
Cone Snail Broad-Transcriptomics Elucidate the Evolutionary Diversification and Anti-Microbial Potential of Conopeptides




Abstract
1. Introduction
2. Materials and Methods
- -
- Criterion A: 100 sequences from at least 30 transcriptomes; the selected sequences would be further annotated according to the database of Tox-Prot;
- -
- Criterion B: 50 sequences from at least 10 transcriptomes; the selected sequences would be further annotated according to the database of UniProt-Trembl.
3. Results
3.1. General Transcriptomes Annotation and Biomedical Predictions
3.2. Shared Transcriptomic Repertoire
3.3. Phylogeny
3.4. AMP Prediction Results
3.4.1. AMP Potential of the Shared Venom Transcripts
3.4.2. Broad Transcriptomes AMP Potential
4. Discussion
4.1. Symbiosis in Venom Organs
4.2. Predation Impact on Venom Evolution
4.3. Biomedical Findings in Conus Venoms
4.4. ACE2 Similarity
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin-Converting Enzyme 2 |
| AMP(s) | Anti-Microbial Peptide(s) |
| Mb | Megabytes |
| PTM(s) | Post-Transcriptional Modification(s) |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO ID | GO Category | GO Term |
|---|---|---|
| GO:0003081 | Biological process | Regulation of systemic arterial blood pressure by renin-angiotensin |
| GO:0003084 | Biological process | Positive regulation of systemic arterial blood pressure |
| GO:0019882 | Biological process | Antigen processing and presentation |
| GO:0030212 | Biological process | Hyaluronan metabolic process |
| GO:0031288 | Biological process | Sorocarp morphogenesis |
| GO:0071577 | Biological process | Zinc ion transmembrane transport |
| GO:0097067 | Biological process | Cellular response to thyroid hormone stimulus |
| GO:0140206 | Biological process | Dipeptide import across plasma membrane |
| GO:1903052 | Biological process | Positive regulation of proteolysis involved in protein catabolic process |
| GO:1903665 | Biological process | Negative regulation of asexual reproduction |
| GO:1903669 | Biological process | Positive regulation of chemorepellent activity |
| GO:0016532 | Molecular function | Superoxide dismutase copper chaperone activity |
| GO:0016671 | Molecular function | Oxidoreductase activity; acting on a sulphur group of donors; disulphide as acceptor |
| GO:0031545 | Molecular function | Peptidyl-proline 4-dioxygenase activity |
Appendix B
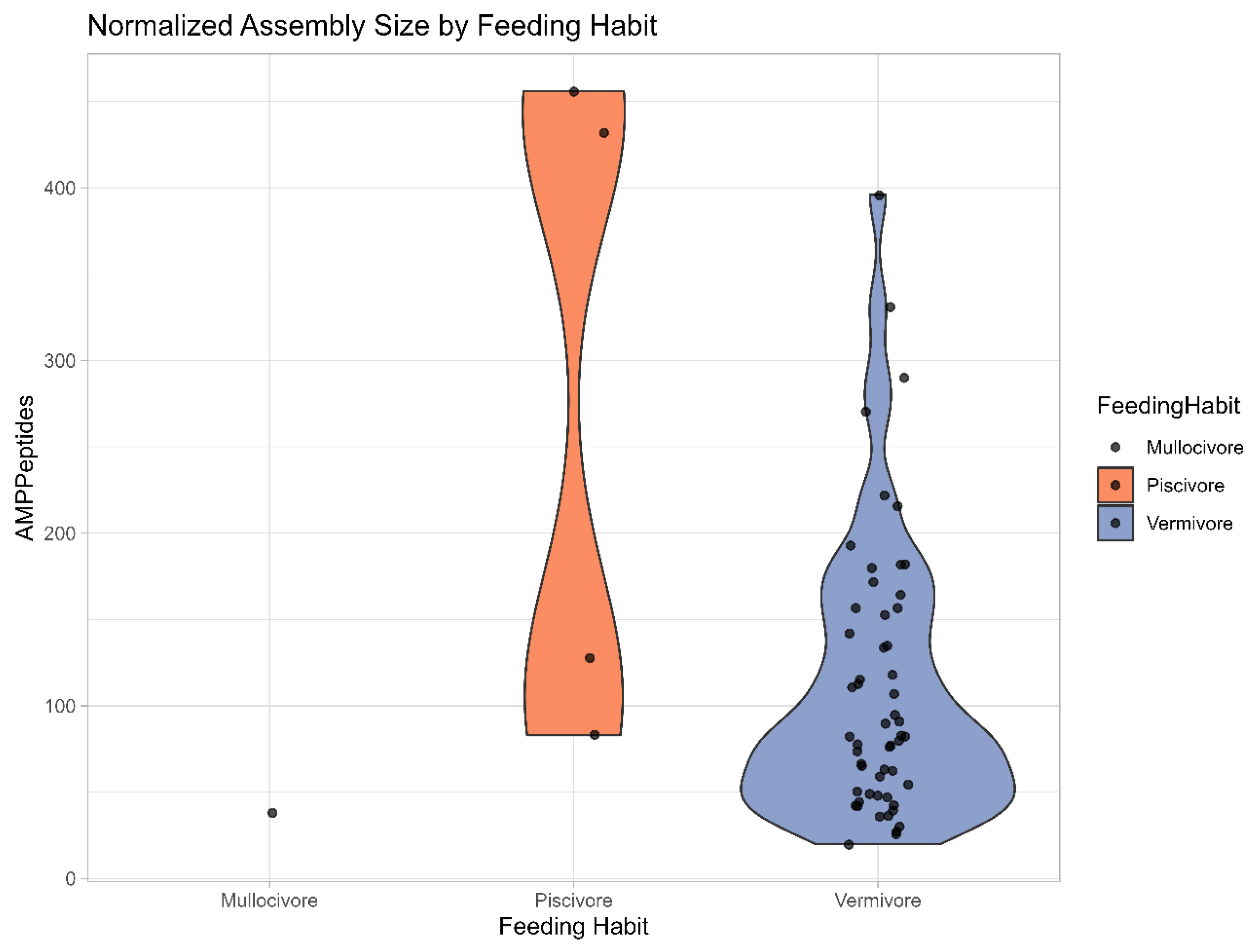
Appendix C
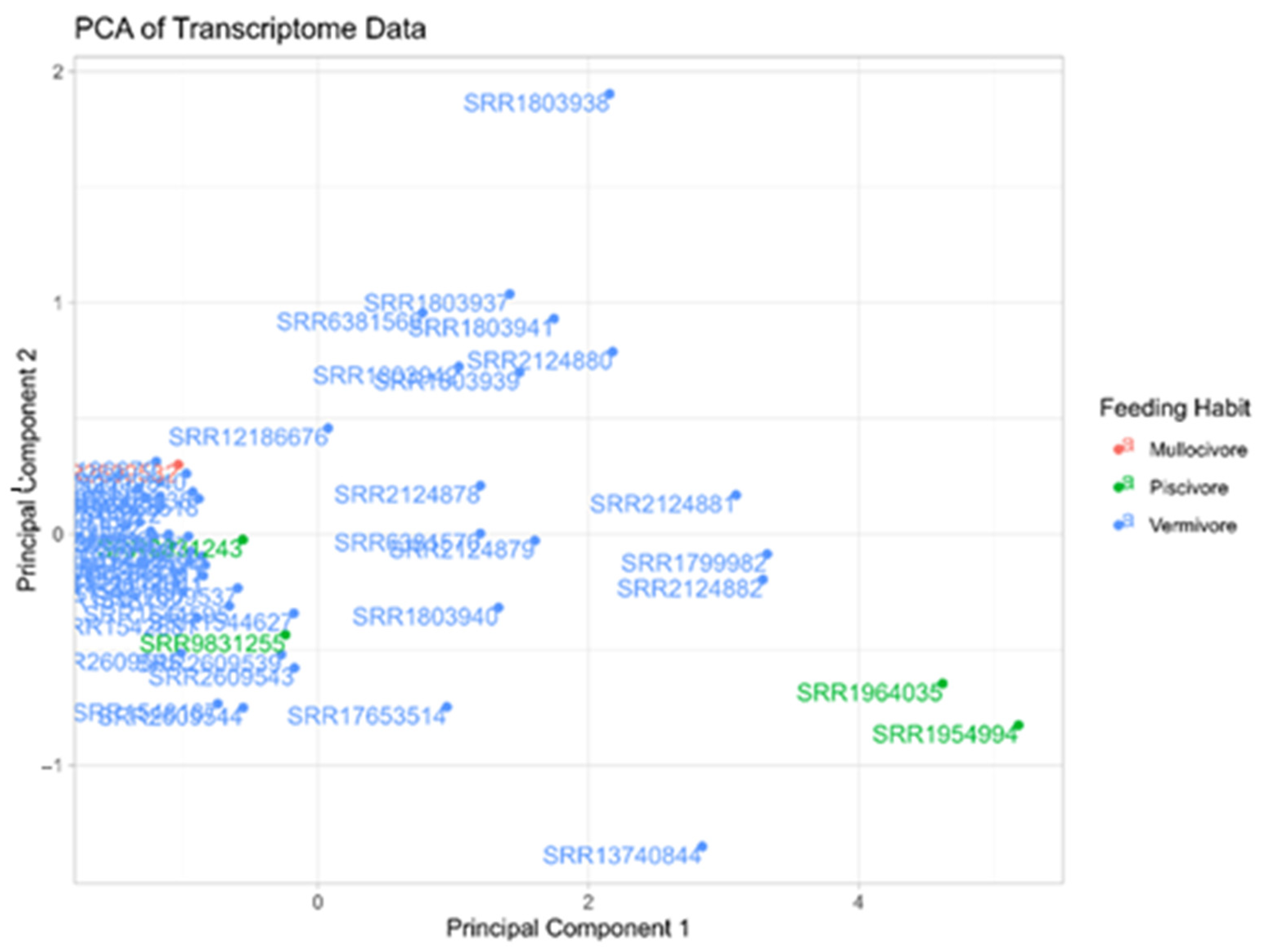
Appendix D
| Sequences | Percentage of Identity |
|---|---|
| >SRR1544119_TRINITY_DN22969_c0_g1_i1 | 51.5% LOC106073291 Biomphalaria glabrata (Bloodfluke planorb) (Freshwater snail) |
| CGGCACTGAAGGACGAGAACAAAGTGGCACGGTTTAACGAGCTAGTGGCCAAGATGTCGGAGATCTACAGCACCGCTCAAGTGTGCTTTACGGAAGGCAACTGTATCTCCCTGGATCCAGACCTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGACTGAAGCCTGGGAGCTGTGGAGAAAGGCCACTGGAGGAAAAATGAAAGCCCTGTACGAGGAGTATGTTGAGCTGGCGAACGAAGGCGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGATGACTACGAAGACGAA | |
| >SRR1544120_TRINITY_DN2907_c0_g1_i1 | 50% LOC106073291 Biomphalaria glabrata (Bloodfluke planorb) (Freshwater snail) |
| TTGCCAACGCCACCCAGCGGCGCCTGCTGAAGCAAATCCGCAAGATCGGCACGGCGGCACAGAAGGACGAGAACAAAGTGGCACGGTTTAACGAGCTAGTGGCCAAGATGTCGGAGATCTACAGCACCGCTCAGGTGTGCTTTACGGAAGACAACTGTATCTCCCTGGATCCAGACCTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGACTGAAGCCTGGGATCTGTGGAGAAAGGCCACTGGAGGGAAAATGAAAGCCCTGTACGAGGAGTATGTTGAGCTGGCGAACGAAGGCGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGATGACTACGAAGACGAA | |
| >SRR1544137_TRINITY_DN16819_c0_g1_i1 | 51.5% LOC106073291 Biomphalaria glabrata (Bloodfluke planorb) (Freshwater snail) |
| CGGCACTGAAGGACGAGAACAAAGTGGCACGGTTTAACGAGCTAGTGGCCAAGATGTCGGAGATCTACAGCACCGCTCAAGTGTGCTTTACGGAAGGCAACTGTATCTCCCTGGATCCAGACCTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGACTGAAGCCTGGGAGCTGTGGAGAAAGGCCACTGGAGGAAAAATGAAAGCCCTGTACGAGGAGTATGTTGAGCTGGCGAACGAAGGCGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGATGACTACGAAGACGAA | |
| >SRR1544140_TRINITY_DN13380_c0_g1_i1 | 52.9% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| AGAACCTGGCGAGGAGCTGGCTACAGAAATACAACCAAGAGCACAAAGACATTTTCTCCAAGTCCTCAGAAATGACCTGGAACTACGCTACCAACGTCACGGACGAAATTCAACAAAAACAAGTGAATGCAGAGCTGCAGGTGGCGCAATGGCAACAGGAGAAGGCAGCAGAAGTGGAACATTACGACTGGGAGCATTTTTCCGACAGCAGTCTTGTACGTCAGTTCCGTTTTGCGAGGAATATAGGCACGTCTGCCATG | |
| >SRR1544595_TRINITY_DN5707_c0_g1_i1 | 61.9% C0Q70_16356 Pomacea canaliculata (Golden apple snail) |
| CTGAGGCTGGACACAAACTGAGGGCCATGCTGTCCAAAGGATCGTCTGAGGTGTGGACAGTACCATTCCAGGCCCTGACAGGACAGACCAAGATGAGCGCACAATCACTGATCCAGTACTTCCAGCCCCTCATGGACTACCTGGAGCAGTACACCAAGGACCACGGCGTGGAGGTTGGGTGGAAGGAGGAGTGTTCT | |
| >SRR1544600_TRINITY_DN9395_c0_g1_i1 | 56.3% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| TCGTTTTTCATGGCAGACGTGCCTATATTCCTTGCAAAACGGAACTGACGTACAAGACTGCTGTCGGAAAAATGCTCCCAGTCGTAATGTTCCACTTCCGCTGCCTTCCTCTGTTGCCATTGCGCCACCTGCAGCTCTGCATTCACTTGTTTTTGTTGATTTTCGTCCGTGACGTTGGTAGCGTAGTTCCAGGTCATTTCTGAGGACTTGGAGACAATGTCTTTGTGCTCTTGGTTGTATTTCTGTAGCCAGCTCCTCGCCAGGTTCTC | |
| >SRR1544692_TRINITY_DN15875_c0_g1_i1 | 55.8% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| TCGTTGCTCAGCGCCACGAACTCCTCGTAATCGCTCTTCATCAGAGGCCCTGTGACGTCACGCCACTCCTTCCACGCCATCAGCAGTTCGTCATAGTCACGCGATGACGCCATCAGTTTGGTCAGTTCAGGATCCAGATTCAAAATGGCCCCGGTCTTTGGATCCTTCACTTTAGCTTTGGCGTAGATACCTTCAATGGTCGACTGTAGCTCTTTTAGCTCTTTCAGTTTAGTTTCGTTTTTCATGGCAGACGTGCCTATATTCCTTGCAAAACGGAACTGACGTACAAGACTGCTGTCGGAAAAATGCTCCCAGTCGTAATGTTCCACTTCCGCTGCCTTCCTCTGTTGCCATTGCGCCACCTGCAGCTCTGCATTCACTTGTTTTTGTTGATTTTCGTCCGTGACGTTGGTAGCGTAGTTCCAGGTCATTTCTGAGGACTTGGAGATAATGTCTTTGTGCTCTTTGTTGTATTTCTGTAGCCAGCTCCTCGCCAGGTTCTC | |
| >SRR1544692_TRINITY_DN2117_c0_g1_i1 | 68.3% LOC110975981 Acanthaster planci (Crown-of-thorns starfish) |
| TCCTCACTCCAGCCAACGGGCTGGCCGGCGTTCTGCTCCTCCAGCCAATCCTGAAGCGGCCTGAAGTACTCCAGCAGCGGCCTCACGTCCATGTGTCTGGTGCCCGTGATCTGCTCCAGGGCCTC | |
| >SRR6983166_TRINITY_DN44676_c0_g1_i1 | 56.8% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| ACAGCACCGCTCGAGTGTGCTTTACGGAAGACAACTGTATTCCCCTGGACCCGGACGTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGGCTGAAGCCTGGGATCTGTGGAGAGAGGCCACTGGAGGAGAAATGAAAGCCCTGTACGAGGAGTATGTTCAGCTGGGAAACGAAGGAGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGACGAGTATGAAGACGAA | |
| >SRR6983168_TRINITY_DN6724_c0_g2_i1 | 49% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| CGGACAGCGAGGCAGTGGAAGCCTTCTTGGAGACGCACGACAAGGAGACTAAGAAGAAGCATGAAAAGTACGAGATTCTGTCCTGGAATCACGAAACCAATATCACCGACTACAATCAGGAGCTGAAGGTCAACTACAGCGTAGAGATGTCAGAATTTGCTAAAGAGCACGCCAGGCAGTCGGCCATGTTTGACCTTGATCACCTTGCCAACGCCACCCAGCGGCGCCTGCTGAAGAAAATCGGCAAAATCGGCACGGCGGCACAGAAGGACGAGAACAAAGTGGCACGGTTTAACGAGCTGGTGGCCAAGATGTCGGAGATCTACAGCACCGCTCGAGTGTGCTTTACGGAAGACAACTGTATTCCCCTGGACCCGGACGTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGGCTGAAGCCTGGGATCTGTGGAGAGAGGCCACTGGAGGAGAAATGAAAGCCCTGTACGAGGAGTATGTTCAGCTGGGAAACGAAGGCGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGACGAGTATGAAGACGAAAATCTGCAAGAAGAGTTAGCGGCCCTGATGGAACAGCTCCGTCCTTTGTACGTGAAGCTCCAC | |
| >SRR6983169_TRINITY_DN17529_c0_g1_i1 | 51% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| TTCTGTCCTGGAATCACGAAACCAATATCACCGACTACAATCAGGAGCTGAAGGTCAACTACAGCGTAGAGATGTCAGAATTTGCTAAAGAGCACGCCAGGCAGTCGGCCATGTTTGACCTTGATCACCTTGCCAACGCCACCCAGCGGCGCCTGCTGAAGAAAATCGGCAAAATCGGCACGGCGGCACAGAAGGACGAGAACAAAGTGGCACGGTTTAACGAGCTGGTGGCCAAGATGTCGGAGATCTACAGCACCGCTCGAGTGTGCTTTACGGAAGACAACTGTATTCCCCTGGACCCGGACGTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGGCTGAAGCCTGGGATCTGTGGAGAGAGGCCACTGGAGGAGAAATGAAAGCCCTGTACGAGGAGTATGTTCAGCTGGGAAACGAAGGCGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGACGAGTATGAAGACGAA |
Appendix E
| Protein ID | AMP ID | Abundance | Protein ID | AMP ID | Abundance |
|---|---|---|---|---|---|
| P83578|IKP1_PHYSA | AMP_11771 | 7 | P20160|CAP7_HUMAN | AMP_14282 | 20 |
| P61095|SFI1_SEGFL | AMP_15699 | 4 | P0DKT2|TU92_GEMSO | AMP_25599 | 20 |
