Abstract
Bacteria are one of the causes of green rot disease (GRD) in Saccharina japonica mariculture, which may lead to complete failure of seedling production. However, the association between bacterial community and host disease severity remains largely unknown. Therefore, in this study, the bacterial communities associated with GRD-infected seedlings with naturally varying disease severity from two seedling hatcheries in Northern China were analyzed to investigate the interactions between bacterial communities and GRD. The results indicated incorrect nutrient supply in both sites. Gammaproteobacteria, Alphaproteobacteria, and Bacteroidetes were prevalent in all samples. Significant structural alterations were detected for epibacterial communities, which were further evidenced by differently abundant bacterial taxa associated with seedlings with varying disease severity. The predicted pathways of bacterial adhesion and antimicrobial compounds biosynthesis were significantly enriched in less severely diseased seedlings, whereas glutathione metabolism and lipopolysaccharide biosynthesis were significantly increased in more severely diseased seedlings. The predicted categories of a two-component system, flagellar assembly, bacterial chemotaxis, and biofilm formation were significantly enriched in the bacterioplankton in more severely infected seawater. The differential bacterial community compositions and predicted functions provide new clues to elucidate the mechanism underlying the interaction between GRD occurrence and bacterial communities.
1. Introduction
Epiphytic microorganisms, dominated by bacteria that closely interact with macroalgal species, are crucial for the sustainability of macroalgal health [1,2,3]. The functional capability of epiphytic microbes is known to have a direct influence on host health by providing CO2 and fixed nitrogen [1,2], growth or morphological factors [4,5,6], adaptation to different environments [7], and resistance to pathogens [8,9]. However, exposure to stressful environments (e.g., elevated temperature) could lead to unbalanced interactions between host defense, epiphytes, and environments, thus favoring disease occurrence, which has been well evidenced by the bleaching disease of the red alga Delisea pulchra [10]. Nevertheless, it is often difficult to attribute a particular macroalgal disease to a single pathogen, owing to the opportunistic nature of pathogens [11], polymicrobial infections in disease [12], the confusion between pathogens and saprophytes [1], etc. Moreover, some physical or chemical measures used for controlling macroalgal diseases, especially for the economically important macroalgal species, are neither sufficiently effective nor environmentally friendly [13]. In contrast, epiphytic bacterial strains exert promising protective effects on macroalgae against pathogens and/or dysbiosis [14,15]. Therefore, understanding the relationship between epiphytic microbiomes and macroalgal diseases is a current ambitious challenge in microbial ecology with applied consequences for the worldwide production of economically important macroalgal species [16].
Disease occurrence is often accompanied by changes in the microbial community, such as altered microbial abundance, composition, and function [12,17,18,19,20]. Bacterial community shifts, arising before physical signs of macroalgal diseases, can also be characterized by the enrichment of pathogenic bacteria and/or virulence-related functional genes [12]. Such bacterial virulence traits are typically composed of several aspects, including toxins, exoenzymes, adhesins, and secretion systems [21]. Moreover, the virulence factors might also include enzymes responsible for resistance to reactive oxygen species (ROS) [11]. The detection of functional genes related to bacterial pathogenicity in the microbiome of bleached red alga D. pulchra confirms the above-mentioned observations and the distribution of these genes in multiple bacterial species also suggests polymicrobial infections [12].
The kelp Saccharina japonica (Areschoug) C. E. Lane, C. Mayes, Druehl and G. W. Saunders 2006, is a commercially important Laminariales alga in the sea farming cultivation industry. China is the largest producer of S. japonica, contributing almost 90% of the world’s commercial volume (approximately more than 10 million tons) (FAO, 2021). The culture process of S. japonica is divided into two stages: indoor cultivation of seedlings and outdoor cultivation of mature sporophytes. The indoor seedling production usually takes about 2 months, including zoospore collection and development, gametophyte development and reproduction, and young sporophytes (juvenile sporeling) growth [22]. In recent years, the cultivated seedlings have been frequently affected by diseases owing to the continuous expansion of aquaculture and deterioration of the marine ecological environment, with green rot disease (GRD) being one of the major infections [18,23]. The symptoms of GRD are characterized by the loss of brown pigmentation at the tip of the seedling and gradual rotting of the whole frond, which quickly spreads to other seedlings causing massive losses or a complete failure of seedling production [18]. At present, GRD is considered to be caused by both environmental factors (i.e., insufficient sunlight) and bacteria. Although alginic acid decomposing bacteria are usually regarded as opportunistic pathogens during GRD infection owing to their ability to decompose alginate [23,24,25], specific information on GRD pathogenesis is still limited because of the detection of diverse bacterial genera with the ability to degrade alginate and induce similar symptoms to field observation [23,26]. Therefore, there is still a lack of in-depth research on the microbiome of GRD, especially analysis using in situ diseased samples.
In the present study, the bacterial communities associated with GRD-infected seedlings with varying disease severity from two distinct sites were investigated. Based on bacterial 16S rRNA gene amplicon sequencing, a series of analyses were implemented to (i) determine the composition and predicted functional profiles of the bacterial communities and (ii) explain the interactions between the bacterial communities and GRD.
2. Materials and Methods
2.1. Sample Collection
Samples were collected during a GRD outbreak from a seedling hatchery in Weihai City (WH), Shandong Province, China, in late September 2018, and from a seedling hatchery in Yantai City (YT), Shandong Province, China, in the middle of September 2020. The general information about the hatcheries is provided in Table S1.
The GRD occurrence in WH hatchery lasted approximately 15–20 days and caused seedlings to rot. Samples were approved to be collected on the 10th day when the seedlings had rotted to varying degrees in all the 200 tanks. Disease severity was evaluated based on the overall greenish degree of each tank, and five tanks with relatively lesser greenish level were grouped and identified as L, and five tanks with relatively more greenish level were grouped and identified as H. From each tank, approximately 3 L of seawater were collected (WH_Lw and WH_Hw for L and H tanks, respectively), after which several segments of palm ropes were cut from seedling collectors using sterilized dissecting scissors to obtain the seedling samples (WH_Lc and WH_Hc for L and H tanks, respectively).
Unlike WH hatchery, GRD in YT hatchery had caused seedlings to rot on several seedling collectors in only 1 of 60 tanks during the period of sample collection. Hence, only seedling samples were considered appropriate for sampling of biological replicates and epiphytic bacterial community analysis. Three types of segments of vinylon ropes were cut from seedling collectors to obtain the seedling samples. Segments without visible signs of rotten seedlings were identified as YT_Lc, those rotted to hold fewer seedlings were identified as YT_Mc, and those rotted to hold much fewer seedlings on vinylon ropes were identified as YT_Hc. A total of four YT_Lc, three YT_Mc, and three YT_Hc segments were collected for epibacterial community analysis. Seawater samples were collected from triplicate tanks, including the diseased tank, for parameter analysis.
