
Variation in Structure and Functional Diversity of Surface Bacterioplankton Communities in the Eastern East China Sea

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
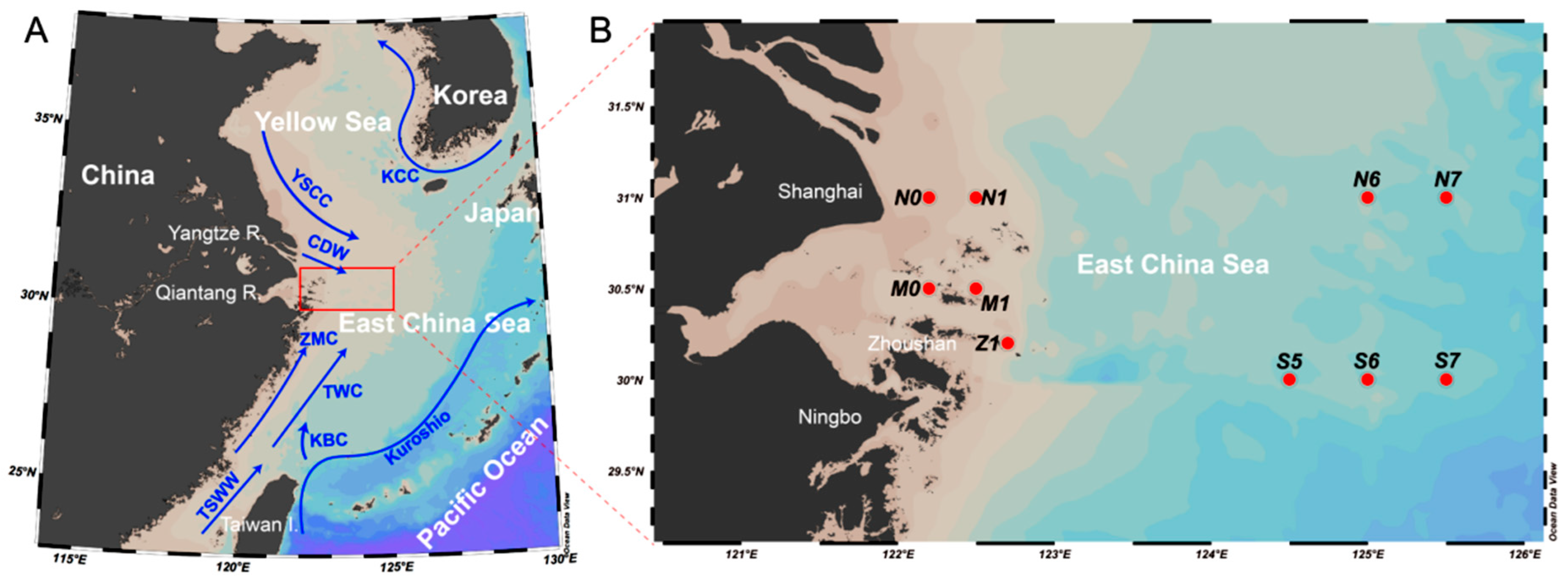
2.1. Sample and Environmental Data Collection
2.2. DNA Extraction and 16S rRNA Gene Pyrosequencing
2.3. Data Analysis
2.3.1. Sequence Processing
2.3.2. Analysis of the α- and β-Diversity
2.3.3. Statistical Analysis and Data Visualization
2.3.4. Partial Least Squares Discriminant Analysis
2.3.5. Co-Occurrence Network Analysis
2.3.6. Redundancy Analysis
3. Results
3.1. Sequencing Data Assessment
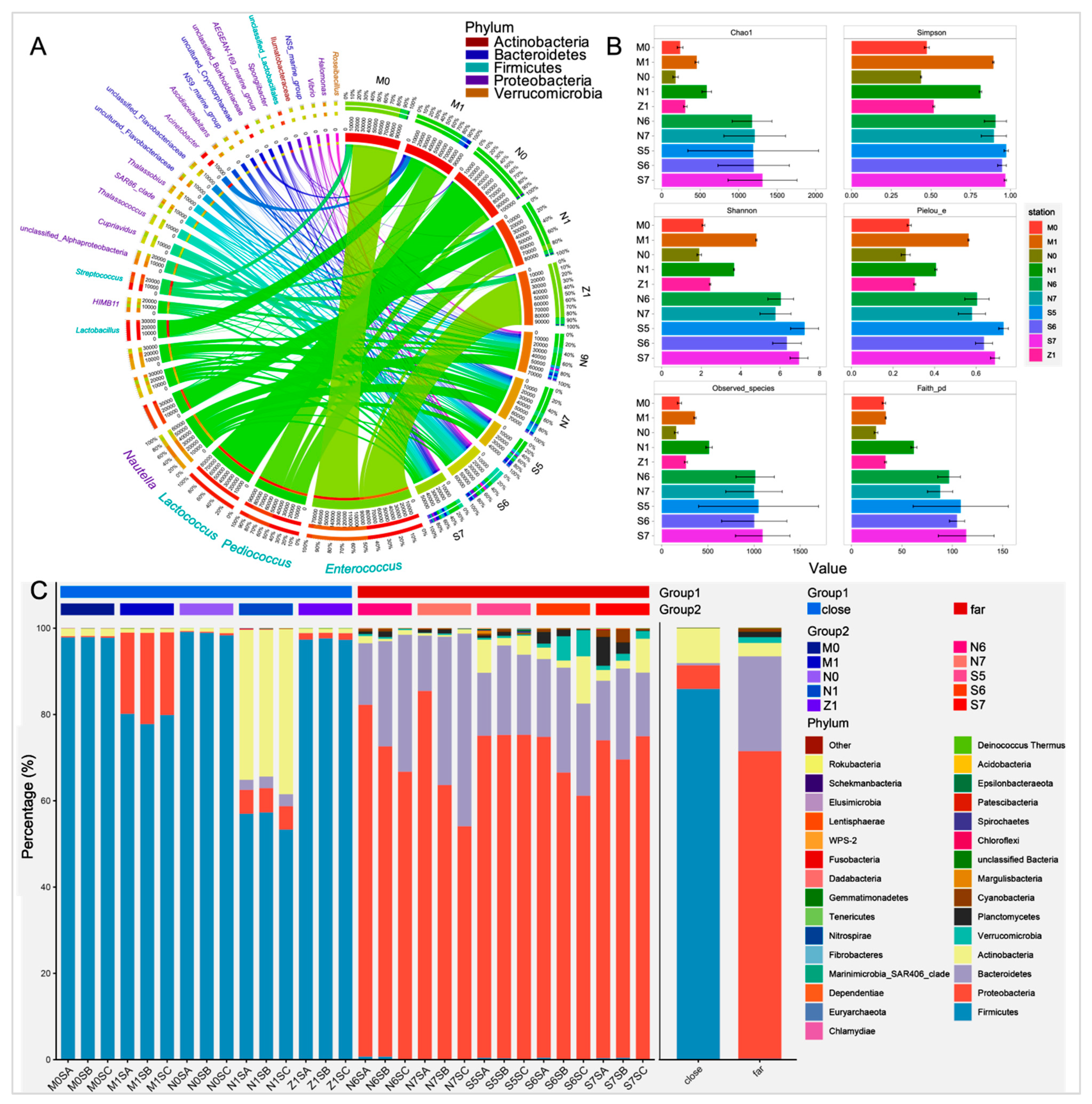
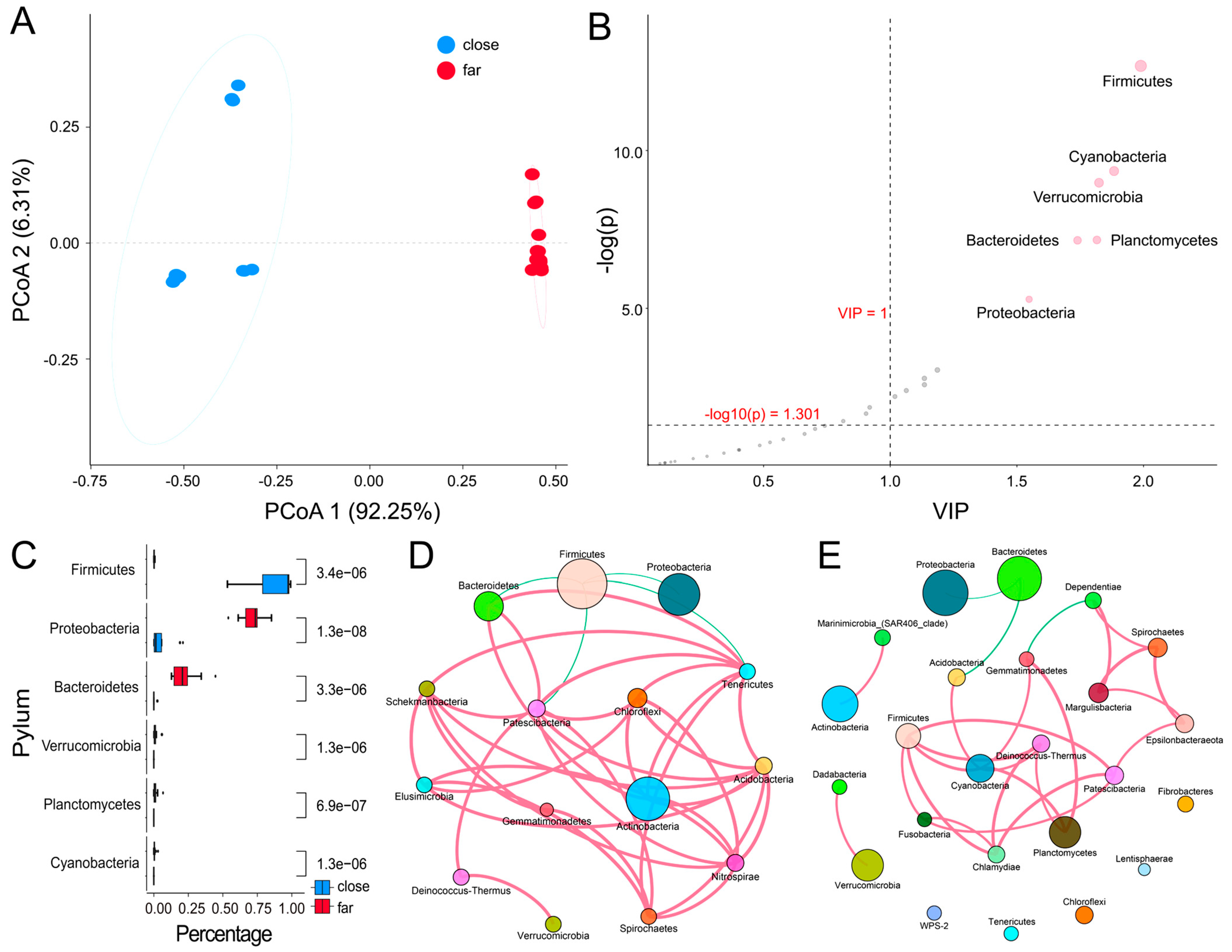
3.2. Taxonomy-Based Comparisons of Surface Bacterioplankton Communities at Phylum and Genus Levels
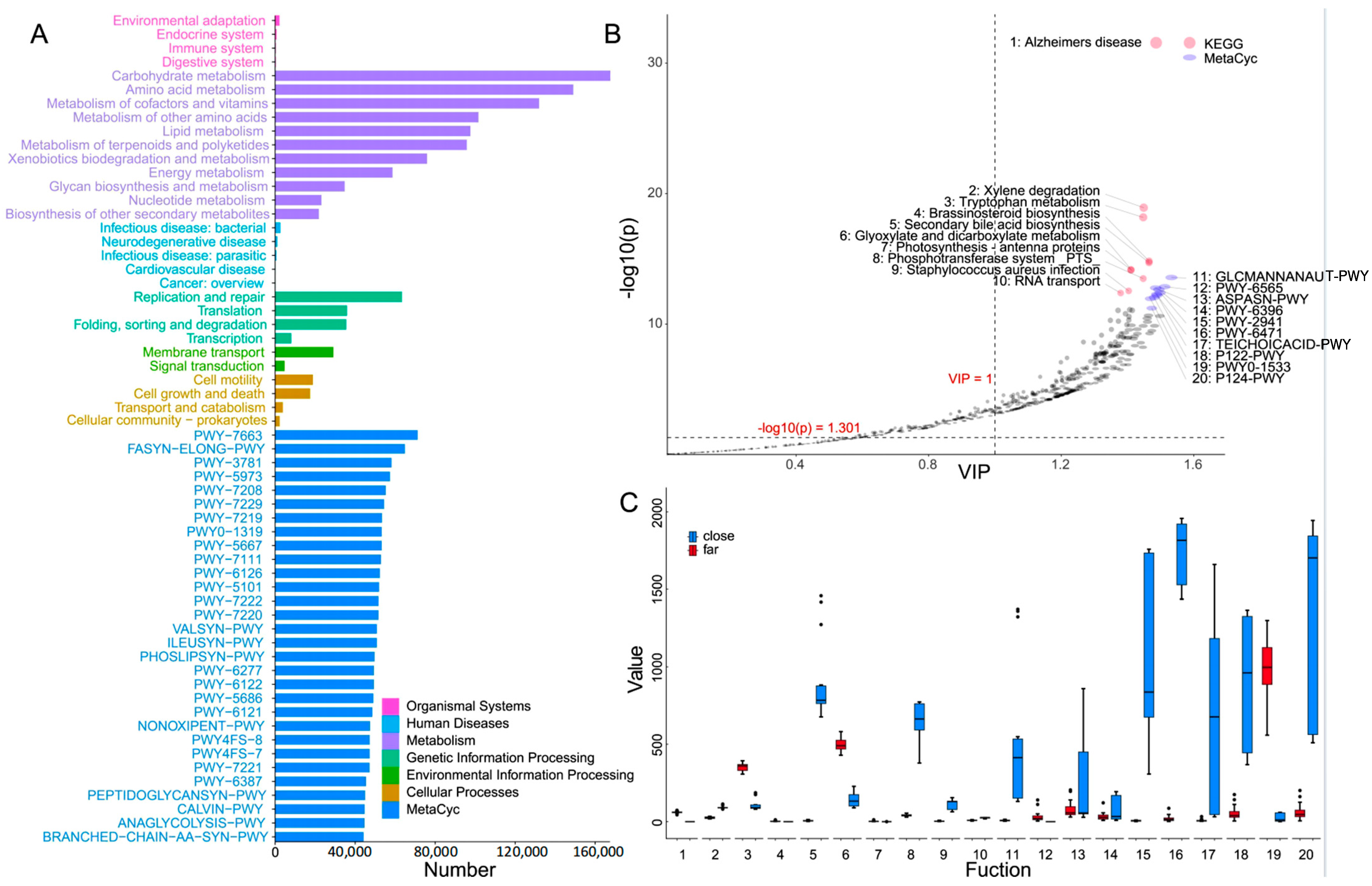
3.3. Functional Prediction of Surface Bacterioplankton Communities
3.4. Comparison of Bacterial Biodiversity between the Nearshore and Offshore Groups
3.5. Correlation Analysis of Surface Bacterioplankton Biodiversity with Environmental Factors
4. Discussion
4.1. Taxa Classification and Functional Characteristics of Surface Bacterioplankton Communities
4.2. Spatial Distribution and Shaping Factors of Surface Bacterioplankton Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fu, X.; Ding, W.; Dadd, K.; Li, J.; Zhu, W.; Feng, K.; Geng, J.; Xu, X. An exotic origin of the eastern East China Sea basement before-150 Ma. Sci. Bull. 2022, 67, 1939–1942. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, X.; Zhou, L.; Tao, S.; Liu, X.; Shi, G. A preliminary study of microbial diversity of the surface layer sediments from the East China Sea. Oceanol. Limnol. Sin. 2012, 43, 805–813. [Google Scholar]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Shiah, F.K.; Chiang, K.P.; Gong, G.C.; Kemp, W.M. Effects of the Changjiang (Yangtze) River discharge on planktonic community respiration in the East China Sea. J. Geophys. Res. Ocean. 