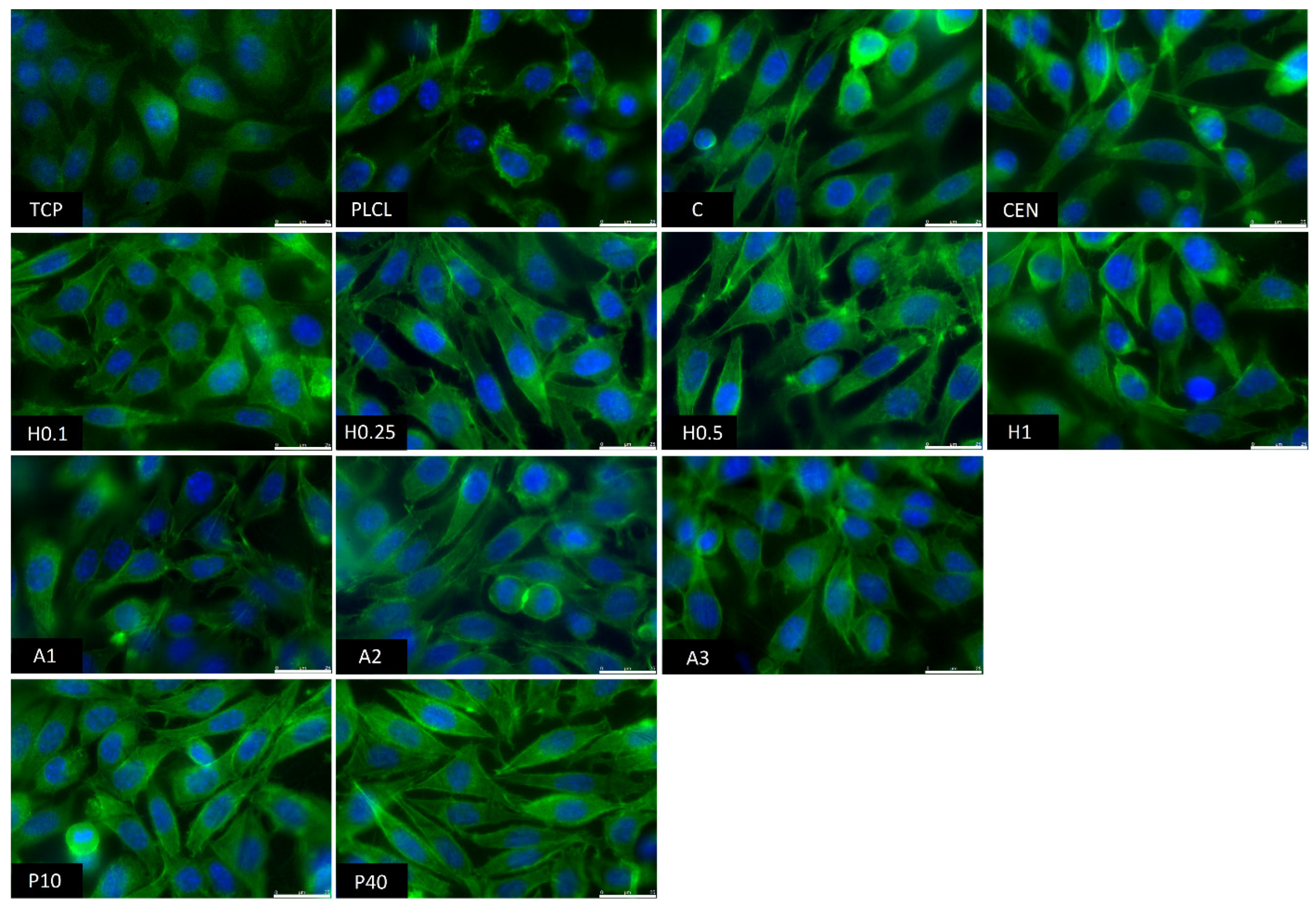
3.2.4. Cellular Studies
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gunatillake, P.A.; Adhikari, R.; Gadegaard, N. Biodegradable synthetic polymers for tissue engineering. Eur. Cells Mater. 2003, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bassani, F.; Liedl, G.L.; Wyder, P. Biological Structures. In Encyclopedia of Condensed Matter Physics; Ottenbrite, R.M., Javan, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 99–108. [Google Scholar]
- Richbourg, N.R.; Peppas, N.A.; Sikavitsas, V.I. Tuning the biomimetic behavior of scaffolds for regenerative medicine through surface modifications. J. Tissue Eng. Regen. Med. 2019, 13, 1275–1293. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y. A brief description of textile fibers. In Medical Textile Materials; Woodhead Publishing: Sawston, UK, 2016; pp. 23–42. [Google Scholar]
- Dulnik, J.; Kołbuk, D.; Denis, P.; Sajkiewicz, P. The effect of a solvent on cellular response to PCL/gelatin and PCL/collagen electrospun nanofibres. Eur. Polym. J. 2018, 104, 147–156. [Google Scholar] [CrossRef]
- Grémare, A.; Guduric, V.; Bareille, R.; Heroguez, V.; Latour, S.; L’heureux, N.; Fricain, J.C.; Catros, S.; Le Nihouannen, D.J. Characterization of printed PLA scaffolds for bone tissue engineering. J. Biomed. Mater. Res. Part A 2018, 106, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Budnicka, M.; Kołbuk, D.; Ruśkowski, P.; Gadomska-Gajadhur, A. Poly-L-lactide scaffolds with super pores obtained by freeze-extraction method. J. Biomed. Mater. Res. Part B Appl. Biomater. 2020, 108, 3162–3173. [Google Scholar] [CrossRef]
- Felfel, R.M.; Poocza, L.; Gimeno-Fabra, M.; Milde, T.; Hildebrand, G.; Ahmed, I.; Scotchford, C.; Sottile, V.; Grant, D.M.; Liefeith, K. In vitro degradation and mechanical properties of PLA-PCL copolymer unit cell scaffolds generated by two-photon polymerization. Biomed. Mater. 2016, 11, 015011. [Google Scholar] [CrossRef]
- Takayama, T.; Todo, M.; Tsuji, H. Effect of annealing on the mechanical properties of PLA/PCL and PLA/PCL/LTI polymer blends. J. Mech. Behav. Biomed. Mater. 2011, 4, 255–260. [Google Scholar] [CrossRef]
- Kolbuk, D.; Jeznach, O.; Wrzecionek, M.; Gadomska-Gajadhur, A. Poly(Glycerol Succinate) as an Eco-Friendly Component of PLLA and PLCL Fibres towards Medical Applications. Polymers 2020, 12, 1731. [Google Scholar] [CrossRef]