All the samples were stored at low temperature (about 4 °C) and transported to the laboratory within 3 h, during which the seedlings were submerged in in situ seawater. The seedlings were washed thrice with sterilized seawater and stored at −80 °C for DNA extraction after histopathological observation. With regard to seawater from WH hatchery, 2 L from each tank were filtered through a 0.2 μm polycarbonate membrane (Isopore, Merck Millipore Ltd., Tullagreen, Ireland) and stored at −80 °C for epiphytic bacterial community DNA extraction.
2.2. Physicochemical Analysis
Seawater temperature, pH, and salinity were recorded at middle depth with appropriate sensors using a water quality sampling and monitoring meter (YSI Life Sciences, Yellow Springs, OH, United States). The concentrations of NO3−-N, PO43−-P, NO2−-N, and NH4+-N in each tank were determined with 1 L of seawater in three technical replicates using standard methods (GB/T 12763.4-2007) with colorimetric assays on a spectrophotometer (Mapada Ltd., Shanghai, China).
2.3. DNA Extraction and Amplification
DNA was extracted from seawater samples as described previously [19]. For epiphytic bacteria, DNA was selectively isolated from seedlings on the segments of the ropes (approximately 5–6 cm in length), according to a previously reported method [27]. The DNA quality and integrity were assessed by gel electrophoresis, and the DNA samples were stored at −20 °C prior to amplification.
The primers 515F and 926R were used to amplify the V4–V5 hypervariable regions of the 16S rRNA gene [28]. The amplicon libraries were constructed using the TruSeq Nano DNA LT Library Prep Kit (Illumina, San Diego, CA, United States), following the manufacturer’s recommendations, and index codes were added. Subsequently, the amplicon libraries were sequenced on a MiSeq PE300 sequencer (Illumina, San Diego, CA, United States), and 300-bp paired-end reads were generated.
2.4. Raw Sequence Processing
The resulting paired sequence reads were merged, truncated, filtered, and clustered into operational taxonomic units (OTUs) using USEARCH version 10.0.240 [29]. Sequences with similarity ≥97% were assigned to the same OTU by the UPARSE-OTU algorithm in USEARCH using the “cluster_otus” command. Chimeras and singletons were removed in USEARCH for further analysis. Taxonomy assignment for representative OTU sequence was performed in QIIME version 1.9.0 [30] using the Ribosomal Database Project (RDP) classifier [31] against the SILVA_132 database (bootstrap confidence 0.8). Sequences of plastids and mitochondria, as well as those not classified in the domain bacteria, were discarded. A rarefied OTU table at the same depth was used to calculate α- and β-diversities in QIIME. Bacterial α-diversity was evaluated using Good’s coverage, Observed species (richness), and Pielou index (evenness), while β-diversity was measured using Bray-Curtis dissimilarity metrics.
2.5. Statistical Analyses
General statistical analyses were performed in R version 4.0.3 [32]. An unpaired t-test was used to examine the significant difference in the physicochemical parameters between the tanks. Principle coordinate analysis (PCoA) was performed to determine the differences in the bacterial community structures based on Bray-Curtis distances, and permutational multivariate analysis of variance (PERMANOVA) was conducted to quantitatively evaluate the effects of disease severity on the variations in the bacterial communities. One-way analysis of variance (ANOVA) was applied to evaluate the significant differences in the bacterial α-diversity indices between the communities. Redundancy analysis (RDA) was performed to investigate the relationships between the bacterial communities and environmental factors. Environmental variables with a variance inflation factor (VIF) > 10 were eliminated to avoid collinearity among the factors, and/or a forward selection was conducted using the “ordiR2step” function in the “vegan” package (version 2.5-7) [33] to select explanatory variables with significant explaining factors (p < 0.05) for further analyses.
Linear discriminant analysis effect size (LEfSe) [34], a method for biomarker detection, was used to determine the bacterial taxa that best characterized each study group. In the present study, taxa with linear discriminant analysis (LDA) score > 3 and p < 0.05 were considered to be significant.
The OTU table was normalized by dividing the abundance of each OTU by its predicted 16S rRNA gene copy number to produce the KEGG orthology (KO) functional categories by using PICRUSt2 [35]. STAMP version 2.1.3 was used to compare differently abundant functions between groups, and a Welch’s t-test was employed to test difference significance (p < 0.01) [36]. Functional pathways were manually checked, and unrelated categories were excluded from statistical analysis.
3. Results
3.1. Histopathological Observation
In the WH_Lc group, the majority of the seedlings on the palm ropes were brown and appeared normal (Figure 1A). In contrast, In the WH_Hc group, the greenish color spread to most of the seedlings and even to their lower parts (Figure 1A and Figure S1A). In the YT_Lc group, the majority of the seedlings exhibited no visible rotted signs, similar to those in the WH_Lc group (Figure 1B). However, in the YT_Mc group, a number of seedlings were notably greenish at their apical parts (Figure 1B and Figure S1B). In the YT_Hc group, the greenish color spread to most parts of the seedlings, similar to that noted in the WH_Hc group (Figure 1B and Figure S1C). All the observed symptoms were highly consistent with those of GRD.
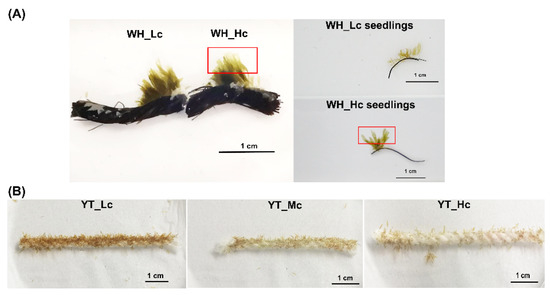
Figure 1.
Comparisons of seedlings from Weihai (WH) (A) and Yantai (YT) (B), respectively. The area marked by the red box indicates the rotten site.
3.2. Physicochemical Characteristics of Seawater
The common physicochemical characteristics of seawater frequently determined in seedling hatcheries are summarized in Table 1. With regard to seawater sampled from WH hatchery, four of the detected parameters were significantly different between the L and H tanks, including water temperature (7.82 °C ± 0.26 °C vs. 8.28 °C ± 0.19 °C), salinity (35.20 ± 0.45‰ vs. 34.00 ± 0.00‰), PO43−-P concentration (2.173 ± 0.088 mg/L vs. 2.202 ± 0.024 mg/L), and NO2−-N concentration (0.434 ± 0.001 mg/L vs. 0.432 ± 0.002 mg/L). However, the concentrations of NO3−-N and PO43−-P in all the tanks were almost eight and five times higher than those usually employed for seedling production (N: 3.5–4.5 mg/L, P: 0.35–0.45 mg/L; Standard protocol DB37/T 1190—2009), respectively. With respect to seawater sampled from the YT hatchery, the PO43−-P concentration (1.288 ± 0.037 mg/L) was also significantly higher than the standard requirements, whereas NO3−-N concentration (0.720 ± 0.126 mg/L) was significantly lower than the standard protocol. Thus, in both of the studied hatcheries, an inaccurate supply of nutrients was determined.