2009, 114, C03005. [Google Scholar] [CrossRef]
- Wu, Y.; Dittmar, T.; Ludwichowski, K.-U.; Kattner, G.; Zhang, J.; Zhu, Z.Y.; Koch, B.P. Tracing suspended organic nitrogen from the Yangtze River catchment into the East China Sea. Mar. Chem. 2007, 107, 367–377. [Google Scholar] [CrossRef]
- Liu, C.; Wang, R.; Gao, H.; Wu, X.; Yin, D. Transport of trace metals and their bioaccumulation in zooplankton from Changjiang (Yangtze River) to the East China Sea. Sci. Total Environ. 2022, 851, 158156. [Google Scholar] [CrossRef] [PubMed]
- Lejeusne, C.; Chevaldonné, P.; Pergent-Martini, C.; Boudouresque, C.F.; Pérez, T. Climate change effects on a miniature ocean: The highly diverse, highly impacted Mediterranean Sea. Trends Ecol. Evol. 2010, 25, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Duarte, C.M. Global change and the future ocean: A grand challenge for marine sciences. Front. Mar. Sci. 2014, 1, 63. [Google Scholar] [CrossRef]
- Cui, Z.; Du, D.; Zhang, X.; Yang, Q. Modeling and Prediction of Environmental Factors and Chlorophyll A Abundance by Machine Learning Based on Tara Oceans Data. J. Mar. Sci. Eng. 2022, 10, 1749. [Google Scholar] [CrossRef]
- Conan, P.; Pujo-Pay, M.; Agab, M.; Calva-Benítez, L.; Chifflet, S.; Douillet, P.; Dussud, C.; Fichez, R.; Grenz, C.; Gutierrez Mendieta, F. Biogeochemical cycling and phyto-and bacterioplankton communities in a large and shallow tropical lagoon (Términos Lagoon, Mexico) under 2009–2010 El Niño Modoki drought conditions. Biogeosciences 2017, 14, 959–975. [Google Scholar] [CrossRef]