- Janmohammadi, M.; Nourbakhsh, M.S.; Bonakdar, S. Electrospun Skin Tissue Engineering Scaffolds Based on Polycaprolactone/Hyaluronic Acid/L-ascorbic Acid. Fibers Polym. 2021, 22, 19–29. [Google Scholar] [CrossRef]
- Leal, B.B.J.; Wakabayashi, N.; Oyama, K.; Kamiya, H.; Braghirolli, D.I.; Pranke, P. Vascular Tissue Engineering: Polymers and Methodologies for Small Caliber Vascular Grafts. Front. Cardiovasc. Med. 2021, 7, 592361. [Google Scholar] [CrossRef]
- Altun, E.; Aydogdu, M.O.; Togay, S.O.; Sengil, A.Z.; Ekren, N.; Haskoylu, M.E.; Oner, E.T.; Altuncu, N.A.; Ozturk, G.; Crabbe-Mann, M.; et al. Bioinspired scaffold induced regeneration of neural tissue. Eur. Polym. J. 2019, 114, 98–108. [Google Scholar] [CrossRef]
- Kim, H.Y.; Chun, S.Y.; Lee, E.H.; Kim, B.; Ha, Y.S.; Chung, J.W.; Lee, J.N.; Kim, B.S.; Oh, S.H.; Kwon, T.G. Bladder Regeneration Using a Polycaprolactone Scaffold with a Gradient Structure and Growth Factors in a Partially Cystectomized Rat Model. J. Korean Med. Sci. 2020, 35, e374. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An Overview of Poly(lactic-co-glycolic) Acid (PLGA)-Based Biomaterials for Bone Tissue Engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- Erisken, C.; Zhang, X.; Moffat, K.L.; Levine, W.N.; Lu, H.H. Scaffold Fiber Diameter Regulates Human Tendon Fibroblast Growth and Differentiation. Tissue Eng. Part A 2013, 19, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.H.; Batchelder, C.A.; Lee, C.I.; Tarantal, A.F. Decellularized rhesus monkey kidney as a three-dimensional scaffold for renal tissue engineering. Tissue Eng. Part A 2010, 16, 2207–2216. [Google Scholar] [CrossRef]
- Gupta, S.K.; Mishra, N.C.; Dhasmana, A. Decellularization Methods for Scaffold Fabrication. Methods Mol. Biol. 2018, 1577, 1–10. [Google Scholar] [CrossRef]
- Taylor, D.A.; Sampaio, L.C.; Ferdous, Z.; Gobin, A.S.; Taite, L.J. Decellularized matrices in regenerative medicine. Acta Biomater. 2018, 74, 74–89. [Google Scholar] [CrossRef]
- Rhee, S.; Puetzer, J.L.; Mason, B.N.; Reinhart-King, C.A.; Bonassar, L.J. 3D Bioprinting of Spatially Heterogeneous Collagen Constructs for Cartilage Tissue Engineering. ACS Biomater. Sci. Eng. 2016, 2, 1800–1805. [Google Scholar] [CrossRef]
- Choi, D.J.; Park, S.J.; Gu, B.K.; Kim, Y.J.; Chung, S.; Kim, C.H. Effect of the pore size in a 3D bioprinted gelatin scaffold on fibroblast proliferation. J. Ind. Eng. Chem. 2018, 67, 388–395. [Google Scholar] [CrossRef]