Table 1.
Physicochemical characteristics of seawater from Weihai (WH) and Yantai (YT).
3.3. Bacterial Diversities and Community Compositions
A total of 549,669 high-quality sequences were obtained for the samples from the WH hatchery. After the removal of archaea, chloroplasts, mitochondria, and unassigned sequences, 470,701 sequences were classified as bacteria, which were clustered into 1482 OTUs. The number of clean reads in each sample ranged from 15,200 to 34,050 (mean ± standard deviation = 47,070 ± 4626 sequences/sample) (Table S2), and 15,200 sequences were randomly selected to standardize the sequencing depth. With regard to samples from the YT hatchery, a total of 274,409 high-quality sequences were acquired, with 72,691 bacterial sequences (7296 ± 3115 sequences/sample) obtained after the removal of archaea, plastids, and unassigned sequences. Moreover, samples with sequences <3000 were also removed for further analysis, which led to 475 binned OTUs and a rarefaction depth of 3788 sequences for the remaining nine samples (i.e., three YT_Lc, three YT_Mc, and three YT_Hc) (Table S2).
The α-diversity was measured using Good’s coverage, Observed species, and Pielou index. Good’s coverage for all the samples was >98% (98.69% ± 0.28%), indicating that all the sequencing depths were reasonable and could reliably reflect the characteristics of the bacterial communities. When considering GRD severity, epibacterial richness showed a slightly decreasing trend, although no significant difference could be observed between the sample groups. In particular, WH_Lc seedlings exhibited higher richness than WH_Hc seedlings (Figure 2A), similar to that noted between YT_Lc seedlings and YT_Mc and YT_Hc seedlings (Figure 2B). With respect to bacterial evenness, a decreasing trend was observed among YT_Lc, YT_Mc, and YT_Hc seedlings (Figure 2B), whereas no obvious trend was noted between WH_Lc and WH_Hc seedlings (Figure 2A). With regard to bacterial communities in WH seawater, both bacterial richness and evenness were lower than those of epibacterial communities, and an increasing trend was observed in parallel with GRD severity (Figure 2A).
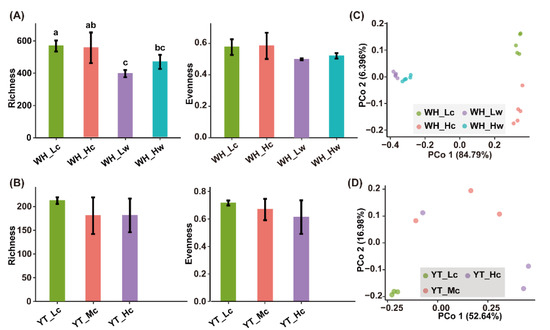
Figure 2.
Bacterial α- and β-diversities associated with seedling and seawater samples. (A,C), α-Diversity and Principal coordinate analysis for WH samples, respectively; (B,D), α-Diversity and Principal coordinate analysis for YT samples, respectively. Letters on the error bars indicate significant difference between sample groups determined by ANOVA with a Tukey’s HSD test (p < 0.05).
The β-diversity was analyzed using Bray-Curtis distance. A clear difference was observed between epiphytic and planktonic bacterial communities in the WH samples, and the PCo1 axis explained 84.79% of the variations (Figure 2C). Moreover, both epiphytic and planktonic bacterial communities in the L tanks were clearly separated from those in the H tanks (Figure 2C), which was also confirmed by the pairwise comparison results (Table S3). Similarly, in the YT samples, the epiphytic bacterial communities were clearly clustered in accordance with disease severity (PERMANOVA, R = 0.542 and p = 0.042 < 0.05), and the PCo1 axis explained 52.64% of the variations (Figure 2D), which was also confirmed by pairwise comparisons (Table S3).
A total of 28 phyla were detected in the WH samples, with 25 phyla detected in both seedlings and seawater. A total of 17 phyla were detected in the YT seedlings. In all the samples, the dominant bacterial taxa were affiliated with classes Gammaproteobacteria (50.23% ± 13.32%), Alphaproteobacteria (25.45% ± 8.92%), and phylum Bacteroidetes (17.85% ± 9.26%) (Figure 3). More importantly, the relative abundances of the most prevalent taxa varied with different disease status of the seedlings, indicating changes in the bacterial community with disease severity (Figure 3).
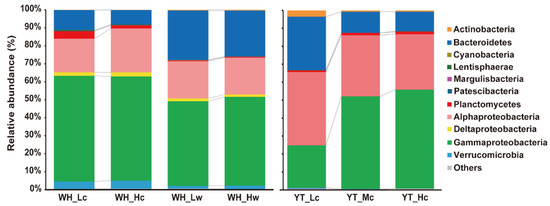
Figure 3.
Dominant phylum or Proteobacterial classes of epiphytic and planktonic bacterial communities. Taxa with relative abundance <0.05% in all datasets and those are not assigned to any known taxa in the current Silva database are merged as “Others”.
3.4. Significant Differences between Bacterial Communities
To further determine the differences in the bacterial taxonomic abundance, the LefSe method was used to compare the bacterial communities in the two study sites (Figure 4, Figures S2 and S3).
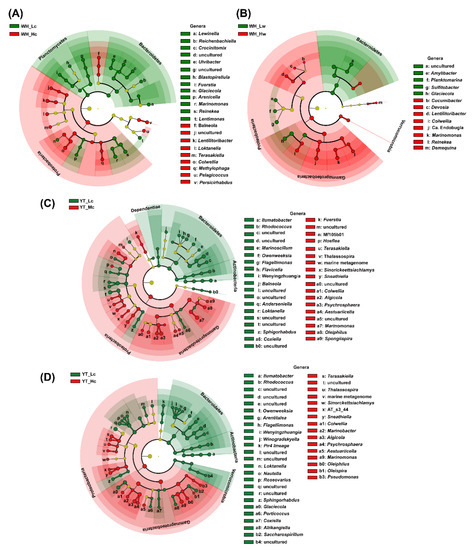
Figure 4.
Significant differences in bacterial community composition showed using LefSe cladogram at the phylum and genus levels. (A) Significantly different taxa between WH_Lc and WH_Hc; (B) Significantly different taxa between WH_Lw and WH_Hw. (C) Significant different taxa between YT_Lc and YT_Mc seedlings; (D) Significantly different taxa between YT_Lc and YT_ Rc seedlings.