- Sun, Y.; Li, H.; Wang, X.; Li, H.; Deng, Y. Kelp Culture Enhances Coastal Biogeochemical Cycles by Maintaining Bacterioplankton Richness and Regulating Its Interactions. Msystems 2023, 8, e00002–e00023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jiang, R.; Zhang, J.; Li, K.; Zhang, J.; Shao, L.; He, W.; He, P. The Impact of IMTA on the Spatial and Temporal Distribution of the Surface Planktonic Bacteria Community in the Surrounding Sea Area of Xiasanhengshan Island of the East China Sea. J. Mar. Sci. Eng. 2023, 11, 476. [Google Scholar] [CrossRef]
- Hu, H.; He, J.; Yan, H.; Hou, D.; Zhang, D.; Liu, L.; Wang, K. Seasonality in spatial turnover of Bacterioplankton along an ecological gradient in the East China Sea: Biogeographic patterns, processes and drivers. Microorganisms 2020, 8, 1484. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, S.J.; Nemergut, D. Microbes ride the current. Science 2014, 345, 1246. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.-P.; Zhuang, G.-C.; Zhang, H.-H.; Dong, Y.; Yang, J. Distribution of dimethylsulfide and dimethylsulfoniopropionate in the Yellow Sea and the East China Sea during spring: Spatio-temporal variability and controlling factors. Mar. Chem. 2012, 138, 21–31. [Google Scholar] [CrossRef]
- Tsai, A.-Y.; Gong, G.-C.; Huang, J.-K.; Lin, Y.-C. Viral and nanoflagellate control of bacterial production in the East China Sea summer 2011. Estuar. Coast. Shelf Sci. 2013, 120, 33–41. [Google Scholar] [CrossRef]
- Falcón, L.I.; Noguez, A.M.; Espinosa-Asuar, L.; Eguiarte, L.E.; Souza, V. Evidence of biogeography in surface ocean bacterioplankton assemblages. Mar. Genom. 2008, 1, 55–61. [Google Scholar] [CrossRef]