| Q90WJ8|AJL2_ANGJA | AMP_00722 | 3 | P20160|CAP7_HUMAN | AMP_14283 | 16 |
| P39060|COIA1_HUMAN | AMP_14664 | 3 | P00738|HPT_HUMAN | AMP_04692 | 15 |
| P01038|CYT_CHICK | AMP_23459 | 2 | P05484|O17A_CONMA | AMP_18775 | 11 |
| P0C2D2|OXLA_CRODC | AMP_00139 | 1 | P05484|O17A_CONMA | AMP_18778 | 11 |
| Q9JHY3|WFD12_MOUSE | AMP_02769 | 1 | P05484|O17A_CONMA | AMP_18779 | 11 |
| P22887|NDKC_DICDI | AMP_09422 | 1 | P05484|O17A_CONMA | AMP_18780 | 11 |
| P22355|PSPB_RAT | AMP_00195 | 1 | P05484|O17A_CONMA | AMP_18781 | 11 |
| Q29075|NKL_PIG | AMP_03456 | 1 | P06702|S10A9_HUMAN | AMP_09443 | 6 |
| Q29075|NKL_PIG | AMP_03457 | 1 | P06702|S10A9_HUMAN | AMP_11387 | 6 |
| Q29075|NKL_PIG | AMP_04409 | 1 | P05484|O17A_CONMA | AMP_27220 | 6 |
| Q29075|NKL_PIG | AMP_21444 | 1 | P05484|O17A_CONMA | AMP_27221 | 6 |
| Q8TDE3|RNAS8_HUMAN | AMP_05220 | 1 | P05484|O17A_CONMA | AMP_27222 | 6 |
| A0A7I2V2E9|A0A7I2V2E9_HUMAN | AMP_19944 | 10 | P83952|WAPA_OXYMI | AMP_04982 | 4 |
| P00974|BPT1_BOVIN | AMP_10608 | 76 | B5G6G7|WAPB_OXYMI | AMP_04983 | 4 |
| I2G9B4|VKT_MACLN | AMP_15720 | 76 | P83240|NDB31_PANIM | AMP_02393 | 2 |
| P20160|CAP7_HUMAN | AMP_04915 | 62 | P83240|NDB31_PANIM | AMP_02394 | 2 |
| P20160|CAP7_HUMAN | AMP_04916 | 62 | P83240|NDB31_PANIM | AMP_02395 | 2 |
| P20160|CAP7_HUMAN | AMP_04917 | 62 | A0A0C4G5K0|NDB3_HETSP | AMP_02396 | 2 |
| P20160|CAP7_HUMAN | AMP_04918 | 60 | P82427|WTX1E_NEOGO | AMP_02397 | 2 |
| P08311|CATG_HUMAN | AMP_04624 | 56 | P0DJ02|NDB49_HETPE | AMP_08335 | 2 |
| P86810|OXLA_SIGCA | AMP_11257 | 50 | A0A0C4G5K0|NDB3_HETSP | AMP_17099 | 2 |
| P86810|OXLA_SIGCA | AMP_11258 | 50 | P83240|NDB31_PANIM | AMP_22673 | 2 |
| Q4JHE1|OXLA_PSEAU | AMP_00142 | 48 | P83240|NDB31_PANIM | AMP_22674 | 2 |
| F8S0Z5|OXLA2_CROAD | AMP_00224 | 48 | P83240|NDB31_PANIM | AMP_22676 | 2 |
| P04284|PR06_SOLLC | AMP_07416 | 45 | P83240|NDB31_PANIM | AMP_22677 | 2 |
| P04284|PR06_SOLLC | AMP_10197 | 45 | P13487|KAX11_LEIHE | AMP_01368 | 1 |
| P81382|OXLA_CALRH | AMP_00135 | 44 | A9XE60|KBX11_MESEU | AMP_02691 | 1 |
| Q6TGQ8|OXLA_BOTMO | AMP_00137 | 44 | P82656|NDB21_HOFAZ | AMP_03049 | 1 |
| Q90W54|OXLA_GLOBL | AMP_00138 | 44 | F1AWB0|NDB27_VAEME | AMP_03190 | 1 |
| Q6STF1|OXLA_GLOHA | AMP_00422 | 44 | F1AWB0|NDB27_VAEME | AMP_03191 | 1 |
| Q6STF1|OXLA_GLOHA | AMP_00423 | 44 | P83239|NDB23_PANIM | AMP_03236 | 1 |
| Q6TGQ9|OXLA1_BOTJR | AMP_08879 | 43 | P83313|NDB24_OPICA | AMP_03238 | 1 |
| Q9U8W7|TL5B_TACTR | AMP_01246 | 42 | P83313|NDB24_OPICA | AMP_03239 | 1 |
| B5AR80|OXLA_BOTPA | AMP_00127 | 41 | P83314|NDB2S_OPICA | AMP_04307 | 1 |
| B5AR80|OXLA_BOTPA | AMP_00128 | 41 | A0A0C4G489|NDB2_HETSP | AMP_04308 | 1 |
| Q9U8W8|TL5A_TACTR | AMP_10191 | 41 | P0C2F4|KBX3_HETLA | AMP_04346 | 1 |
| P00734|THRB_HUMAN | AMP_09738 | 39 | P56972|KBX3_PANIM | AMP_04348 | 1 |
| P00734|THRB_HUMAN | AMP_09739 | 39 | Q0GY40|KBX3_HOFGE | AMP_04390 | 1 |
| P20160|CAP7_HUMAN | AMP_14281 | 29 | Q5WR03|KBX31_OPICA | AMP_05482 | 1 |
| Q1PHZ4|VM3B1_BOTJR | AMP_01520 | 27 | P13487|KAX11_LEIHE | AMP_10082 | 1 |
| Q6IWZ0|OXLA_APLCA | AMP_01042 | 26 | P0C2F4|KBX3_HETLA | AMP_17928 | 1 |
| Q6IWZ0|OXLA_APLCA | AMP_01043 | 26 | A9XE60|KBX11_MESEU | AMP_23221 | 1 |
Appendix F
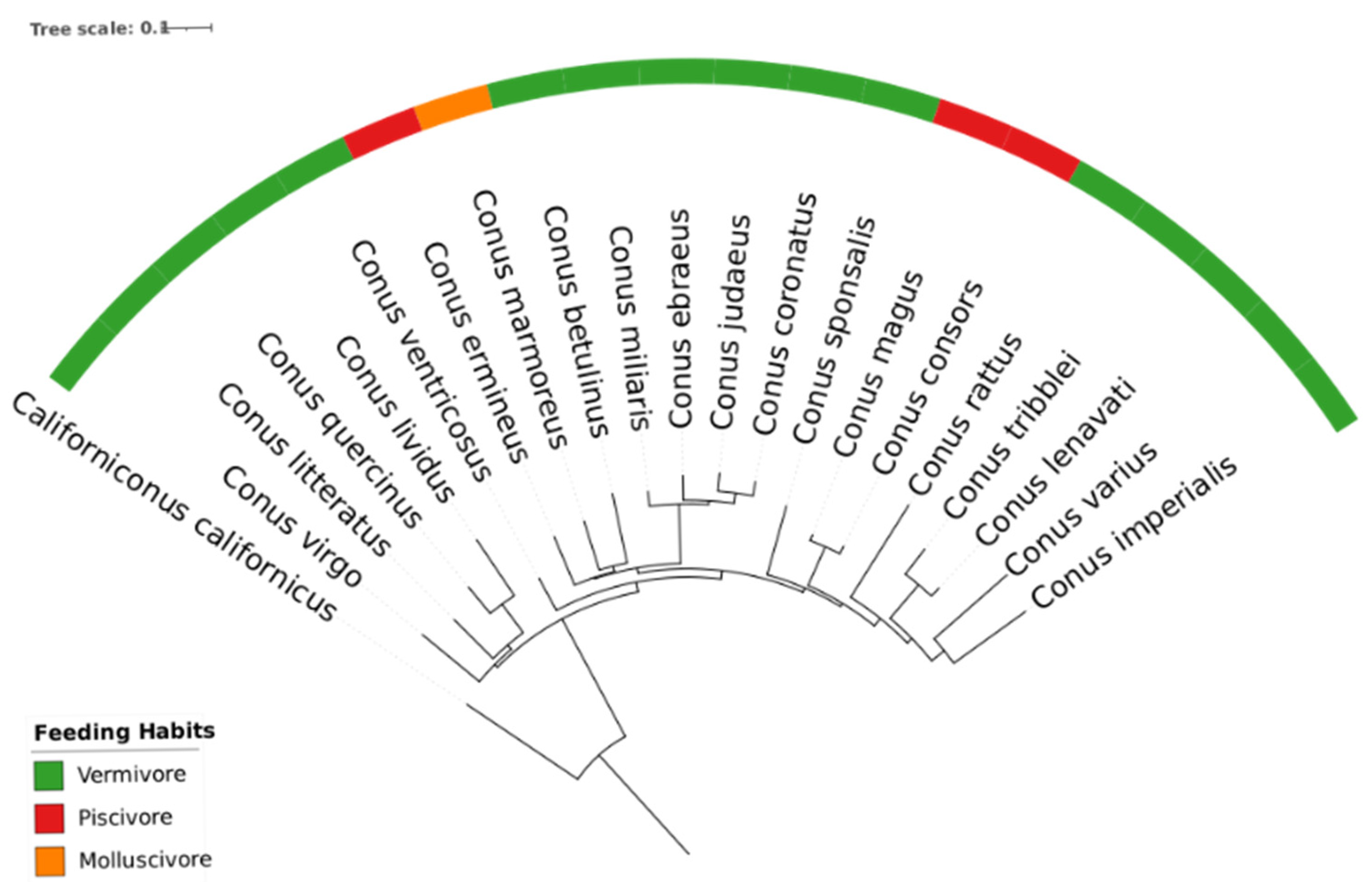
Appendix G
| Species and Tissue | Completed—Total | Complete—Single Copy | Complete—Duplicated | Fragmented | Missing |
|---|---|---|---|---|---|
| C. miliaris—Venom duct | 110 | 94 | 16 | 75 | 769 |
| C. miliaris—Venom duct | 122 | 102 | 20 | 85 | 747 |
| C. coronatus—Venom duct | 122 | 108 | 14 | 74 | 758 |
| C. miliaris—Venom duct | 128 | 109 | 19 | 98 | 728 |
| C. miliaris—Venom duct | 128 | 110 | 18 | 98 | 728 |
| C. miliaris—Venom duct | 142 | 119 | 23 | 116 | 696 |
| C. miliaris—Venom duct | 153 | 129 | 24 | 108 | 693 |
| C. miliaris—Venom duct | 153 | 129 | 24 | 106 | 695 |
| C. miliaris—Venom duct | 166 | 135 | 31 | 118 | 670 |
| C. miliaris—Venom duct | 161 | 136 | 25 | 98 | 695 |