Among the WH seedlings samples, the relative abundances of 55 bacterial taxa significantly varied between WH_Lc and WH_Hc groups (Figure S2). Disease severity led to an enrichment in the proportion of sequences belonging to Proteobacteria (WH_Lc: 79.54% ± 2.76%; WH_Hc: 84.84% ± 3.81%), and a reduction in the proportion of sequences belonging to Bacteroidetes (WH_Lc: 11.12% ± 1.09%; WH_Hc: 8.06% ± 2.05%) and Planctomycetes (WH_Lc: 4.23% ± 1.07%; WH_Hc: 1.79% ± 0.61%) (Figure 4A). At the genus level, 22 genera were found to be significantly different between WH_Lc and WH_Hc seedlings, among which nine and 13 genera were enriched in WH_Hc and WH_Lc seedlings, respectively (Figure 4A). Within these genera, Reinekea, Marinomonas, and Lewinella were more enriched in WH_Lc seedlings, and Colwellia, Loktanella, and Lentilitoribacter were more enriched in WH_Hc seedlings, although all of them were relatively abundant (mean relative abundance > 1%) in all of the seedlings samples (Figure 4A and Table S4). Among the WH seawater samples, the relative abundances of 35 taxa were significantly different between WH_Lw and WH_Hw (Figure 4B). The relative abundances of planktonic Verrucomicrobia (WH_Lw: 1.50% ± 0.19%, WH_Hw: 1.84% ± 0.20%) and Gammaproteobacteria (WH_Lw: 47.09% ± 0.98%; WH_Hw: 49.34% ± 0.89%) significantly increased with the disease severity, whereas the opposite trend was noted with regard to the relative abundance of Bacteroidetes (WH_Lw: 27.45% ± 0.77%; WH_Hw: 25.42% ± 1.08%) (Figure 4B). The relative abundances of 13 genera were significantly different between WH_Lw and WH_Hw (Figure 4B), while the relative abundances of Glaciecola, Lentilitoribacter, Reinekea, Marinomonas, and Colwellia were significantly different between WH_Lc and WH_Hc seedlings (Figure 4).
Among the YT seedlings, the relative abundances of 87 bacterial taxa were significantly different between YT_Lc and YT_Mc seedlings, whereas those of 92 bacterial taxa were significantly different between YT_Lc and YT_Hc seedlings (Figure S3). Disease severity led to an increase in the relative abundance of Proteobacteria, notably, Gammaproteobacteria (YT_Lc: 23.65% ± 0.70%; YT_Mc: 51.62% ± 19.71%; YT_Hc: 55.06% ± 20.03%), and a decrease in the relative abundances of Bacterodetes (YT_Lc: 29.79%; YT_Mc: 11.81%; YT_Hc: 10.87%) and Actinobacteria (YT_Lc: 3.47%; YT_Mc: 0.75%; YT_Hc: 0.72%) (Figure 4C,D). At the genus level, the relative abundances of 37 and 41 genera were significantly different between YT_Lc and YT_Mc seedlings (Figure 4C) and between YT_Lc and YT_Hc seedlings (Figure 4D), respectively. Although the predominant genera such as Marinomonas, Algicola, Sneathiella, Terasakiella, Oleiphilus, and Colwellia, were significantly enriched in both YT_Mc and YT_Hc seedlings (Figure 4 and Table S5), their proportions varied to a certain extent between the two groups. For example, Spongiispira, Fuerstia, Hoeflea, etc., were significantly enriched in YT_Mc seedlings (Figure 4C), whereas Pseudomonas, Olespira, and Marinobacter were significantly enriched in YT_Hc seedlings (Figure 4D).
3.5. Predicted Bacterial Functions
In the epiphytic bacteria datasets for the WH samples, 10 significantly enriched KO functional categories, including metabolism, biosynthesis, signaling, and invasion pathways, were detected in H and L tanks, respectively (Figure 5A). In particular, bacterial adhesion (adherens junction) was significantly enriched in WH_Lc seedlings, while the glutathione (GSH) metabolism pathway was significantly increased in WH_Hc seedlings. Moreover, pathways related to the biosynthesis of secondary metabolites, such as phenazine, stilbenoid, diarylheptanoid, gingerol, and flavonoid, were also enriched in WH_Lc seedlings (Figure 5A). In the planktonic bacteria datasets for the WH samples, 32 KO functional categories were determined to be significantly different between L and H tanks, with 15 categories detected in the WH_Hw group (Figure 5B). Most of the first 10 functional components, such as signal transduction (two-component system), cell mobility (flagellar assembly and bacterial chemotaxis), and biofilm formation (related to Escherichia coli, Pseudomonas aeruginosa, and V. cholerae), etc. were significantly enriched in the H tanks (8 of 10 functional components).
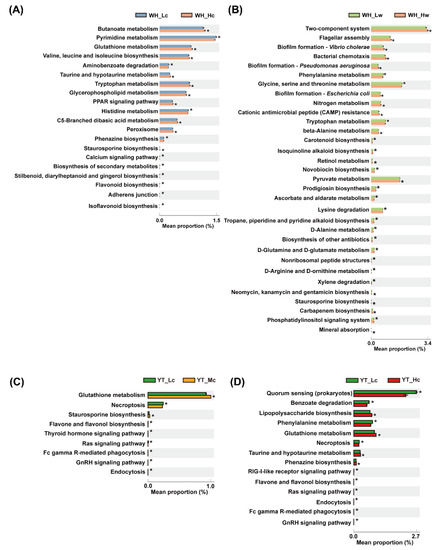
Figure 5.
Significant differences in predicted functional categories between study groups. (A,B) Significantly different functional profiles between WH_Lc and WH_Hc, and WH_Lw and WH_Hw, respectively. (C,D) Significant differences in predicted functional categories between YT_Lc and YT_Mc seedlings, and YT_Lc and YT_Hc seedlings, respectively. The asterisk on the bar represents that the functional pathway is significantly enriched in this experimental group (Welch’s t-test, p < 0.01).
In the epiphytic bacteria datasets for the YT samples, only two functional categories, including GSH metabolism and staurosporine biosynthesis, were significantly enriched in YT_Mc seedlings when compared with those in YT_Lc seedlings (Figure 5C). A total of seven predicted categories, mainly involving signaling pathways and cellular processes (i.e., necroptosis and endocytosis), were significantly enriched in YT_Lc seedlings when compared with those in YT_Mc seedlings (Figure 5C). Furthermore, a total of 14 categories were significantly different between YT_Lc and YT_Hc seedlings (Figure 5D). In particular, functional categories related to quorum sensing (QS) were significantly increased in YT_Lc seedlings, while those associated with lipopolysaccharide (LPS) biosynthesis and GSH metabolism were significantly enriched in YT_Hc seedlings (Figure 5D). Moreover, the pathway of biosynthesis of flavonoids (flavone and flavonol) was significantly enriched in YT_Lc seedlings when compared with that in YT_Hc and YT_Mc seedlings (Figure 5C,D).
