1. Introduction
Anthocyanins are water-soluble pigments localized in vacuoles, primarily comprising cyanidin-3-glucoside, pelargonidin-3-glucoside, and delphinidin-3-glucoside. Among them, cyanidin-3-galactoside is the dominant pigment responsible for the pink-to-purple coloration of chrysanthemum (
Chrysanthemum morifolium) petals [
1]. Additionally, anthocyanins enhance plant resilience to abiotic stresses such as drought, cold, and intense light by scavenging excess reactive oxygen species [
2,
3,
4].
Anthocyanin metabolism in plants involves biosynthesis and degradation, with their dynamic balance determining anthocyanin accumulation. Over the past few decades, the anthocyanin biosynthetic pathway has been extensively studied, revealing four distinct stages. First, phenylalanine is converted into p-coumaroyl-CoA, a key intermediate in various secondary metabolic pathways, catalyzed by phenylalanine ammonia-lyase (PAL), the rate-limiting enzyme. Next, p-coumaroyl-CoA is converted into naringenin, a crucial step in flavonoid metabolism, catalyzed by chalcone synthase (CHS) and chalcone isomerase (CHI), with CHS serving as the rate-limiting enzyme. Subsequently, flavone synthase (FNS) and flavanone 3-hydroxylase (F3H) catalyze the conversion of naringenin into flavone and dihydrokaempferol, respectively. Finally, dihydrokaempferol is a key branch point with two potential fates: it can be converted into flavonols via flavonol synthase (FLS), or it can be transformed into various leucoanthocyanidins. The conversion to leucoanthocyanidins involves the catalytic actions of flavonoid 3′-hydroxylase (F3′H), dihydroflavonol 4-reductase (DFR), and flavonoid 3′,5′-hydroxylase (F3′5′H). These leucoanthocyanidins are then converted into anthocyanins through the actions of anthocyanidin synthase (ANS) and flavonoid 3-O-glycosyltransferase (UFGT) [
5,
6].
Compared to anthocyanin biosynthesis, research on anthocyanin biodegradation remains relatively limited. Evidence from in vitro and in vivo studies suggests that anthocyanin degradation follows three main pathways. The first is coupled oxidation, in which anthocyanins degrade through redox coupling with polyphenolic compounds. During this process, quinones from polyphenols are reduced to their original phenolic forms, while anthocyanins are oxidized into anthocyanin quinones or other degradation products. This reaction is catalyzed by polyphenol oxidase (PPO) and requires oxygen (O
2) as a substrate. The second is two-step degradation, which occurs in two stages. Initially, anthocyanins undergo deglycosylation, catalyzed by β-glucosidase (also known as anthocyanase), producing unstable anthocyanidins. These are subsequently oxidized into colorless or degraded products by polyphenol oxidase (PPO) or peroxidase (POD), which use O
2 or H
2O
2 as substrates, respectively. The third pathway is direct oxidation, where anthocyanins are degraded without prior deglycosylation. This reaction is catalyzed by peroxidase (POD), which utilizes hydrogen peroxide (H
2O
2) to rapidly oxidize and degrade anthocyanins. While all three pathways may occur under specific conditions, POD-mediated direct oxidation is considered the most probable mechanism in planta [
7]. Class III peroxidases (
PRXs) are a plant-specific group of peroxidases that catalyze anthocyanin degradation. To date, several
PRX genes involved in this process have been identified in various plant species. In
Brunfelsia calycina, flower color shifts rapidly from purple to white upon blooming, a process strongly linked to anthocyanin degradation. Studies have shown that the PRX enzyme BcPrx01, localized in petal vacuoles, directly degrades anthocyanins, leading to petal color changes [
8]. In wine grape (
Vitis vinifera L.
cv. Sangiovese), the
PRX gene
VviPRX31 is strongly induced under high temperatures and contributes to fruit discoloration. Overexpression of
VviPRX31 in
Petunia hybrida, followed by heat stress, significantly reduces anthocyanin content in transgenic plants [
9]. The degradation products of anthocyanins vary depending on oxidative conditions, with protocatechuic acid being a common byproduct [
10,
11]. In
Prunus salicina Lindl., exposure to 35 °C for nine days results in a significant decline in anthocyanin content, accompanied by a notable increase in protocatechuic acid levels. In vitro experiments further confirm that the addition of H
2O
2 and horseradish peroxidase (HRP) accelerates anthocyanin (cyanidin-3-glucoside) degradation and promotes protocatechuic acid formation [
12].