- Cram, J.A.; Chow, C.-E.T.; Sachdeva, R.; Needham, D.M.; Parada, A.E.; Steele, J.A.; Fuhrman, J.A. Seasonal and interannual variability of the marine bacterioplankton community throughout the water column over ten years. ISME J. 2015, 9, 563–580. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, R.; He, Z.; Van Nostrand, J.D.; Zheng, Q.; Zhou, J.; Jiao, N. Functional Gene Diversity and Metabolic Potential of the Microbial Community in an Estuary-Shelf Environment. Front. Microbiol. 2017, 8, 1153. [Google Scholar] [CrossRef]
- Ye, Q.; Wu, Y.; Zhu, Z.; Wang, X.; Li, Z.; Zhang, J. Bacterial diversity in the surface sediments of the hypoxic zone near the Changjiang Estuary and in the East China Sea. MicrobiologyOpen 2016, 5, 323–339. [Google Scholar] [CrossRef]
- Wang, K.; Ye, X.; Chen, H.; Zhao, Q.; Hu, C.; He, J.; Qian, Y.; Xiong, J.; Zhu, J.; Zhang, D. Bacterial biogeography in the coastal waters of northern Zhejiang, East China Sea is highly controlled by spatially structured environmental gradients. Environ. Microbiol. 2015, 17, 3898–3913. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, L.; Tian, X.; Huang, H.; Yang, Q. Biodiversity study of intracellular bacteria closely associated with paralytic shellfish poisoning dinoflagellates Alexandrium tamarense and A. minutum. Int. J. Environ. Res. 2015, 4, 23–27. [Google Scholar] [CrossRef]
- Li, S.; Liu, Q.; Duan, C.; Li, J.; Sun, H.; Xu, L.; Yang, Q.; Wang, Y.; Shen, X.; Zhang, L. c-di-GMP inhibits the DNA binding activity of H-NS in Salmonella. Nat. Commun. 2023, 14, 7502. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. Wires. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar]