3.6. Correlation between Bacterial Community Structure and Environmental Factors
By using both canonical correspondence analysis (CCA)-based VIF and forward selection, variables important to community structure distributions in the WH samples were identified. For the epibacterial communities, the variations were significantly correlated with salinity (p = 0.001) and temperature (p = 0.049). In particular, the bacterial communities in WH_Lc samples were positively correlated with salinity, while those in WH_Hc samples were positively correlated with temperature (Figure 6A). With regard to planktonic bacterial communities, temperature (p = 0.020) was the only environmental factor that was significantly associated with bacterial community variations. The RDA biplot showed that the bacterial communities in WH_Hw samples were positively correlated with temperature (Figure 6B).
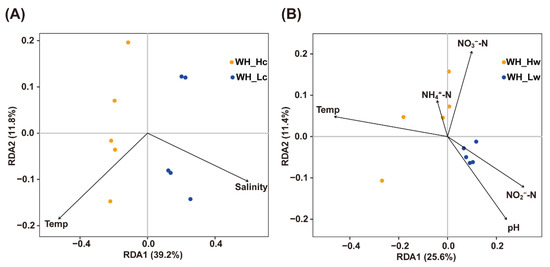
Figure 6.
Redundancy analysis (RDA) for bacterial communities and environmental factors in WH. Community datasets associated with seedlings (A) and seawater (B) were used.
4. Discussion
In the present study, two distinct seedling hatcheries that had reported GRD outbreaks were examined, and the association between bacterial communities and GRD severity was investigated and compared.
Algal diseases are the consequence of the complex interplay between environments, algae, and microorganisms, and the external environments tend to play significant roles in disease onset [37,38]. In S. japonica seedlings, insufficient sunlight irradiation combined with alginate-decomposing bacteria has been proposed as the cause of GRD onset [18,23]. However, it has been proved that incorrect nutrient supply, possibly owing to non-standard operation, is one of the primary causes of GRD [39]. The abnormal nutrient levels observed in the present study possibly expanded the spectrum of environmental stressors that lead to GRD occurrence, with multiple environmental changes being considered as stressors favoring the onset of ice-ice disease [40]. Moreover, significant differences were noted in the detected parameters, such as temperature, salinity, PO43−-P, and NO23−-N, between the L and H tanks in the WH hatchery, indicating to a certain extent, the non-standard management of the hatchery and the correlation between different degrees of community dysbiosis and disease severity (Figure 2 and Figure 6). Furthermore, the isolated alginate-decomposing bacteria did lead to higher disease severity in the seedlings, demonstrating the vital role of such bacteria in GRD [25,39]. However, alginate-decomposing bacteria might not be the true causative agents of GRD because the ability to decompose algal tissues is only one of the aspects associated with the virulence of macroalgal pathogens [37], and these bacteria isolated from diseased samples might possibly be saprophytes. Therefore, to identify seaweed pathogens, research must focus on the relationship between virulence factors and pathogenicity rather than on the decomposing activity alone.
A higher bacterial richness was observed in the WH_Lc and YT_Lc seedlings, which decreased with disease severity. A similar trend had also been reported for the epibacterial communities in the red algae Pyropia yezoensis infected with oomycetes pathogen [19] as well as the soil microbiome of infected potato [41]. Higher bacterial community diversity is often associated with greater resistance to microbial invasions [42]. It is believed that macroalgal hosts might possibly recruit bacteria to resist pathogen invasion. Exposure to stressful environments (such as inappropriate nutrient supply) can compromise the host defense, allowing many more chances of pathogen infections. In such a context, the algal host may require more protective strength from the associated bacteria, which is one of the constitutive defense capabilities of macroalgae [37]. A significant proportion of planktonic bacteria colonize and actively interact with macroalgae and secrete antimicrobial compounds or QS inhibitors, which can assist the algal host in resisting microbial invasion [2,8]. Consequently, the algal host might recruit more of these bacteria to achieve resistance to pathogen invasions. This postulation is, to a certain extent, supported by the findings of the present study, which revealed enrichment of pathways related to secondary metabolites in WH_Lc and YT_Lc seedlings. Secondary metabolites such as flavonoids (particularly flavones and flavonols) are promising antibiotics that are more effective than standard antibiotics because they can protect the host through various strategies, including inhibition of bacterial adhesion and invasion, biofilm formation, multidrug resistance pumps, etc. [43]; diarylheptanoids have both antibacterial and antifungal activities [44]; gingerol and stilbenoid are capable of reducing biofilm formation and/or virulence via inhibiting QS activity of bacterial pathogens [45,46].
Multiple lines of evidence have demonstrated that macroalgal diseases result from the proliferation of opportunistic pathogens when there is dysbiosis in the host microbiome owing to environmental pressures [47,48]. In certain cases, dysbiosis is characterized by the rise of pathogenic bacteria [18,20,49] and/or enrichment of virulence factors [12,17]. In the present study, comparisons of the bacterial community dysbiosis in the two hatcheries were performed. Within the differently abundant genera in WH seedlings, data on their roles as macroalgal pathogens are limited. Nevertheless, most of these bacterial genera, such as Reinekea [50], Marinomonas [51], Lewinella [52], Colwellia [51], and Loktanella [53], are well-known degraders of organic compounds (e.g., algal-polysaccharides), and alterations in their relative abundance from WH_Lc to WH_Hc seedlings indicated that different profiles of degraders might be involved in different stages of GRD. With regard to bacterial communities in the YT samples, structural comparisons revealed two potentially pathogenic bacterial genera, namely, Hoeflea and Pseudomonas, with Hoeflea being associated with coral disease [54,55] and Pseudomonas being the causative agent of seaweed disease in S. japonica and P. yezoensis [56]. However, it must be noted that the pathogenicity of these two bacteria is mostly based on compositional differences or degradation abilities. Therefore, it is still unclear whether these bacteria are only saprophytes or the real causative agents of algal diseases because their degradation abilities have been documented in the CAZY database (http://www.cazy.org) (accessed on 14 February 2022), suggesting that disease severity on algal seedlings contributed to the involvement of different profiles of degraders.
In order to obtain more information on GRD pathogenesis, function prediction was performed in the present study using PICRUSt2 based on the current 16S rRNA data. However, one of the main limitations of PICRUSt2 is that certain environment-related functions are less likely to be identified based on the existing reference genomes [35]. Provided the limited genomic data on seaweed and seagrass-associated microbiome [57,58,59], the predictions in the present study might possibly be biased to a certain extent. Nevertheless, the predictions provided some information regarding potential virulence by comparing samples with varying disease severity. For example, the enriched adherens junction in WH_Lc and YT_Lc seedlings might suggest potential bacterial invasions when there is dysbiosis in the epibacterial communities under stressful environments. In addition, a significant enrichment of pathway involving GSH metabolism in more severely diseased seedlings from both the study sites might also possibly indicate bacterial invasions. GSH is often considered the most potent natural antioxidant, which can be metabolized by a series of enzymes in the GSH system [60]. For instance, the enzymes of the GSH system have been found to detoxify ROS produced by the host during bacterial invasion [37,60], such as the macroalgal pathogen Nautella sp. R11 [11]. Moreover, an increase in LPS biosynthesis in more severely diseased seedlings (i.e., YT_Hc seedlings) might also signify bacterial invasions. LPS, also known as glycolipid, is a pathogenicity determinant in bacteria because of its remarkable endotoxicity to host cells, which can induce host defense responses [61]. These findings indicate that disease occurrence is correlated with bacterial invasions in addition to the altered assembly of epibacterial communities owing to environmental stressors.