| C. miliaris—Venom duct | 174 | 138 | 36 | 113 | 667 |
| C. miliaris—Venom duct | 164 | 140 | 24 | 138 | 652 |
| C. coronatus—Venom duct | 166 | 144 | 22 | 122 | 666 |
| C. miliaris—Venom duct | 174 | 145 | 29 | 95 | 685 |
| C. miliaris—Venom duct | 180 | 151 | 29 | 133 | 641 |
| C. miliaris—Venom duct | 176 | 151 | 25 | 104 | 674 |
| C. miliaris—Venom duct | 191 | 161 | 30 | 135 | 628 |
| C. miliaris—Venom duct | 191 | 162 | 29 | 135 | 628 |
| C. miliaris—Venom duct | 190 | 163 | 27 | 111 | 653 |
| C. consors—Foot | 244 | 163 | 81 | 129 | 581 |
| C. coronatus—Venom duct | 192 | 166 | 26 | 128 | 634 |
| C. sponsalis—Venom duct | 203 | 167 | 36 | 127 | 624 |
| C. miliaris—Venom duct | 196 | 173 | 23 | 115 | 643 |
| C. miliaris—Venom duct | 210 | 176 | 34 | 163 | 581 |
| C. miliaris—Venom duct | 230 | 189 | 41 | 133 | 591 |
| C. imperialis—Venom duct | 220 | 190 | 30 | 114 | 620 |
| C. virgo—Venom duct | 210 | 191 | 19 | 150 | 594 |
| C. imperialis—Venom duct | 227 | 192 | 35 | 162 | 565 |
| C. coronatus—Venom duct | 222 | 193 | 29 | 114 | 618 |
| C. miliaris—Venom duct | 241 | 196 | 45 | 148 | 565 |
| C. ebraeus—Venom duct | 229 | 200 | 29 | 180 | 545 |
| C. imperialis—Venom duct | 240 | 207 | 33 | 148 | 566 |
| C. ermineus—Venom duct | 256 | 214 | 42 | 111 | 587 |
| C. lividus—Venom duct | 259 | 215 | 44 | 189 | 506 |
| C. ermineus—Venom duct | 256 | 219 | 37 | 184 | 514 |
| C. imperialis—Venom duct | 251 | 222 | 29 | 125 | 578 |
| C. marmoreus—Venom duct | 247 | 222 | 25 | 151 | 556 |
| C. miliaris—Venom duct | 268 | 224 | 44 | 184 | 502 |
| C. ermineus—Venom duct | 271 | 228 | 43 | 110 | 573 |
| C. ebraeus—Venom duct | 286 | 232 | 54 | 86 | 582 |
| C. rattus—Venom duct | 282 | 234 | 48 | 186 | 486 |
| C. quercinus—Venom duct | 267 | 237 | 30 | 161 | 526 |
| C. ermineus—Venom duct | 301 | 249 | 52 | 130 | 523 |
| C. varius—Venom duct | 297 | 250 | 47 | 163 | 494 |
| C. ermineus—Venom duct | 294 | 251 | 43 | 175 | 485 |
| C. tribblei—Venom duct | 307 | 253 | 54 | 281 | 366 |
| C. tribblei—Venom duct | 313 | 256 | 57 | 264 | 377 |
| C. magus—Venom duct | 315 | 258 | 57 | 106 | 533 |
| C. tribblei—Venom duct | 318 | 267 | 51 | 256 | 380 |
| C. lenavati—Venom duct | 343 | 274 | 69 | 262 | 349 |
| C. lenavati—Venom duct | 334 | 274 | 60 | 207 | 413 |
| C. ermineus—Venom duct | 356 | 274 | 82 | 104 | 494 |
| C. magus—Venom duct | 344 | 279 | 65 | 113 | 497 |
| C. lenavati—Venom duct | 365 | 284 | 81 | 253 | 336 |
| C. betulinus—Venom duct | 355 | 286 | 69 | 263 | 336 |
| C. ermineus—Venom duct | 364 | 287 | 77 | 103 | 487 |
| C. betulinus—Venom duct | 388 | 301 | 87 | 268 | 298 |
| C. ermineus—Venom duct | 390 | 314 | 76 | 116 | 448 |
| C. judaeus—Venom duct | 421 | 337 | 84 | 123 | 410 |
| C. litteratus—Venom duct | 453 | 357 | 96 | 230 | 271 |
| C. litteratus—Venom duct | 500 | 371 | 129 | 218 | 236 |
| C. ventricosus—Foot | 471 | 382 | 89 | 121 | 362 |
| C. imperialis—Venom duct | 460 | 385 | 75 | 170 | 324 |
| C. ventricosus—Foot | 544 | 424 | 120 | 120 | 290 |
| C. ermineus—Venom duct | 574 | 429 | 145 | 133 | 247 |
| C. consors—Nervous ganglions | 771 | 446 | 325 | 94 | 89 |
| C. consors—Venom duct | 750 | 458 | 292 | 90 | 114 |
| C. ventricosus—Venom gland | 584 | 463 | 121 | 124 | 246 |
| C. betulinus—Venom duct | 568 | 464 | 104 | 208 | 178 |
| C. consors—Osphradium | 836 | 465 | 371 | 65 | 53 |
| C. consors—Salivary glands | 694 | 480 | 214 | 130 | 130 |
| C. betulinus—Venom duct | 662 | 485 | 177 | 154 | 138 |
| C. consors—Proboscis | 826 | 489 | 337 | 63 | 65 |
| C. consors—Venom bulb | 751 | 489 | 262 | 99 | 104 |
| C. betulinus—Venom duct | 600 | 517 | 83 | 205 | 149 |
| C. tribblei—Venom duct | 667 | 542 | 125 | 165 | 122 |
References