Amid global climate warming, the adverse effects of heat stress on the yield and quality of economic crops have drawn increasing attention. Heat stress inhibits anthocyanin accumulation in plants, significantly affecting the ornamental quality and commercial value of chrysanthemums and other important flowering plants [
13,
14,
15]. Most studies have focused on elucidating how heat stress suppresses anthocyanin biosynthesis. For example, in tea (
Camellia sinensis), the MBW complex, formed by CsMYB75, the bHLH transcription factor CsTT8, and the WD40 protein CsTTG1, promotes anthocyanin biosynthesis by directly activating the structural genes
CsDFR and
CsANS. Under heat stress, however, the E3 ubiquitin ligase
CsCOP1 is upregulated and accumulates. This leads to the ubiquitin-mediated degradation of CsbZIP1, an upstream activator of
CsMYB75, subsequently preventing CsbZIP1 from activating
CsMYB75 and thereby disrupting MBW complex formation and reducing anthocyanin accumulation [
16]. In
Chrysanthemum morifolium, heat stress induces the accumulation of the atypical subgroup 7 (SG7) R2R3-MYB transcription factor CmMYB012. This transcription factor suppresses anthocyanin biosynthesis by repressing the transcription of the structural genes
CmCHS,
CmDFR,
CmANS, and
CmUFGT, thereby reducing anthocyanin accumulation in petals [
17]. A previous study subjected ‘Nannong Ziyunying’ and ‘Nannong Zizhu’ chrysanthemums to 38 °C heat stress from the bud stage to full bloom. Transcriptomic analysis revealed that heat stress inhibits anthocyanin biosynthesis and accumulation in petals by downregulating key anthocyanin biosynthetic genes [
18]. Despite these insights, the impact of heat stress on anthocyanin metabolism in chrysanthemum petals, particularly its degradation process, remains poorly understood.
Here, we focused on the
Chrysanthemum cultivar ‘Nannong Fencui’. Our previous research indicates that the flowering process of the ‘Nannong Fencui’ chrysanthemum consists of six stages: flower bud, broken bud, dew color, first bloom, full bloom, and post-flowering. Anthocyanin accumulation in the petals first appears at the dew color stage, gradually increases, peaks at full bloom, and declines during post-flowering [
19]. This peak accumulation phase is a critical point for examining color stability under heat stress. Therefore, in this study, fully bloomed ‘Nannong Fencui’ chrysanthemums, having reached peak anthocyanin accumulation, were exposed to 35 °C for five days, with a parallel control maintained at 22 °C. Samples were collected for transcriptomic and untargeted metabolomic sequencing to comprehensively assess the effects of heat stress on anthocyanin metabolism, including both biosynthesis and degradation. Our results indicate that anthocyanin biosynthesis is largely inactive in fully bloomed chrysanthemums, and heat stress has minimal impact on this process. This suggests that petal fading at this stage is unlikely to result from the direct inhibition of anthocyanin biosynthesis. Ultra-high-performance liquid chromatography (UHPLC) and untargeted metabolomics revealed a significant reduction in anthocyanin (cyanidin-3-glucoside) levels in heat-treated petals. While protocatechuic acid, a common anthocyanin degradation product, was undetectable in both heat-treated and control groups, its O-methylated oxidation derivative, 5-carboxyvanillic acid, accumulated significantly after heat exposure. Furthermore, multiple
PRX enzyme genes potentially associated with anthocyanin degradation were significantly upregulated under heat stress. Similarly,
O-methyltransferase genes involved in the O-methylation of protocatechuic acid were also significantly upregulated. These findings suggest that anthocyanin degradation plays a major role in petal fading in fully bloomed chrysanthemums under heat stress. Our study provides new insights into the regulation of anthocyanin metabolism in chrysanthemums and offers potential strategies for maintaining flower color quality during summer production.