Furthermore, the present study also found changes in the planktonic bacterial communities with GRD occurrence. In addition to differently abundant bacterial taxa coinciding with epibacterial members, a certain number of predicted functional categories that were more enriched in the WH_Hw samples were also found to be involved in bacterial virulence. For example, the flagellum is capable of helping the bacteria to reach the optimal host site, colonize or invade, remain at the infection site, and achieve post-infection dispersal [62]; chemotaxis aids in the initial stages of infection (e.g., host tissue penetration) by various types of human, animal, and plant pathogens [63]; two-component signal transduction systems in bacteria regulate the expression of metabolic and virulence genes in response to changing environments [64]; and biofilm formation elicits bacterial resistance to antibiotic treatment, thus ensuring their pathogenicity [65]. Based on these observations, it can be hypothesized that a certain proportion of planktonic bacteria might be involved in the progress of GRD. Nevertheless, further studies are still required to verify the extent to which planktonic bacterial communities are involved in GRD and their roles in disease progression.
5. Conclusions
This study explored the bacterial communities associated with S. japonica seedlings infected in situ with GRD at varying severity. By using 16S rRNA gene amplicon sequencing, epibacterial communities were determined to be markedly clustered in parallel with disease severity. The significant structural changes were also evidenced by variations in the abundance of bacterial taxa in seedlings exhibiting different disease severity. The predicted pathways related to bacterial adhesion and biosynthesis of antimicrobial compounds were significantly enriched in less severely diseased seedlings, whereas pathways indicating bacterial invasions, such as GSH metabolism and LPS biosynthesis, were significantly enriched in more severely diseased seedlings. The potential virulence-associated categories, such as two-component system, flagellar assembly, bacterial chemotaxis, and biofilm formation, were also significantly enriched in the bacterioplankton in more severely infected tanks. These findings broaden the understanding of the relationships between GRD occurrence and bacterial communities and provide novel insights into GRD occurrence. However, as the present study performed comparisons only based on predictions with 16S rRNA gene sequences, further comprehensive metagenomic analysis is necessary to reveal the underlying microbial ecological mechanisms related to GRD. Moreover, an in-depth functional analysis is also required to determine pathogen(s) and invasion stages of GRD because symptom observation is insufficient for such investigations.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jmse10060730/s1, Figure S1: Microscopic observation of diseased seedlings; Figure S2: Significant differences in bacterial community composition between L and H tanks in Weihai (WH) determined using LefSe. Figure S3: Significant differences in bacterial community composition between different seedlings in Yantai (YT) determined using LefSe; Table S1: General information for the hatcheries in Weihai (WH) and Yantai (YT), respectively; Table S2: General statistics for analyzed 16s rRNA gene sequences of all samples; Table S3: Pairwise comparison results based on Bray-Curtis distances for bacterial communities in WH and YT, respectively; Table S4: Proportions of the top 30 bacterial genera in seedlings and seawater from the WH hatchery; Table S5: Proportions of the top 30 bacterial genera in seedlings from the YT hatchery.
Author Contributions
Conceptualization, Y.Y. and Z.M.; methodology, Y.Y. and Z.M.; validation, Z.M.; investigation, Y.Y., S.W., F.L. and J.L.; resources, S.W. and F.L.; formal analysis, Y.Y.; visualization, Y.Y. and S.W.; data curation, Y.Y. and J.L.; writing—original draft preparation, Y.Y.; writing—review and editing, S.W., F.L. and Z.M.; project administration, Z.M.; funding acquisition, Z.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by AGRICULTURE RESEARCH SYSTEM (CARS-50), the FUNDAMENTAL RESEARCH FUNDS FOR THE CENTRAL UNIVERSITIES (202064006), and SHANDONG PROVINCIAL NATURAL SCIENCE FOUNDATION, CHINA (ZR2019BD060). The APC was funded by AGRICULTURE RESEARCH SYSTEM.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive [66] in National Genomics Data Center [67], China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA006338) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa (accessed on 11 March 2022).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Egan, S.; Harder, T.; Burke, C.; Steinberg, P.; Kjelleberg, S.; Thomas, T. The seaweed holobiont: Understanding seaweed-bacteria interactions. FEMS Microbiol. Rev. 2013, 37, 462–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goecke, F.; Labes, A.; Wiese, J.; Imhoff, J.F. Chemical interactions between marine macroalgae and bacteria. Mar. Ecol. Prog. Ser. 2010, 409, 267–299. [Google Scholar] [CrossRef]
- Singh, R.P.; Reddy, C.R.K. Unraveling the functions of the macroalgal microbiome. Front. Microbiol. 2016, 6, 1488. [Google Scholar] [CrossRef] [PubMed]
- Dimitrieva, G.Y.; Crawford, R.L.; Yuksel, G.U. The nature of plant growth-promoting effects of a pseudoalteromonad associated with the marine algae Laminaria japonica and linked to catalase excretion. J. Appl. Microbiol. 2006, 100, 1159–1169. [Google Scholar] [CrossRef] [Green Version]
- Spoerner, M.; Wichard, T.; Bachhuber, T.; Stratmann, J.; Oertel, W. Growth and thallus morphogenesis of Ulva mutabilis (Chlorophyta) depends on a combination of two bacterial species excreting regulatory factors. J. Phycol. 2012, 48, 1433–1447. [Google Scholar] [CrossRef]
- Yong, J.J.J.Y.; Chew, K.W.; Khoo, K.S.; Show, P.L.; Chang, J. Prospects and development of algal-bacterial biotechnology in environmental management and protection. Biotechnol. Adv. 2021, 47, 107684. [Google Scholar] [CrossRef]
- Dittami, S.M.; Duboscq-Bidot, L.; Perennou, M.; Gobet, A.; Corre, E.; Boyen, C.; Tonon, T. Host–microbe interactions as a driver of acclimation to salinity gradients in brown algal cultures. ISME J. 2016, 10, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Busetti, A.; Maggs, C.A.; Gilmore, B.F. Marine macroalgae and their associated microbiomes as a source of antimicrobial chemical diversity. Eur. J. Phycol. 2017, 52, 452–465. [Google Scholar] [CrossRef]