- Peng, C.; Huang, Y.; Bian, C.; Li, J.; Liu, J.; Zhang, K.; You, X.; Lin, Z.; He, Y.; Chen, J.; et al. The first Conus genome assembly reveals a primary genetic central dogma of conopeptides in C. betulinus. Cell Discov. 2021, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Pardos-Blas, J.R.; Irisarri, I.; Abalde, S.; Afonso, C.M.; Tenorio, M.J.; Zardoya, R. The genome of the venomous snail Lautoconus ventricosus sheds light on the origin of conotoxin diversity. Gigascience 2021, 10, giab037. [Google Scholar] [CrossRef]
- Herráez-Pérez, A.; Pardos-Blas, J.R.; Afonso, C.M.L.; Tenorio, M.J.; Zardoya, R. Chromosome-level genome of the venomous snail Kalloconus canariensis: A valuable model for venomics and comparative genomics. GigaScience 2023, 12, giad075. [Google Scholar] [CrossRef]
- Duda, T.F., Jr.; Kohn, A.J.; Palumbi, S.R. Origins of diverse feeding ecologies within Conus, a genus of venomous marine gastropods. Biol. J. Linn. Soc. 2001, 73, 391–409. [Google Scholar] [CrossRef]
- Puillandre, N.; Duda, T.F.; Meyer, C.; Olivera, B.M.; Bouchet, P. One, four or 100 genera? A new classification of the cone snails. J. Molluscan Stud. 2015, 81, 1–23. [Google Scholar] [CrossRef]
- Gao, B.; Peng, C.; Yang, J.; Yi, Y.; Zhang, J.; Shi, Q. Cone snails: A big store of conotoxins for novel drug discovery. Toxins 2017, 9, 397. [Google Scholar] [CrossRef]
- Prashanth, J.R.; Dutertre, S.; Jin, A.H.; Lavergne, V.; Hamilton, B.; Cardoso, F.C.; Griffin, J.; Venter, D.J.; Alewood, P.F.; Lewis, R.J. The role of defensive ecological interactions in the evolution of conotoxins. Mol. Ecol. 2016, 25, 598–615. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.T.; Craik, D.J.; Kaas, Q. Bibliometric Review of the Literature on Cone Snail Peptide Toxins from 2000 to 2022. Mar. Drugs 2023, 21, 154. [Google Scholar] [CrossRef]
- Richard, G.; Rabiller, M. Panorama sur La Diversite des Conidae 110 Espèces Prédatrices des Plus Efficaces; CRIOBE Tahiti: Perpignan, France, 2021. [Google Scholar]
- Endean, R.; Duchemin, C. The venom apparatus of Conus magus. Toxicon 1967, 4, 275–284. [Google Scholar] [CrossRef]
- Jin, A.-H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and Biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure: Activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Buczek, O.; Bulaj, G.; Olivera, B.M. Conotoxins and the posttranslational modification of secreted gene products. Cell. Mol. Life Sci. 2005, 62, 3067–3079. [Google Scholar] [CrossRef] [PubMed]
- Neves, J.L.B.; Lin, Z.; Imperial, J.S.; Antunes, A.; Vasconcelos, V.; Olivera, B.M.; Schmidt, E.W. Small Molecules in the Cone Snail Arsenal. Org. Lett. 2015, 17, 4933–4935. [Google Scholar] [CrossRef]
- Lu, A.; Yang, L.; Xu, S.; Wang, C. Various Conotoxin Diversifications Revealed by a Venomic Study of Conus flavidus. Mol. Cell. Proteom. 2014, 13, 105–118. [Google Scholar] [CrossRef]
- Jin, A.-H.; Dutertre, S.; Kaas, Q.; Lavergne, V.; Kubala, P.; Lewis, R.J.; Alewood, P.F. Transcriptomic messiness in the venom duct of Conus miles contributes to conotoxin diversity. Mol. Cell. Proteom. 2013, 12, 3824–3833. [Google Scholar] [CrossRef]
- Jakubowski, J.A.; Kelley, W.P.; Sweedler, J.V. Screening for post-translational modifications in conotoxins using liquid chromatography/mass spectrometry: An important component of conotoxin discovery. Toxicon 2006, 47, 688–699. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef]
- Laht, S.; Koua, D.; Kaplinski, L.; Lisacek, F.; Stöcklin, R.; Remm, M. Identification and classification of conopeptides using profile Hidden Markov Models. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2012, 1824, 488–492. [Google Scholar] [CrossRef]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2011, 40, D325–D330. [Google Scholar] [CrossRef]
- Rivera-Ortiz, J.A.; Cano, H.; Marí, F. Intraspecies variability and conopeptide profiling of the injected venom of Conus ermineus. Peptides 2011, 32, 306–316. [Google Scholar] [CrossRef]