- Gustavsson, E.K.; Zhang, D.; Reynolds, R.H.; Garcia-Ruiz, S.; Ryten, M. ggtranscript: An R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 2022, 38, 3844–3846. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Web and Social Media, San Jose, CA, USA, 17–20 May 2009; pp. 361–362. [Google Scholar]
- Landry, Z.C.; Vergin, K.; Mannenbach, C.; Block, S.; Yang, Q.; Blainey, P.; Carlson, C.; Giovannoni, S.J. Optofluidic Single-Cell Genome Amplification of Sub-micron Bacteria in the Ocean Subsurface. Front. Microbiol. 2018, 9, 1152. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.J.; Kirchman, D.L. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J. 2013, 7, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Rippy, M.A.; Franks, P.J.; Feddersen, F.; Guza, R.T.; Moore, D.F. Physical dynamics controlling variability in nearshore fecal pollution: Fecal indicator bacteria as passive particles. Mar. Pollut. Bull. 2013, 66, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; He, R.; Cheng, Z.; Han, M.; Zha, Y.; Yang, P.; Yao, Q.; Zhou, H.; Zhong, C.; Ning, K. The seasonal dynamics and the influence of human activities on campus outdoor microbial communities. Front. Microbiol. 2019, 10, 1579. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zeng, L.; Boot, K.; Liu, Q. Satellite observed spatial and temporal variabilities of particulate organic carbon in the East China Sea. Remote Sens. 2022, 14, 1799. [Google Scholar] [CrossRef]
- Coclet, C.; Garnier, C.; Durrieu, G.; Omanović, D.; D’Onofrio, S.; Le Poupon, C.; Mullot, J.U.; Briand, J.F.; Misson, B. Changes in bacterioplankton communities resulting from direct and indirect interactions with trace metal gradients in an urbanized marine coastal area. Front. Microbiol. 2019, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, M.T.; Kirchman, D.L. Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low-and high-molecular-weight dissolved organic matter. Appl. Environ. Microbiol. 2000, 66, 1692–1697. [Google Scholar] [CrossRef]