- Irawan, V.; Sung, T.C.; Higuchi, A.; Ikoma, T. Collagen Scaffolds in Cartilage Tissue Engineering and Relevant Approaches for Future Development. Tissue Eng. Regen. Med. 2018, 15, 673–697. [Google Scholar] [CrossRef]
- Dai, N.T.; Williamson, M.R.; Khammo, N.; Adams, E.F.; Coombes, A.G.A. Composite cell support membranes based on collagen and polycaprolactone for tissue engineering of skin. Biomaterials 2004, 25, 4263–4271. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Varshney, R.; Das, N.; Sircar, D.; Roy, P. Synthesis and characterization of gelatin-PVP polymer composite scaffold for potential application in bone tissue engineering. Eur. Polym. J. 2019, 119, 155–168. [Google Scholar] [CrossRef]
- Theodoridis, K.; Aggelidou, E.; Manthou, M.; Demiri, E.; Bakopoulou, A.; Kritis, A. Assessment of cartilage regeneration on 3D collagen-polycaprolactone scaffolds: Evaluation of growth media in static and in perfusion bioreactor dynamic culture. Colloids Surf. B Biointerfaces 2019, 183, 110403. [Google Scholar] [CrossRef] [PubMed]
- Huettner, N.; Dargaville, T.R.; Forget, A. Discovering Cell-Adhesion Peptides in Tissue Engineering: Beyond RGD. Trends Biotechnol. 2018, 36, 372–383. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.E.; Ginsberg, M.H.; Plow, E.F. Arginyl-glycyl-aspartic acid (RGD): A cell adhesion motif. Trends Biochem. Sci. 1991, 16, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Khew, S.T.; Tong, Y.W. The specific recognition of a cell binding sequence derived from type I collagen by Hep3B and L929 cells. Biomacromolecules 2007, 8, 3153–3161. [Google Scholar] [CrossRef]
- Ma, Z.; He, W.; Yong, T.; Ramakrishna, S. Grafting of Gelatin on Electrospun Poly(caprolactone) Nanofibers to Improve Endothelial Cell Spreading and Proliferation and to Control Cell Orientation. Tissue Eng. 2005, 11, 1149–1158. [Google Scholar] [CrossRef]
- Paredes, J.A.U.; Polini, A.; Chrzanowski, W. Protein-based Biointerfaces to Control Stem Cell Differentiation. RSC Smart Mater. 2015, 2015, 3–29. [Google Scholar] [CrossRef]
- He, L.; Shi, Y.; Han, Q.; Zuo, Q.; Ramakrishna, S.; Xue, W.; Zhou, L. Surface Modification of Electrospun Nanofibrous Scaffolds via Polysaccharide-Protein Assembly Multilayer for Neurite Outgrowth. J. Mater. Chem. 2012, 22, 13187–13196. [Google Scholar] [CrossRef]