- Prator, C.A.; Murayama, K.M.; Schulz, J.R. Venom variation during prey capture by the cone snail, Conus textile. PLoS ONE 2014, 9, e98991. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.H.; Dutertre, S.; Dutt, M.; Lavergne, V.; Jones, A.; Lewis, R.J.; Alewood, P.F. Transcriptomic-Proteomic Correlation in the Predation-Evoked Venom of the Cone Snail, Conus imperialis. Mar. Drugs 2019, 17, 177. [Google Scholar] [CrossRef]
- Junqueira-de-Azevedo, I.L.; Bastos, C.M.V.; Ho, P.L.; Luna, M.S.; Yamanouye, N.; Casewell, N.R. Venom-related transcripts from Bothrops jararaca tissues provide novel molecular insights into the production and evolution of snake venom. Mol. Biol. Evol. 2015, 32, 754–766. [Google Scholar] [CrossRef]
- Reyes-Velasco, J.; Card, D.C.; Andrew, A.L.; Shaney, K.J.; Adams, R.H.; Schield, D.R.; Casewell, N.R.; Mackessy, S.P.; Castoe, T.A. Expression of venom gene homologs in diverse python tissues suggests a new model for the evolution of snake venom. Mol. Biol. Evol. 2015, 32, 173–183. [Google Scholar] [CrossRef]
- Hargreaves, A.D.; Swain, M.T.; Hegarty, M.J.; Logan, D.W.; Mulley, J.F. Restriction and recruitment—Gene duplication and the origin and evolution of snake venom toxins. Genome Biol. Evol. 2014, 6, 2088–2095. [Google Scholar] [CrossRef]
- Duda, T.F., Jr.; Palumbi, S.R. Molecular genetics of ecological diversification: Duplication and rapid evolution of toxin genes of the venomous gastropod Conus. Biol. Sci. 1999, 96, 6820–6823. [Google Scholar] [CrossRef]
- Puillandre, N.; Watkins, M.; Olivera, B.M. Evolution of Conus Peptide Genes: Duplication and Positive Selection in the A-Superfamily. J. Mol. Evol. 2010, 70, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Duda, T.F., Jr. Extensive and Continuous Duplication Facilitates Rapid Evolution and Diversification of Gene Families. Mol. Biol. Evol. 2012, 29, 2019–2029. [Google Scholar] [CrossRef]
- Whittington, C.M.; Belov, K. Platypus venom genes expressed in non-venom tissues. Aust. J. Zool. 2009, 57, 199–202. [Google Scholar] [CrossRef]
- Fry, B.G.; Undheim, E.A.B.; Ali, S.A.; Jackson, T.N.W.; Debono, J.; Scheib, H.; Ruder, T.; Morgenstern, D.; Cadwallader, L.; Whitehead, D.; et al. Squeezers and Leaf-cutters: Differential Diversification and Degeneration of the Venom System in Toxicoferan Reptiles. Mol. Cell. Proteom. 2013, 12, 1881–1899. [Google Scholar] [CrossRef]
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.A.; King, G.F.; Nevalainen, T.J.; Norman, J.A.; Lewis, R.J.; Norton, R.S.; et al. The Toxicogenomic Multiverse: Convergent Recruitment of Proteins Into Animal Venoms. Annu. Rev. Genom. Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.S.W.; Belov, K. Venom evolution through gene duplications. Gene 2012, 496, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Helsen, J.; Voordeckers, K.; Vanderwaeren, L.; Santermans, T.; Tsontaki, M.; Verstrepen, K.J.; Jelier, R. Gene Loss Predictably Drives Evolutionary Adaptation. Mol. Biol. Evol. 2020, 37, 2989–3002. [Google Scholar] [CrossRef]
- Sharma, V.; Hecker, N.; Roscito, J.G.; Foerster, L.; Langer, B.E.; Hiller, M. A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nat. Commun. 2018, 9, 1215. [Google Scholar] [CrossRef]
- Casewell, N.R. Venom Evolution: Gene Loss Shapes Phenotypic Adaptation. Curr. Biol. 2016, 26, R849–R851. [Google Scholar] [CrossRef]
- Von Reumont, B.M. Studying Smaller and Neglected Organisms in Modern Evolutionary Venomics Implementing RNASeq (Transcriptomics)—A Critical Guide. Toxins 2018, 10, 292. [Google Scholar] [CrossRef]
- Zancolli, G.; von Reumont, B.M.; Anderluh, G.; Caliskan, F.; Chiusano, M.L.; Fröhlich, J.; Hapeshi, E.; Hempel, B.F.; Ikonomopoulou, M.P.; Jungo, F.; et al. Web of venom: Exploration of big data resources in animal toxin research. GigaScience 2024, 13, giae054. [Google Scholar] [CrossRef]
- Peraud, O.; Biggs, J.S.; Hughen, R.W.; Light, A.R.; Concepcion, G.P.; Olivera, B.M.; Schmidt, E.W. Microhabitats within venomous cone snails contain diverse actinobacteria. Appl. Environ. Microbiol. 2009, 75, 6820–6826. [Google Scholar] [CrossRef]