- Singh, R.P.; Kumari, P.; Reddy, C.R.K. Antimicrobial compounds from seaweeds-associated bacteria and fungi. Appl. Microbiol. Biot. 2015, 99, 1571–1586. [Google Scholar] [CrossRef]
- Campbell, A.H.; Harder, T.; Nielsen, S.; Kjelleberg, S.; Steinberg, P.D. Climate change and disease: Bleaching of a chemically defended seaweed. Global Change Biol. 2011, 17, 2958–2970. [Google Scholar] [CrossRef]
- Fernandes, N.; Case, R.J.; Longford, S.R.; Seyedsayamdost, M.R.; Steinberg, P.D.; Kjelleberg, S.; Thomas, T. Genomes and virulence factors of novel bacterial pathogens causing bleaching disease in the marine red alga Delisea pulchra. PLoS ONE 2011, 6, e27387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zozaya Valdés, E.; Roth Schulze, A.J.; Egan, S.; Thomas, T. Microbial community function in the bleaching disease of the marine macroalgae Delisea pulchra. Environ. Microbiol. 2017, 19, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Moon, K.; Kim, J.; Shim, J.; Klochkova, T.A. A revaluation of algal diseases in Korean Pyropia (Porphyra) sea farms and their economic impact. Algae 2014, 29, 249–265. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Majzoub, M.E.; Marzinelli, E.M.; Dai, Z.; Thomas, T.; Egan, S. Bacterial controlled mitigation of dysbiosis in a seaweed disease. ISME J. 2021, 16, 378–387. [Google Scholar] [CrossRef]
- Saha, M.; Weinberger, F. Microbial “gardening” by a seaweed holobiont: Surface metabolites attract protective and deter pathogenic epibacterial settlement. J. Ecol. 2019, 107, 2255–2265. [Google Scholar] [CrossRef]
- Gachon, C.M.M.; Sime-Ngando, T.; Strittmatter, M.; Chambouvet, A.; Kim, G.H. Algal diseases: Spotlight on a black box. Trends Plant Sci. 2010, 15, 633–640. [Google Scholar] [CrossRef]
- Fernandes, N.; Steinberg, P.; Rusch, D.; Kjelleberg, S.; Thomas, T. Community structure and functional gene profile of bacteria on healthy and diseased thalli of the red seaweed Delisea pulchra. PLoS ONE 2012, 7, e50854. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Pang, S.; Shan, T.; Su, L. Changes of microbial community structures associated with seedlings of Saccharina japonica at early stage of outbreak of green rotten disease. J. Appl. Phycol. 2020, 32, 1323–1327. [Google Scholar] [CrossRef]
- Yan, Y.; Yang, H.; Tang, L.; Li, J.; Mao, Y.; Mo, Z. Compositional shifts of bacterial communities associated with Pyropia yezoensis and surrounding seawater co-occurring with red rot disease. Front. Microbiol. 2019, 10, 1666. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Chang, L.; Xiao, L.; Zhang, X.; Han, Q.; Li, N.; Egan, S.; Wang, G. Diversity of the epiphytic bacterial communities associated with commercially cultivated healthy and diseased Saccharina japonica during the harvest season. J. Appl. Phycol. 2020, 32, 2071–2080. [Google Scholar] [CrossRef]
- Brown, S.P.; Cornforth, D.M.; Mideo, N. Evolution of virulence in opportunistic pathogens: Generalism, plasticity, and control. Trends Microbiol. 2012, 20, 336–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Sun, X.; Wang, G.; Xu, P.; Wang, X.; Lin, Z.; Wang, F. Effect of blue light on indoor seedling culture of Saccharina japonica (Phaeophyta). J. Appl. Phycol. 2010, 22, 737–744. [Google Scholar] [CrossRef]
- Wang, G.; Lu, B.; Shuai, L.; Li, D.; Zhang, R. Microbial diseases of nursery and field-cultivated Saccharina japonica (Phaeophyta) in China. Algol. Stud. 2014, 145, 39–51. [Google Scholar] [CrossRef]
- Lin, W.; Zhang, W.; Yan, X.; Duan, D. Distribution and reinfection of alginic acid decomposing bacteria on juvenile Laminaria japonica. Oceanologia Et Limnologia Sinica 2004, 35, 562–567. [Google Scholar]
- Wang, L.; Tang, X.; Wang, M.; Zhang, P. The roles played by alginic acid decomposing bacteria during the time of green decay disease of Laminaria japonica. J. Ocean Univ. Qingdao 2003, 32, 245–248. [Google Scholar]
- Ahmad, R.; Chen, Y.; Zhuang, Y.; Qiu, Q.; Chen, D.; Saha, M.; Wu, H.; Wang, G. Isolation and identification of a pathogenic bacterium, Exiguobacterium oxidotolerans XP-2, from the abnormal diseased mature sporophytes of a commercially cultivated brown seaweed Saccharina japonica. J. Appl. Phycol. 2021, 33, 3239–3249. [Google Scholar] [CrossRef]
- Burke, C.; Kjelleberg, S.; Thomas, T. Selective extraction of bacterial DNA from the surfaces of macroalgae. Appl. Environ. Microbiol. 2009, 75, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 2016, 1, e00009-15. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. Available online: http://www.drive5.com/usearch (accessed on 28 April 2022). [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. Available online: http://qiime.org (accessed on 28 April 2022). [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2014. Available online: https://CRAN.R-project.org (accessed on 28 April 2022).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Community Ecology Package. Available online: https://CRAN.R-project.org/package=vegan (accessed on 28 April 2022).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. Available online: http://huttenhower.sph.harvard.edu/galaxy (accessed on 28 April 2022). [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. Available online: https://github.com/picrust/picrust2 (accessed on 28 April 2022). [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. Available online: https://beikolab.cs.dal.ca/software/STAMP (accessed on 28 April 2022). [CrossRef] [PubMed] [Green Version]
- Egan, S.; Fernandes, N.D.; Kumar, V.; Gardiner, M.; Thomas, T. Bacterial pathogens, virulence mechanism and host defence in marine macroalgae. Environ. Microbiol. 2014, 16, 925–938. [Google Scholar] [CrossRef] [Green Version]
- Egan, S.; Gardiner, M. Microbial dysbiosis: Rethinking disease in marine ecosystems. Front. Microbiol. 2016, 7, 991. [Google Scholar] [CrossRef]
- Bai, L.; Yan, Y.; Wang, S.; Li, J.; Zhang, W.; Yang, H.; Wang, P.; Mo, Z. Etiology analysis of green rot of kelp (Saccharina japonica) seedlings in a nursery farm. J. Anhui Agr. Univ. 2020, 47, 921–926. [Google Scholar]