- Katti, D.; Vasita, R.; Shanmugam, K. Improved Biomaterials for Tissue Engineering Applications: Surface Modification of Polymers. Curr. Top. Med. Chem. 2008, 8, 341–353. [Google Scholar] [CrossRef]
- Croll, T.I.; O’Connor, A.J.; Stevens, G.W.; Cooper-White, J.J. Controllable surface modification of poly(lactic-co-glycolic acid) (PLGA) by hydrolysis or aminolysis I: Physical, chemical, and theoretical aspects. Biomacromolecules 2004, 5, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Mao, Z.; Gao, C. Surface modification and property analysis of biomedical polymers used for tissue engineering. Colloids Surf. B Biointerfaces 2007, 60, 137–157. [Google Scholar] [CrossRef] [PubMed]
- Durrieu, M.C.; Pallu, S.; Guillemot, F.; Bareille, R.; Amédée, J.; Baquey, C.; Labrugre, C.; Dard, M. Grafting RGD containing peptides onto hydroxyapatite to promote osteoblastic cells adhesion. J. Mater. Sci. Mater. Med. 2004, 15, 779–786. [Google Scholar] [CrossRef]
- Barbosa, M.; Vale, N.; Costa, F.M.; Martins, M.C.L.; Gomes, P. Tethering antimicrobial peptides onto chitosan: Optimization of azide-alkyne “click” reaction conditions. Carbohydr. Polym. 2017, 165, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Mao, Z.; Zhao, Y.; Zhang, J.; Guo, Q.; Gou, Z.; Gao, C. Polycaprolactone scaffold modified with galactosylated chitosan for hepatocyte culture. Macromol. Res. 2012, 20, 283–291. [Google Scholar] [CrossRef]
- Gong, Y.; Zhu, Y.; Liu, Y.; Ma, Z.; Gao, C.; Shen, J. Layer-by-layer assembly of chondroitin sulfate and collagen on aminolyzed poly(l-lactic acid) porous scaffolds to enhance their chondrogenesis. Acta Biomater. 2007, 3, 677–685. [Google Scholar] [CrossRef]
- Korogiannaki, M.; Zhang, J.; Sheardown, H. Surface modification of model hydrogel contact lenses with hyaluronic acid via thiol-ene “click” chemistry for enhancing surface characteristics. J. Biomater. Appl. 2017, 32, 446–462. [Google Scholar] [CrossRef]
- Rasal, R.M.; Janorkar, A.V.; Hirt, D.E. Poly(lactic acid) modifications. Prog. Polym. Sci. 2010, 35, 338–356. [Google Scholar] [CrossRef]
- Becker, J.M.; Pounder, R.J.; Dove, A.P. Synthesis of poly (lactide) s with modified thermal and mechanical properties. Macromol. Rapid Commun. 2010, 31, 1923–1937. [Google Scholar] [CrossRef]
- Salhi, S.; Mahfoudh, J.; Abid, S.; Atanase, L.I.; Popa, M.; Delaite, C. Random poly(ε-caprolactone-L-alanine) by direct melt copolymerization. Polym. Int. 2020, 69, 1161–1168. [Google Scholar] [CrossRef]