- Torres, J.P.; Tianero, M.D.; Robes, J.M.D.; Kwan, J.C.; Biggs, J.S.; Concepcion, G.P.; Olivera, B.M.; Haygood, M.G.; Schmidt, E.W. Stenotrophomonas-Like Bacteria Are Widespread Symbionts in Cone Snail Venom Ducts. Appl. Environ. Microbiol. 2017, 83, e01418-17. [Google Scholar] [CrossRef]
- Giglio, M.L.; Salcedo, P.F.; Watkins, M.; Olivera, B. Insights into a putative polychaete-gastropod symbiosis from a newly identified annelid worm that predates upon Conus ermineus eggs. Contrib. Zool. 2023, 92, 97–111. [Google Scholar] [CrossRef]
- Agüero-Chapin, G.; Domínguez-Pérez, D.; Marrero-Ponce, Y.; Castillo-Mendieta, K.; Antunes, A. Unveiling Encrypted Antimicrobial Peptides from Cephalopods’ Salivary Glands: A Proteolysis-Driven Virtual Approach. ACS Omega 2024, 43, 43353–43367. [Google Scholar] [CrossRef] [PubMed]
- Barroso, R.A.; Agüero-Chapin, G.; Sousa, R.; Marrero-Ponce, Y.; Antunes, A. Unlocking Antimicrobial Peptides: In Silico Proteolysis and Artificial Intelligence-Driven Discovery from Cnidarian Omics. Molecules 2025, 30, 550. [Google Scholar] [CrossRef]
- Morim, J. Deciphering the Transcriptomics of the Conus Species’ Natural Venoms. Master’s Thesis, University of Porto, Porto, Portugal, 2022. [Google Scholar]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 8, 1166–1167. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024, 52, 78–82. [Google Scholar] [CrossRef]
- Yang, S.; Yang, Z.; Ni, X. AMPFinder: A computational model to identify antimicrobial peptides and their functions based on sequence-derived information. Anal. Biochem. 2023, 673, 115196. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Patro, S.G.K.; Sahu, K.K. Normalization: A Preprocessing Stage. arXiv 2015, arXiv:1503.06462. [Google Scholar] [CrossRef]
- McDougal, O.M.; Turner, M.W.; Ormond, A.J.; Poulter, C.D. Three-dimensional structure of conotoxin tx3a: An m-1 branch peptide of the M-superfamily. Biochemistry 2008, 47, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.B.; McDougal, O.M. The M-superfamily of conotoxins: A review. Cell. Mol. Life Sci. 2010, 67, 17–27. [Google Scholar] [CrossRef]
- Corpuz, G.P.; Jacobsen, R.B.; Jimenez, E.C.; Watkins, M.; Walker, C.; Colledge, C.; Garrett, J.E.; McDougal, O.; Li, W.; Gray, W.R.; et al. Definition of the M-conotoxin superfamily: Characterization of novel peptides from molluscivorous Conus venoms. Biochemistry 2005, 44, 8176–8186. [Google Scholar] [CrossRef]
- Zhao, Y.; Antunes, A. Biomedical Potential of the Neglected Molluscivorous and Vermivorous Conus Species. Mar. Drugs 2022, 20, 105. [Google Scholar] [CrossRef]
- Olivera, B.M.; Seger, J.; Horvath, M.P.; Fedosov, A.E. Prey-Capture Strategies of Fish-Hunting Cone Snails: Behavior, Neurobiology and Evolution. Brain Behav. Evol. 2015, 86, 58–74. [Google Scholar] [CrossRef]
- Abalde, S.; Tenorio, M.J.; Afonso, C.M.L.; Zardoya, R. Conotoxin Diversity in ChelyConus ermineus (Born, 1778) and the Convergent Origin of Piscivory in the Atlantic and Indo-Pacific Cones. Genome Biol. Evol. 2018, 10, 2643–2662. [Google Scholar] [CrossRef]
- Rogalski, A.; Himaya, S.W.A.; Lewis, R.J. Coordinated adaptations define the ontogenetic shift from worm- to fish-hunting in a venomous cone snail. Nat. Commun. 2023, 14, 3287. [Google Scholar] [CrossRef]
- Gotsiridze-Columbus, N.S. (Ed.) Snails: Biology, Ecology and Conservation; Nova Science Publishers: Hauppauge, NY, USA, 2011; pp. 85–104. [Google Scholar]
- Dutertre, S.; Jin, A.H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 3521. [Google Scholar] [CrossRef]