- Ward, G.M.; Kambey, C.S.B.; Faisan, J.P.; Tan, P.L.; Daumich, C.C.; Matoju, I.; Stentiford, G.D.; Bass, D.; Lim, P.E.; Brodie, J.; et al. Ice-Ice disease: An environmentally and microbiologically driven syndrome in tropical seaweed aquaculture. Rev. Aquacult. 2021, 14, 414–439. [Google Scholar] [CrossRef]
- Shi, W.; Li, M.; Wei, G.; Tian, R.; Li, C.; Wang, B.; Lin, R.; Shi, C.; Chi, X.; Zhou, B.; et al. The occurrence of potato common scab correlates with the community composition and function of the geocaulosphere soil microbiome. Microbiome 2019, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Mallon, C.A.; Elsas, J.D.V.; Salles, J.F. Microbial invasions: The process, patterns, and mechanisms. Trends Microbiol. 2015, 23, 719–729. [Google Scholar] [CrossRef]
- Farhadi, F.; Khameneh, B.; Iranshahi, M.; Iranshahy, M. Antibacterial activity of flavonoids and their structure–activity relationship: An update review. Phytother. Res. 2019, 33, 13–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novaković, M.; Novaković, I.; Cvetković, M.; Sladić, D.; Tešević, V. Antimicrobial activity of the diarylheptanoids from the black and green alder. Braz. J. Bot. 2015, 38, 441–446. [Google Scholar] [CrossRef]
- Kim, H.; Lee, S.; Byun, Y.; Park, H. 6-Gingerol reduces Pseudomonas aeruginosa biofilm formation and virulence via quorum sensing inhibition. Sci. Rep. 2015, 5, 8656. [Google Scholar] [CrossRef]
- Mattio, L.M.; Catinella, G.; Dallavalle, S.; Pinto, A. Stilbenoids: A natural arsenal against bacterial pathogens. Antibiotics 2020, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Stratil, S.B.; Neulinger, S.C.; Knecht, H.; Friedrichs, A.K.; Wahl, M. Temperature-driven shifts in the epibiotic bacterial community composition of the brown macroalga Fucus vesiculosus. MicrobiologyOpen 2013, 2, 338–349. [Google Scholar] [CrossRef] [Green Version]
- Zozaya-Valdés, E.; Roth-Schulze, A.J.; Thomas, T. Effects of temperature stress and aquarium conditions on the red macroalga Delisea pulchra and its associated microbial community. Front. Microbiol. 2016, 7, 161. [Google Scholar] [CrossRef]
- Zozaya-Valdes, E.; Egan, S.; Thomas, T. A comprehensive analysis of the microbial communities of healthy and diseased marine macroalgae and the detection of known and potential bacterial pathogens. Front. Microbiol. 2015, 6, 146. [Google Scholar] [CrossRef] [Green Version]
- Hakamada, Y.; Ohkubo, Y.; Ohashi, S. Purification and characterization of β-Mannanase from Reinekea sp. KIT-YO10 with transglycosylation activity. Biosci. Biotech. Bioch. 2014, 78, 722–728. [Google Scholar] [CrossRef]
- Martin, M.; Barbeyron, T.; Martin, R.; Portetelle, D.; Michel, G.; Vandenbol, M. The cultivable surface microbiota of the brown alga Ascophyllum nodosum is enriched in macroalgal-polysaccharide-degrading bacteria. Front. Microbiol. 2015, 6, 1487. [Google Scholar] [CrossRef]
- Kang, H.; Kim, H.; Joung, Y.; Joh, K. Lewinella maritima sp. nov., and Lewinella lacunae sp. nov., novel bacteria from marine environments. Int. J. Syst. Evol. Microbiol. 2017, 67, 3603–3609. [Google Scholar] [CrossRef]
- Bengtsson, M.M.; Sjøtun, K.; Storesund, J.E.; Øvreås, J. Utilization of kelp-derived carbon sources by kelp surface-associated bacteria. Aquat. Microb. Ecol. 2011, 62, 191–199. [Google Scholar] [CrossRef]
- Ng, J.C.; Chan, Y.; Tun, H.M.; Leung, F.C.; Shin, P.K.; Chiu, J.M. Pyrosequencing of the bacteria associated with Platygyra carnosus corals with skeletal growth anomalies reveals differences in bacterial community composition in apparently healthy and diseased tissues. Front. Microbiol. 2015, 6, 1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quéré, G.; Intertaglia, L.; Payri, C.; Galand, P.E. Disease specific bacterial communities in a coralline algae of the northwestern Mediterranean Sea: A combined culture dependent and -independent approach. Front. Microbiol. 2019, 10, 1850. [Google Scholar] [CrossRef] [PubMed]
- Ward, G.M.; Faisan, J.P.; Cottier Cook, E.J.; Gachon, C.; Hurtado, A.Q.; Lim, P.E.; Matoju, I.; Msuya, F.E.; Bass, D.; Brodie, J. A review of reported seaweed diseases and pests in aquaculture in Asia. J. World Aquacult. Soc. 2020, 51, 815–828. [Google Scholar] [CrossRef]
- Karimi, E.; Geslain, E.; KleinJan, H.; Tanguy, G.; Legeay, E.; Corre, E.; Dittami, S.M. Genome sequences of 72 bacterial strains isolated from Ectocarpus subulatus: A resource for algal microbiology. Genome Biol. Evol. 2020, 12, 3647–3655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Tang, X.; Mo, Z.; Mao, Y. Metagenome-assembled genomes from Pyropia haitanensis microbiome provide insights into the potential metabolic functions to the seaweed. Front. Microbiol. 2022, 13, 857901. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.M.; Nishimura, M.; Haider, M.N.; Sano, M.; Ijichi, M.; Kogure, K.; Yoshizawa, S. Diversity and composition of microbial communities in an eelgrass (Zostera marina) bed in Tokyo Bay, Japan. Microbes Environ. 2021, 36, ME21037. [Google Scholar] [CrossRef]
- Staerck, C.; Gastebois, A.; Vandeputte, P.; Calenda, A.; Larcher, G.; Gillmann, L.; Papon, N.; Bouchara, J.; Fleury, M.J.J. Microbial antioxidant defense enzymes. Microb. Pathogenesis 2017, 110, 56–65. [Google Scholar] [CrossRef]
- Bertani, B.; Ruiz, N.; Slauch, J.M. Function and biogenesis of lipopolysaccharides. Ecosal Plus 2018, 8, 1–19. [Google Scholar] [CrossRef]
- Chaban, B.; Hughes, H.V.; Beeby, M. The flagellum in bacterial pathogens: For motility and a whole lot more. Semin. Cell Dev. Biol. 2015, 46, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Matilla, M.A.; Krell, T.S. The effect of bacterial chemotaxis on host infection and pathogenicity. FEMS Microbiol. Rev. 2017, 42, 40–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Senadheera, D.B.; Cvitkovitch, D.G. An intimate link: Two-component signal transduction systems and metal transport systems in bacteria. Future Microbiol. 2014, 9, 1283–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, R.; Tiwari, M.; Donelli, G.; Tiwari, V. Strategies for combating bacterial biofilms: A focus on anti-biofilm agents and their mechanisms of action. Virulence 2018, 9, 522–554. [Google Scholar] [CrossRef] [PubMed]
- The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinf. 2021, 19, 578–583. [CrossRef] [PubMed]
- Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).