- Vilay, V.; Mariatti, M.; Ahmad, Z.; Pasomsouk, K.; Todo, M. Characterization of the mechanical and thermal properties and morphological behavior of biodegradable poly(L-lactide)/poly(ε-caprolactone) and poly(L-lactide)/poly(butylene succinate-co-L-lactate) polymeric blends. J. Appl. Polym. Sci. 2009, 114, 1784–1792. [Google Scholar] [CrossRef]
- Hiljanen-Vainio, M.; Karjalainen, T.; Seppala, J. Biodegradable lactone copolymers. I. Characterization and mechanical behavior of ε-caprolactone and lactide copolymers. J. Appl. Polym. Sci. 1996, 59, 1281–1288. [Google Scholar] [CrossRef]
- Vieira, A.C.; Guedes, R.M.; Marques, A.T. Development of ligament tissue biodegradable devices: A review. J. Biomech. 2009, 42, 2421–2430. [Google Scholar] [CrossRef] [PubMed]
- Hiljanen-Vainio, M.P.; Orava, P.A.; Seppala, J.V. Properties of ϵ-caprolactone/DL-lactide (ϵ-CL/DL-LA) copolymers with a minor ϵ-CL content. J. Biomed. Mater. Res. 1997, 34, 39–46. [Google Scholar] [CrossRef]
- Lu, X.L.; Cai, W.; Gao, Z.Y. Shape-memory behaviors of biodegradable poly(L-lactide-co-ϵ-caprolactone) copolymers. J. Appl. Polym. Sci. 2008, 108, 1109–1115. [Google Scholar] [CrossRef]
- Laurent, C.P.; Vaquette, C.; Liu, X.; Schmitt, J.F.; Rahouadj, R. Suitability of a PLCL fibrous scaffold for soft tissue engineering applications: A combined biological and mechanical characterisation. J. Biomater. Appl. 2018, 32, 1276–1288. [Google Scholar] [CrossRef]
- Eastoe, J.E. The amino acid composition of mammalian collagen and gelatin. Biochem. J. 1955, 61, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Jeznach, O.; Kołbuk, D.; Marzec, M.; Bernasik, A.; Sajkiewicz, P. Aminolysis as a surface functionalization method of aliphatic polyester nonwovens: Impact on material properties and biological response. RSC Adv. 2022, 12, 11303–11317. [Google Scholar] [CrossRef]
- Gupta, B.; Plummer, C.; Bisson, I.; Frey, P.; Hilborn, J. Plasma-induced graft polymerization of acrylic acid onto poly(ethylene terephthalate) films: Characterization and human smooth muscle cell growth on grafted films. Biomaterials 2002, 23, 863–871. [Google Scholar] [CrossRef]
- Nuuttila, M. Gel Permeation Chromatography Methods in the Analysis of Lactide-Based Polymer. Master’s Thesis, University of Jyväskylä, Jyväskylä, Finland, 2018. Available online: http://urn.fi/URN:NBN:fi:jyu-201811054624 (accessed on 15 June 2022).