- Koch, T.L.; Robinson, S.D.; Salcedo, P.F.; Chase, K.; Biggs, J.; Fedosov, A.E.; Yandell, M.; Olivera, B.M.; Safavi-Hemami, H. Prey Shifts Drive Venom Evolution in Cone Snails. Mol. Biol. Evol. 2024, 41, msae120. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.J.; Saunders, P.R.; Wiener, S. Preliminary studies on the venom of the marine snail Conus. Ann. N. Y. Acad. Sci. 1960, 90, 706–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, 1087–1093. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Oliveira, A.S.F.; Ibarra, A.A.; Bermudez, I.; Casalino, L.; Gaieb, Z.; Shoemark, D.K.; Gallagher, T.; Sessions, R.B.; Amaro, R.E.; Mulholland, A.J. A potential interaction between the SARS-CoV-2 spike protein and nicotinic acetylcholine receptors. Biophys. J. 2021, 120, 983–993. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Wang, X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Changeux, J.P.; Amoura, Z.; Rey, F.A.; Miyara, M. A nicotinic hypothesis for COVID-19 with preventive and therapeutic implications. Comptes Rendus. Biologies 2020, 343, 33–39. [Google Scholar] [CrossRef]
- Santiago, U.; Camacho, C.J. Omicron and Alpha P680H block SARS-CoV2 spike protein from accessing cholinergic inflammatory pathway via α9-nAChR mitigating the risk of MIS-C. bioRxiv 2022. [Google Scholar] [CrossRef]
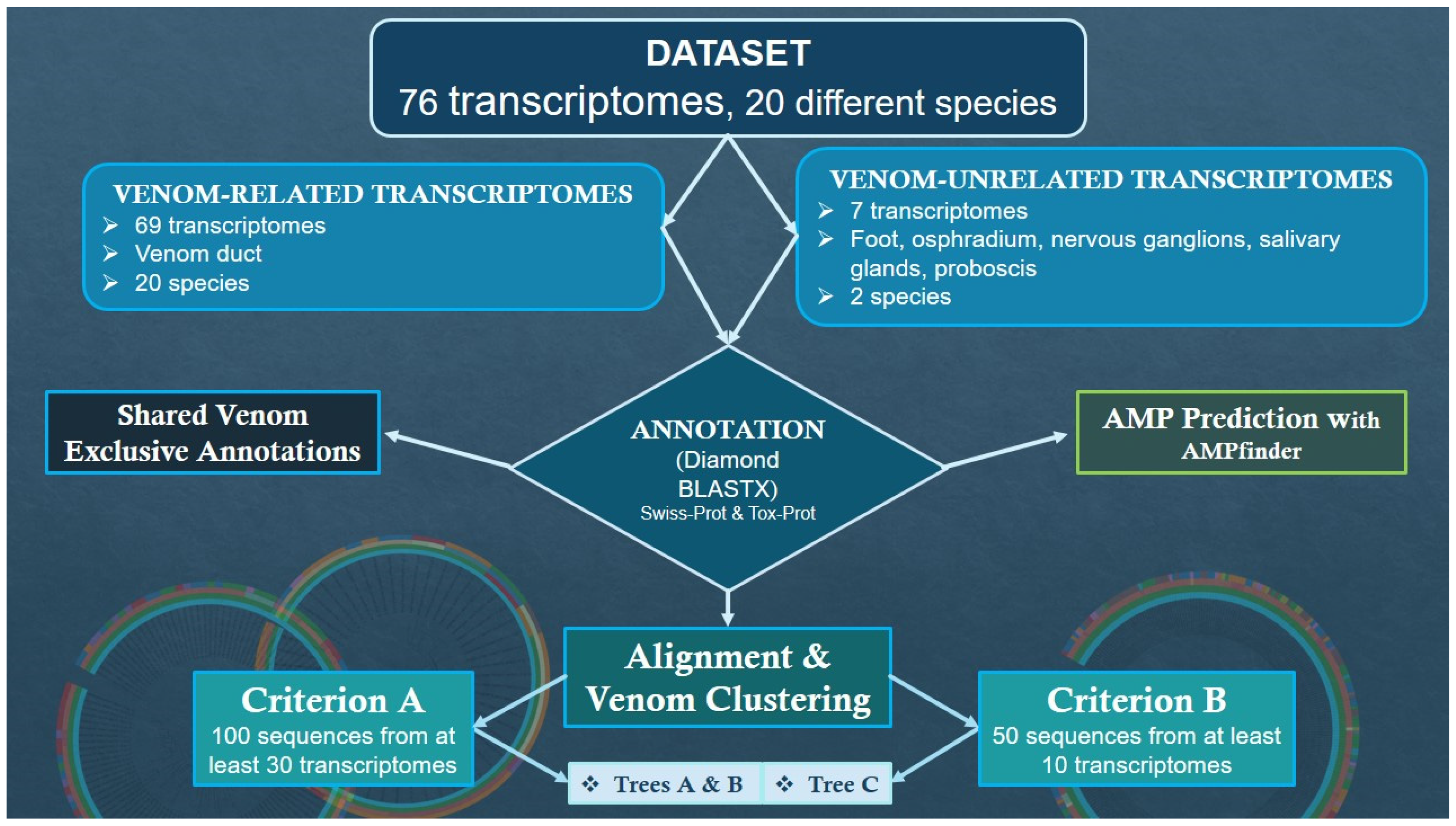
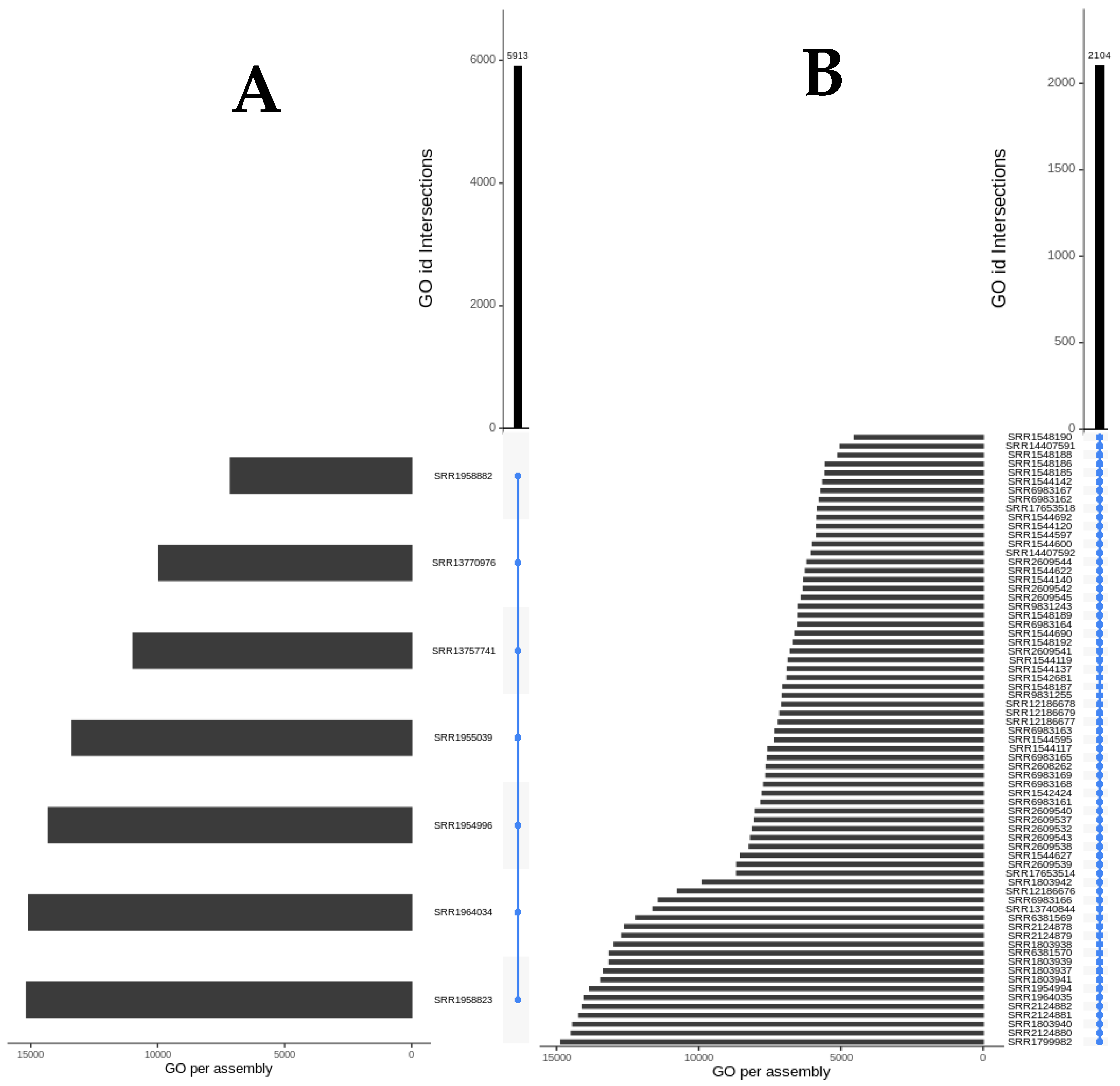

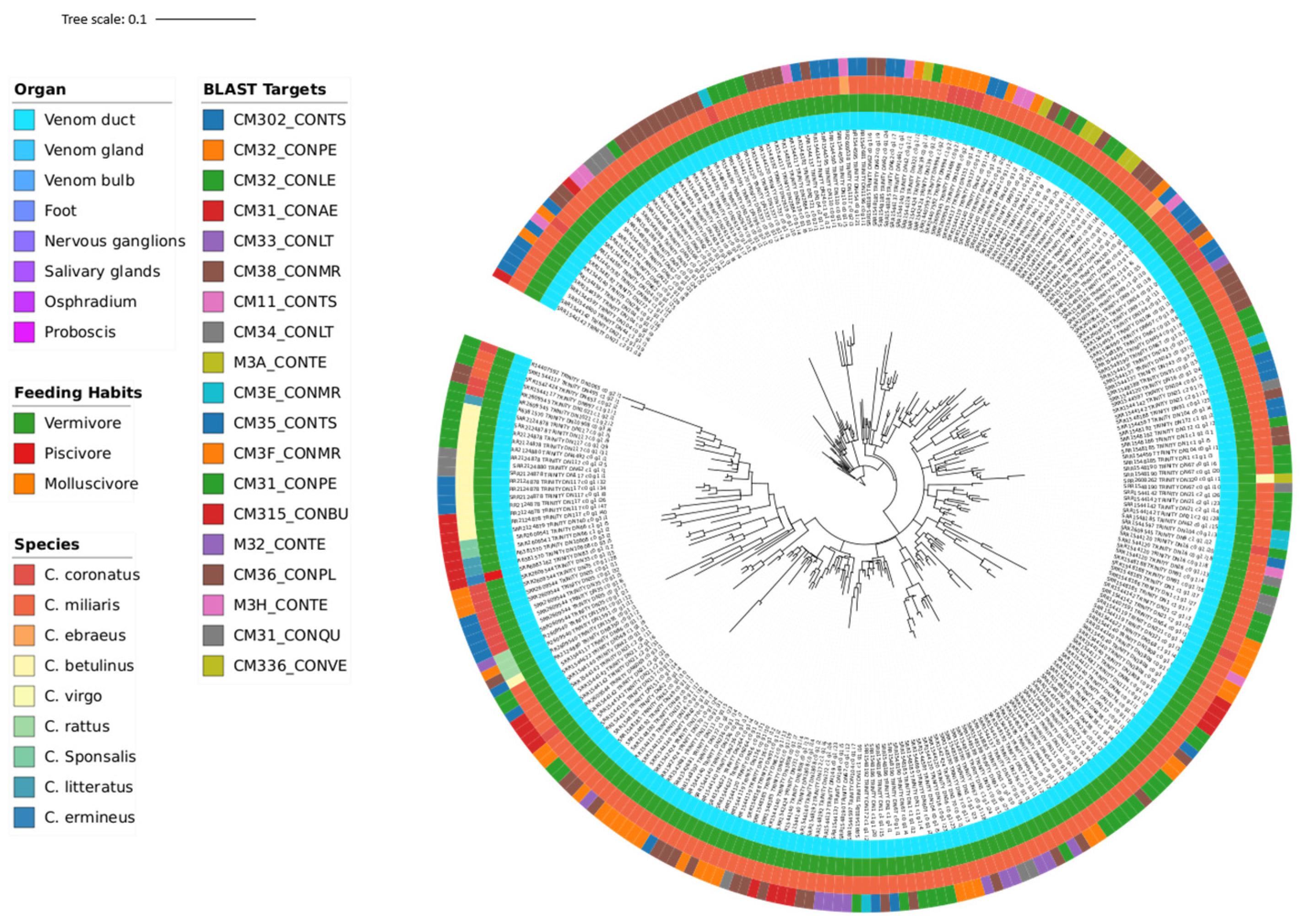
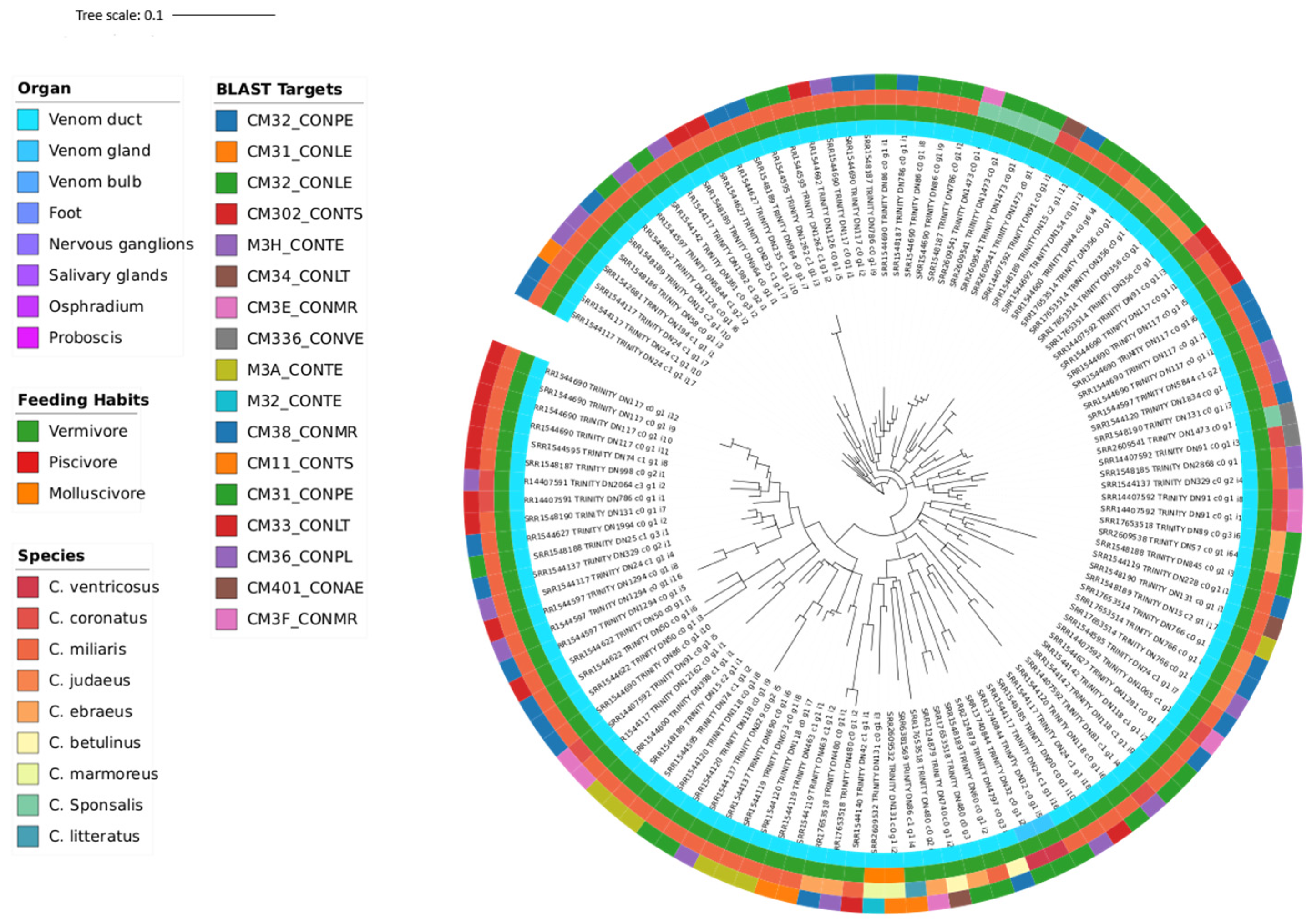
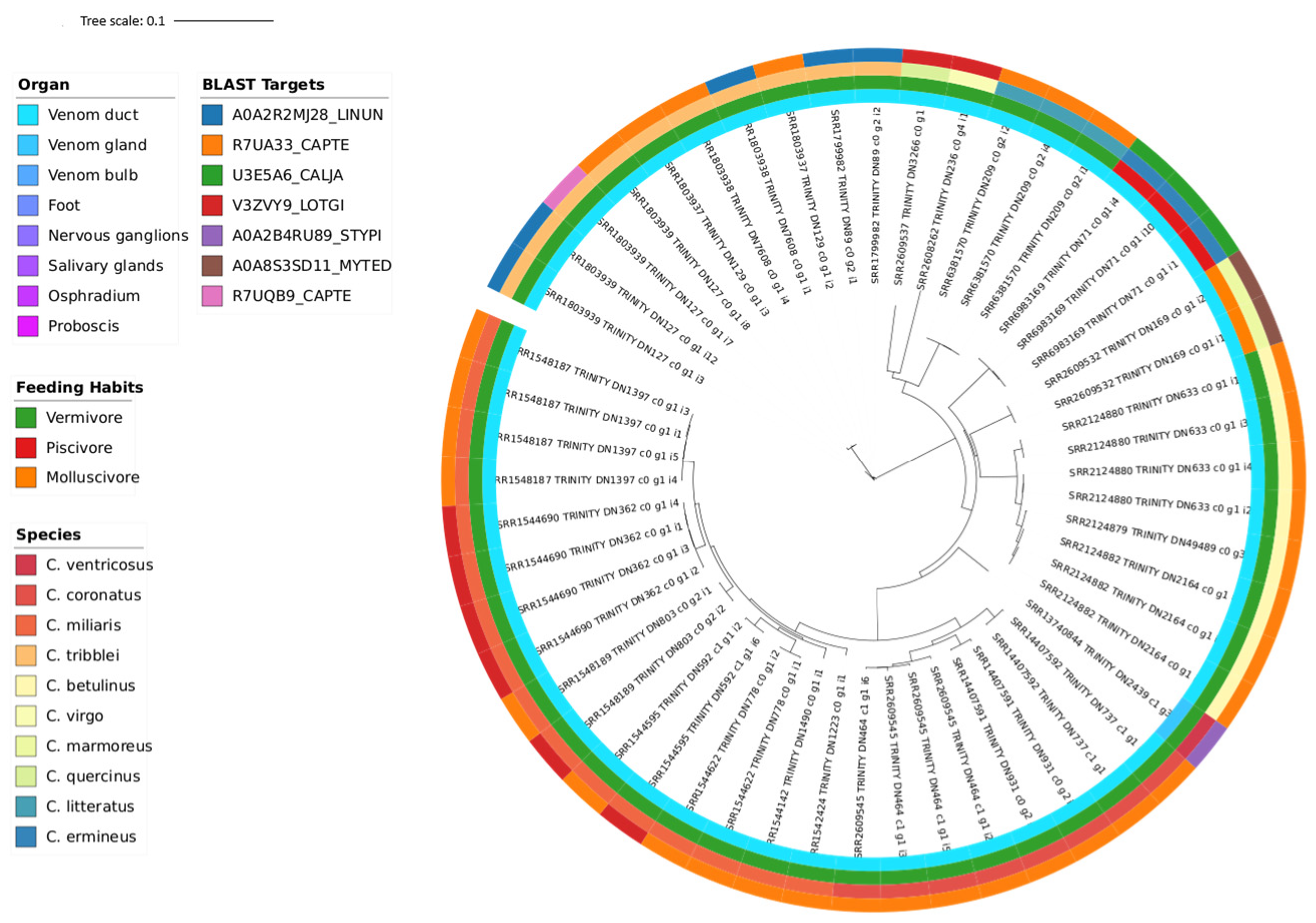
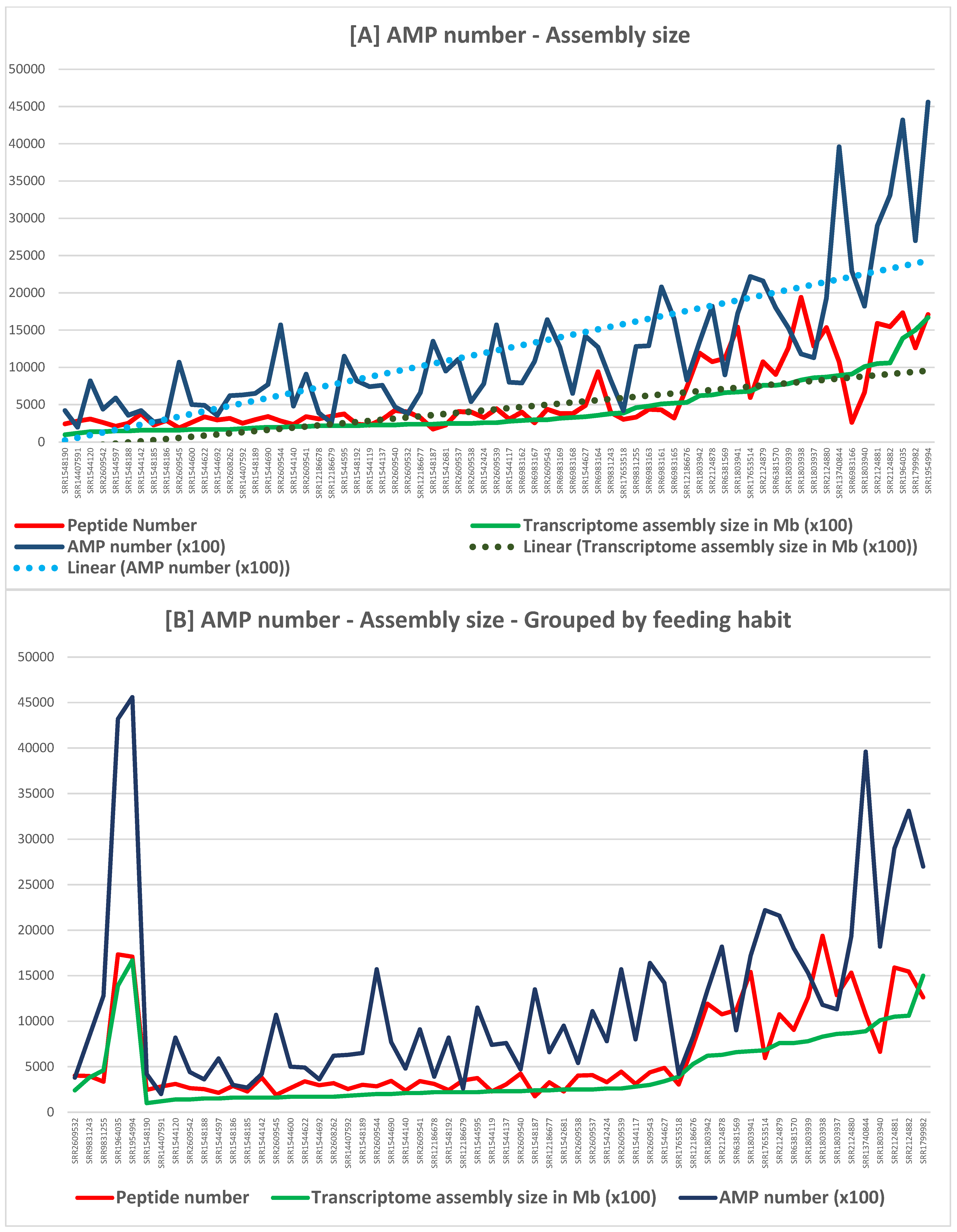
| Transcriptomic Group | Number of Peptides | Number of Toxins | AMP Predictions |
|---|---|---|---|
| Venom-related | 553,878 | 28,805 | 71 (15 unique) |
| Venom-unrelated | 108,466 | 1902 | 73 (17 unique) |
| Data Index | File | Species (Diet) | Abundance | Data Base Hit | Description |
|---|---|---|---|---|---|
| AMP_00195 | SRR2124881 | C. betulinus vermivorous | 1 | AGANDLCQECEDIVHLLTKMTKEDAFQDTIRKFLEQECDILPLKLLVPRCRQVLDVYLPLVIDYFQGQIKPKAICSHVGLC | Pulmonary protein B precursor |
| AMP_03456 | SRR6983168 | C. ermineus piscivorous | 1 | GLICESCRKIIQKLEDMVGPQPNEDTVTQAASQVCDKLKILRGLCKKIMRSFLRRISWDILTGKKPQAICVDIKICKEKT | Antimicrobial peptide NK-lysin precursor |
| AMP_03457 | SRR6983168 | 1 | GLICESCRKIIQKLEDMVGPQPNEDTVTQAASQVCDKLKILRGLCKKIMRSFLRRISWDILTGKKPQAICVDIKICKEKTGLI | ||
| AMP_04409 | SRR6983168 | 1 | GYFCESCRKIIQKLEDMVGPQPNEDTVTQAASQVCDKLKILRGLCKKIMRSFLRRISWDILTGKKPQAICVDIKICKE | ||
| AMP_21444 | SRR6983168 | C. ermineus piscivorous | 1 | YFCESCRKIIQKLEDMVGPQPNEDTVTQAASQVCDKLKILRGLCKKIMRSFLRRISWDILTGKKPQAICVDIKICKE | |
| AMP_05220 | SRR17653518 | C. ebraeus vermivorous | 1 | KPKDMTSSQWFKTQHVQPSPQACNSAMSIINKYTERCKDLNTFLHEPFSSVAITCQTPNIACKNSCKNCHQSHGPMSLTMGELTSGKYPNCRYKEKHLNTPYIVACDPPQQGDPGYPLVPVHLDKVV | Ribonuclease, RNase A family, 8 |
| AMP_19944 | SRR6381569 SRR6381570 | C. literatus vermivorous | 4 | EEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNITEE (See Appendix D for more transcripts sharing similarities with ACE protein) | ACE2 |
| SRR1542424 SRR1544142 | C. miliaris vermivorous | 2 | |||
| SRR12186679 | C. imperialis vermivorous | 2 | |||
| SRR13740844 | C. ventricosus vermivorous | 1 | |||
| SRR2609537 | C. quercinus vermivorous | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morim, J.; Zhao, Y.; Huang, L.; Antunes, A. Cone Snail Broad-Transcriptomics Elucidate the Evolutionary Diversification and Anti-Microbial Potential of Conopeptides. J. Mar. Sci. Eng. 2025, 13, 1006. https://doi.org/10.3390/jmse13061006
Morim J, Zhao Y, Huang L, Antunes A. Cone Snail Broad-Transcriptomics Elucidate the Evolutionary Diversification and Anti-Microbial Potential of Conopeptides. Journal of Marine Science and Engineering. 2025; 13(6):1006. https://doi.org/10.3390/jmse13061006
Chicago/Turabian StyleMorim, José, Yihe Zhao, Lei Huang, and Agostinho Antunes. 2025. "Cone Snail Broad-Transcriptomics Elucidate the Evolutionary Diversification and Anti-Microbial Potential of Conopeptides" Journal of Marine Science and Engineering 13, no. 6: 1006. https://doi.org/10.3390/jmse13061006
APA StyleMorim, J., Zhao, Y., Huang, L., & Antunes, A. (2025). Cone Snail Broad-Transcriptomics Elucidate the Evolutionary Diversification and Anti-Microbial Potential of Conopeptides. Journal of Marine Science and Engineering, 13(6), 1006. https://doi.org/10.3390/jmse13061006