- Wu, D.; Dai, Q.; Liu, X.; Fan, Y.; Wang, J. Comparison of bacterial community structure and potential functions in hypoxic and non-hypoxic zones of the Changjiang Estuary. PLoS ONE 2019, 14, e0217431. [Google Scholar] [CrossRef]
- DeLong, E.F.; Franks, D.G.; Alldredge, A.L. Phylogenetic diversity of aggregate-attached vs. Free-living marine bacterial assemblages. Limnol. Oceanogr. 1993, 38, 924–934. [Google Scholar] [CrossRef]
- Pinhassi, J.; Sala, M.M.; Havskum, H.; Peters, F.; Guadayol, O.; Malits, A.; Marrasé, C. Changes in bacterioplankton composition under different phytoplankton regimens. Appl. Environ. Microbiol. 2004, 70, 6753–6766. [Google Scholar] [CrossRef]
- Kirchman, D.L.; Keel, R.G.; Simon, M.; Welschmeyer, N.A. Biomass and production of heterotrophic bacterioplankton in the oceanic subarctic Pacific. Deep Sea Res. Part I Oceanogr. Res. Pap. 1993, 40, 967–988. [Google Scholar] [CrossRef]
- Karlusich, J.J.P.; Ibarbalz, F.M.; Bowler, C. Phytoplankton in the Tara Ocean. Ann. Rev. Mar. Sci. 2020, 12, 233–265. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, C.S.; Herfort, L.; Zuber, P.; Baptista, A.M.; Crump, B.C. Spatial variability overwhelms seasonal patterns in bacterioplankton communities across a river to ocean gradient. ISME J. 2012, 6, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.F.; Long, S.R. Rhizobium–plant signal exchange. Nature 1992, 357, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, B.; Yang, H.; Zhao, M.; He, B.; Zhang, X. Phylogenetic shifts of bacterioplankton community composition along the Pearl Estuary: The potential impact of hypoxia and nutrients. Front. Microbiol. 