- Bu, Y.; Ma, J.; Bei, J.; Wang, S. Surface Modification of Aliphatic Polyester to Enhance Biocompatibility. Front. Bioeng. Biotechnol. 2019, 7, 1–10. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Naskar, D.; Kim, H.W.; Maiti, T.K.; Bhattacharya, D.; Kundu, S.C. Non-mulberry silk fibroin grafted PCL nanofibrous scaffold: Promising ECM for bone tissue engineering. Eur. Polym. J. 2015, 71, 490–509. [Google Scholar] [CrossRef]
- Monnier, A.; Al Tawil, E.; Nguyen, Q.T.; Valleton, J.M.; Fatyeyeva, K.; Deschrevel, B. Functionalization of poly (lactic acid) scaffold surface by aminolysis and hyaluronan immobilization: How it affects mesenchymal stem cell proliferation. Eur. Polym. J. 2018, 107, 202–217. [Google Scholar] [CrossRef]
- Toledo, A.L.M.M.; Ramalho, B.S.; Picciani, P.H.S.; Baptista, L.S.; Martinez, A.M.B.; Dias, M.L. Effect of three different amines on the surface properties of electrospun polycaprolactone mats. Int. J. Polym. Mater. Polym. Biomater. 2021, 70, 1258–1270. [Google Scholar] [CrossRef]
- Polini, A.; Petre, D.G.; Iafisco, M.; de Lacerda Schickert, S.; Tampieri, A.; van den Beucken, J.; Leeuwenburgh, S.C.G. Polyester Fibers Can Be Rendered Calcium Phosphate-Binding by Surface Functionalization with Bisphosphonate Groups. J. Biomed. Mater. Res. Part A 2017, 105, 2335–2342. [Google Scholar] [CrossRef]
- Mistry, B.D. A Handbook of Spectroscopic Data Chemistry (UV, JR, PMR, JJCNMR and Mass Spectroscopy); Oxford Book Company: Oxford, UK, 2009. [Google Scholar]
- Zhu, Y.; Mao, Z.; Shi, H.; Gao, C. In-Depth Study on Aminolysis of Poly(ε-Caprolactone): Back to the Fundamentals. Sci. China Chem. 2012, 55, 2419–2427. [Google Scholar] [CrossRef]
- Weidner, S.; Kühn, G.; Decker, R.; Roessner, D.; Friedrich, J. Influence of Plasma Treatment on the Molar Mass of Poly(Ethylene Terephthalate) Investigated by Different Chromatographic and Spectroscopic Methods. J. Polym. Sci. Part A Polym. Chem. 1998, 36, 1639–1648. [Google Scholar] [CrossRef]
- Jeznach, O.; Kolbuk, D.; Sajkiewicz, P. Aminolysis of Various Aliphatic Polyesters in a Form of Nanofibers and Films. Polymers 2019, 11, 1669. [Google Scholar] [CrossRef]
- Oliveira, S.; Felizardo, T.; Amorim, S.; Mithieux, S.M.; Pires, R.A.; Reis, R.L.; Martins, A.; Weiss, A.S.; Neves, N.M. Tubular Fibrous Scaffolds Functionalized with Tropoelastin as a Small-Diameter Vascular Graft. Biomacromolecules 2020, 21, 3582–3595. [Google Scholar] [CrossRef]
- Abercrombie, M. Fibroblasts. J. Clin. Pathol. Suppl. (R. Coll. Pathol.) 1978, 12, 1–6. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1347118/ (accessed on 15 June 2022).
- Kurashina, Y.; Miyata, S.; Komotori, J.; Koyama, T. Proliferation and Adhesion of L929 Fibroblasts on Surface with Different Microtopography. Mater. Res. Soc. Symp. Proc. 2014, 1648, 101. [Google Scholar] [CrossRef]
- Stephansson, S.N.; Byers, B.A.; García, A.J. Enhanced expression of the osteoblastic phenotype on substrates that modulate fibronectin conformation and integrin receptor binding. Biomaterials 2002, 23, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Keselowsky, B.G.; Collard, D.M.; García, A.J. Surface chemistry modulates fibronectin conformation and directs integrin binding and specificity to control cell adhesion. J. Biomed. Mater. Res. Part A 2003, 66, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Vallières, K.; Chevallier, P.; Sarra-Bournet, C.; Turgeon, S.; Laroche, G. AFM imaging of immobilized fibronectin: Does the surface conjugation scheme affect the protein orientation/conformation? Langmuir 2007, 23, 9745–9751. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mahara, A.; Kambe, Y.; Hsu, Y.-I.; Yamaoka, T. Endothelial cell adhesion and blood response to hemocompatible peptide 1 (HCP-1), REDV, and RGD peptide sequences with free N-terminal amino groups immobilized on a biomedical expanded polytetrafluorethylene surface. Biomater. Sci. 2021, 9, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.G.; He, J.; Aparicio, C. Surface Immobilization Chemistry of a Laminin-Derived Peptide Affects Keratinocyte Activity. Coatings 2020, 10, 560. [Google Scholar] [CrossRef]
- Vida, Y.; Collado, D.; Najera, F.; Claros, S.; Becerra, J.; Andrades, J.A.; Perez-Inestrosa, E. Dendrimer surface orientation of the RGD peptide affects mesenchymal stem cell adhesion. RSC Adv. 2016, 6, 49839–49844. [Google Scholar] [CrossRef]
- Yuan, L.; Yu, Q.; Li, D.; Chen, H. Surface Modification to Control Protein/Surface Interactions. Macromol. Biosci. 2011, 11, 1031–1040. [Google Scholar] [CrossRef]
- Hern, D.L.; Hubbell, J.A. Incorporation of adhesion peptides into nonadhesive hydrogels useful for tissue resurfacing. J. Biomed. Mater. Res. 1998, 39, 266–276. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activation Method | Optimisation Conditions | |
|---|---|---|
| Treatment Time | Reagent Concentration | |
| Cold oxygen plasma | 5, 10, 15, 40, 60 (s) | - |
| Alkaline hydrolysis | 10, 30, 60, 180 (min) | 0.05, 0.1, 0.25, 0.5, 1 (M NaOH) |
| Aminolysis | 5, 10, 15, 30 (min) | 2, 6, 10 (% w/v EDA in isopropanol) |
| Surface Activation Method | Time | Reagent Concentration | Crosslinking Method | Name |
|---|---|---|---|---|
| Hydrolysis | 3 h | 1 M | EDC/NHS | H1 |
| 0.5 M | H0.5 | |||
| 0.25 M | H0.25 | |||
| 0.1 M | H0.1 | |||
| Plasma | 10 s | - | EDC/NHS | P10 |
| 40 s | P40 | |||
| Aminolysis | 5 min | 2% w/v | GTA | A1 |
| 10 min | 2% w/v | GTA | A2 | |
| 5 min | 6% w/v | GTA | A3 | |
| Control (no activation) | - | - | EDC/NHS | CEN |
| - | C |
| Sample | Mn (kDa) | Mw (kDa) | % MnPLCL | % MwPLCL | PDI |
|---|---|---|---|---|---|
| PLCL | 58.7 | 88.3 | 100% | 100% | 1.50 |
| A1 | 49.8 | 81.3 | 85% | 92% | 1.63 |
| A2 | 47.8 | 75.9 | 81% | 86% | 1.59 |
| A3 | 30.6 | 58.4 | 52% | 66% | 1.91 |
| P10 | 54.5 | 82.1 | 93% | 93% | 1.51 |
| P40 | 52.5 | 77.5 | 89% | 88% | 1.48 |
| P60 | 46.0 | 71.3 | 78% | 81% | 1.55 |
| H1 | 57.7 | 85.3 | 98% | 97% | 1.48 |
| H0.5 | 57.3 | 85.9 | 97% | 97% | 1.50 |
| H0.25 | 59.6 | 87.7 | 100% | 99% | 1.47 |
| H0.1 | 56.4 | 85.2 | 96% | 97% | 1.51 |
| H0.05 | 57.3 | 86.2 | 97% | 98% | 1.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dulnik, J.; Jeznach, O.; Sajkiewicz, P. A Comparative Study of Three Approaches to Fibre’s Surface Functionalization. J. Funct. Biomater. 2022, 13, 272. https://doi.org/10.3390/jfb13040272
Dulnik J, Jeznach O, Sajkiewicz P. A Comparative Study of Three Approaches to Fibre’s Surface Functionalization. Journal of Functional Biomaterials. 2022; 13(4):272. https://doi.org/10.3390/jfb13040272
Chicago/Turabian StyleDulnik, Judyta, Oliwia Jeznach, and Paweł Sajkiewicz. 2022. "A Comparative Study of Three Approaches to Fibre’s Surface Functionalization" Journal of Functional Biomaterials 13, no. 4: 272. https://doi.org/10.3390/jfb13040272
APA StyleDulnik, J., Jeznach, O., & Sajkiewicz, P. (2022). A Comparative Study of Three Approaches to Fibre’s Surface Functionalization. Journal of Functional Biomaterials, 13(4), 272. https://doi.org/10.3390/jfb13040272