2015, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Dai, T.; Tang, Y.; Tao, Y.; Huang, B.; Mu, Q.; Wen, D. Sediment bacterial community structures and their predicted functions implied the impacts from natural processes and anthropogenic activities in coastal area. Mar. Pollut. Bull. 2018, 131, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hong, Y.; Zada, S.; Hu, Z.; Wang, H. Spatial variability and co-acclimation of phytoplankton and bacterioplankton communities in the pearl river estuary, China. Front. Microbiol. 2018, 9, 2503. [Google Scholar] [CrossRef]
- Wang, Z.; Juarez, D.L.; Pan, J.F.; Blinebry, S.K.; Gronniger, J.; Clark, J.S.; Johnson, Z.I.; Hunt, D.E. Microbial communities across nearshore to offshore coastal transects are primarily shaped by distance and temperature. Environ. Microbiol. 2019, 21, 3862–3872. [Google Scholar] [CrossRef]
- Yang, W.; Zheng, S.; Zhou, S.; Zhao, L.; Zhu, J.; Lukwambe, B.; Nicholaus, R.; Li, C.; Zheng, Z. Structure and Functional Diversity of Surface Bacterioplankton Communities in an Overwintering Habitat for Large Yellow Croaker, Pseudosciaena Crocea, of the Southern East China Sea. Front. Mar. Sci. 2020, 7, 472. [Google Scholar] [CrossRef]
- Zhang, X.L.; Tian, X.Q.; Ma, L.Y.; Feng, B.; Liu, Q.H.; Yuan, L.D. Biodiversity of the symbiotic bacteria associated with toxic marine dinoflagellate Alexandrium tamarense. J. Biosci. Med. 2015, 3, 23–28. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Station No. | Latitude | Longitude | Offshore Distance (km) | Depth/m | Temperature/°C | Salinity/psu | Oxygen/mg/L |
|---|---|---|---|---|---|---|---|
| N0 | 31.00 E | 122.20 N | 32.50 | 1.99 | 28.21 | 5.24 | 4.70 |
| N1 | 31.00 E | 122.50 N | 20.50 | 1.94 | 26.59 | 10.92 | 4.85 |
| M0 | 30.50 E | 122.20 N | 5.10 | 3.97 | 25.93 | 20.57 | 4.27 |
| M1 | 30.50 E | 122.50 N | 3.30 | 3.48 | 24.53 | 25.53 | 4.13 |
| Z1 | 30.20 E | 122.70 N | 1.00 | 1.49 | 25.26 | 30.06 | 5.77 |
| N6 | 31.00 E | 125.00 N | 242.30 | 3.49 | 29.40 | 28.64 | 5.62 |
| N7 | 31.00 E | 125.50 N | 297.80 | 3.97 | 28.95 | 29.21 | 4.72 |
| S5 | 30.00 E | 124.50 N | 191.80 | 2.94 | 29.29 | 28.54 | 5.43 |
| S6 | 30.00 E | 125.00 N | 247.30 | 3.01 | 29.77 | 30.36 | 4.70 |
| S7 | 30.00 E | 125.50 N | 302.90 | 2.03 | 29.56 | 30.99 | 4.63 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Xie, P.; Dai, J.; Zhang, L.; Yang, Q.; Zhang, X.; Yang, X. Variation in Structure and Functional Diversity of Surface Bacterioplankton Communities in the Eastern East China Sea. J. Mar. Sci. Eng. 2024, 12, 69. https://doi.org/10.3390/jmse12010069
Wang Z, Xie P, Dai J, Zhang L, Yang Q, Zhang X, Yang X. Variation in Structure and Functional Diversity of Surface Bacterioplankton Communities in the Eastern East China Sea. Journal of Marine Science and Engineering. 2024; 12(1):69. https://doi.org/10.3390/jmse12010069
Chicago/Turabian StyleWang, Zuochun, Pengfei Xie, Jun Dai, Lei Zhang, Qiao Yang, Xiaoling Zhang, and Xi Yang. 2024. "Variation in Structure and Functional Diversity of Surface Bacterioplankton Communities in the Eastern East China Sea" Journal of Marine Science and Engineering 12, no. 1: 69. https://doi.org/10.3390/jmse12010069
APA StyleWang, Z., Xie, P., Dai, J., Zhang, L., Yang, Q., Zhang, X., & Yang, X. (2024). Variation in Structure and Functional Diversity of Surface Bacterioplankton Communities in the Eastern East China Sea. Journal of Marine Science and Engineering, 12(1), 69. https://doi.org/10.3390/jmse12010069