2. Materials and Methods
2.1. Plant Material and Growth Conditions
The chrysanthemum cultivar ‘Nannong Fencui’ was provided by the Chrysanthemum Germplasm Resource Preservation Center at Nanjing Agricultural University, China. Initially, chrysanthemum cuttings were rooted in trays for 15 days and then transferred to an intelligent greenhouse for 45 days of cultivation, maintained at 24 °C/18 °C (day/night), under a 16/8 h light/dark photoperiod, and 70% relative humidity. Subsequently, seedlings exhibiting consistent growth were selected, and new cuttings were taken and rooted in trays for another 15 days. These cuttings were then transplanted into pots and placed in an intelligent glass greenhouse for three months to induce flowering under conditions of 24 °C/18 °C (day/night), an 8/16 h light/dark cycle, and 70% relative humidity. Finally, 48 uniformly vigorous, fully bloomed ‘Nannong Fencui’ plants were selected and randomly divided into six groups of eight plants each for further experiments conducted in Nanjing City, Jiangsu Province, China, in November 2024.
For the high-temperature treatment, two groups of eight plants each (totaling 16 plants) were transferred to plant incubators under two conditions: the treatment group (35 °C/22 °C day/night temperatures, 8/16 h light/dark cycle, 30,000 lx light intensity, and 75% relative humidity) and the control group (CK) (22 °C/18 °C day/night temperatures, 8/16 h light/dark cycle, 30,000 lx light intensity, and 75% relative humidity). The 35 °C daytime temperature for the treatment group was specifically selected because it represents a significant heat stress level for chrysanthemum, a cool-season plant with an optimal growth range of 17–22 °C. This temperature also reflects challenging high-temperature conditions commonly encountered during summer horticultural production. Both groups were watered every two days. After five days of treatment, one flower was randomly sampled from each plant per group, and their ray flower petals were pooled to form a biological replicate. Each treatment was performed in triplicate, yielding three biological replicates per condition. Samples were flash-frozen in liquid nitrogen for anthocyanin quantification and subsequently analyzed using multi-omics approaches at Shanghai Bioprofile (
www.bioprofile.cn (accessed on 20 November 2024)).
2.2. Determination of Anthocyanin Content by Ultra-Performance Liquid Chromatography (UPLC)
For anthocyanin content determination, ray floret samples were collected in three biological replicates from both the high-temperature treatment and control groups. For each replicate, 0.1 g of powdered sample (pulverized in liquid nitrogen) was transferred to a test tube containing 5 mL of extraction solution (70% methanol, 27% ultrapure water, 2% formic acid, and 1% trifluoroacetic acid), wrapped in aluminum foil to prevent light exposure, and incubated at 4 °C for 24 h. The supernatant was then filtered through an organic membrane. A 1.5 mL portion of the filtrate was collected into a brown volumetric flask for subsequent analysis. Cyanidin standard solutions (100, 50, 10, 5, and 2.5 μg/mL) were prepared using 25 mg/mL DMSO and standard compounds from Med Chem Express. The mobile phase consisted of ultrapure water, methanol, 0.1% aqueous formic acid, and 0.1% formic acid in acetonitrile. All prepared solutions were filtered through organic or inorganic membranes and subjected to ultrasonic treatment for 10 min. Chromatographic analysis was conducted using a T3 column maintained at 35 °C, with cyanidin detected at 525 nm. Anthocyanin concentration was calculated using a standard curve derived from linear regression, plotting absorbance (y-axis) against concentration (x-axis). The equation was y = 5.468x − 0.08888, where x represents the peak area.
2.3. Transcriptome Sequencing
Total RNA was extracted from ray floret samples collected in three biological replicates from both the high-temperature treatment and control groups. mRNA with a poly(A) tail was enriched using Oligo(dT) magnetic beads, followed by RNA fragmentation into ~300 bp fragments via ion fragmentation. First-strand cDNA was synthesized using RNA as a template, random hexamer primers, and reverse transcriptase. The second-strand cDNA was then synthesized using the first-strand cDNA as a template. After library construction, PCR amplification was performed to enrich library fragments, and quality control was conducted using an Agilent 2100 Bioanalyzer. Libraries with different index sequences were proportionally mixed, diluted to 2 nM, and denatured to generate single-stranded libraries. Next-generation sequencing (NGS) was performed on the Illumina platform using paired-end (PE) sequencing. Raw sequencing data were filtered to obtain high-quality clean reads, which were then aligned to the reference genome of
C. morifolium [
20]. Gene expression levels were quantified based on the alignment results, followed by differential expression analysis, enrichment analysis, and clustering analysis. The aligned reads were further assembled to reconstruct transcript sequences, using the full-length transcriptome of
C. morifolium as the reference genome.
2.4. Identification of Differentially Expressed Genes (DEGs) and Functional Annotation Analysis
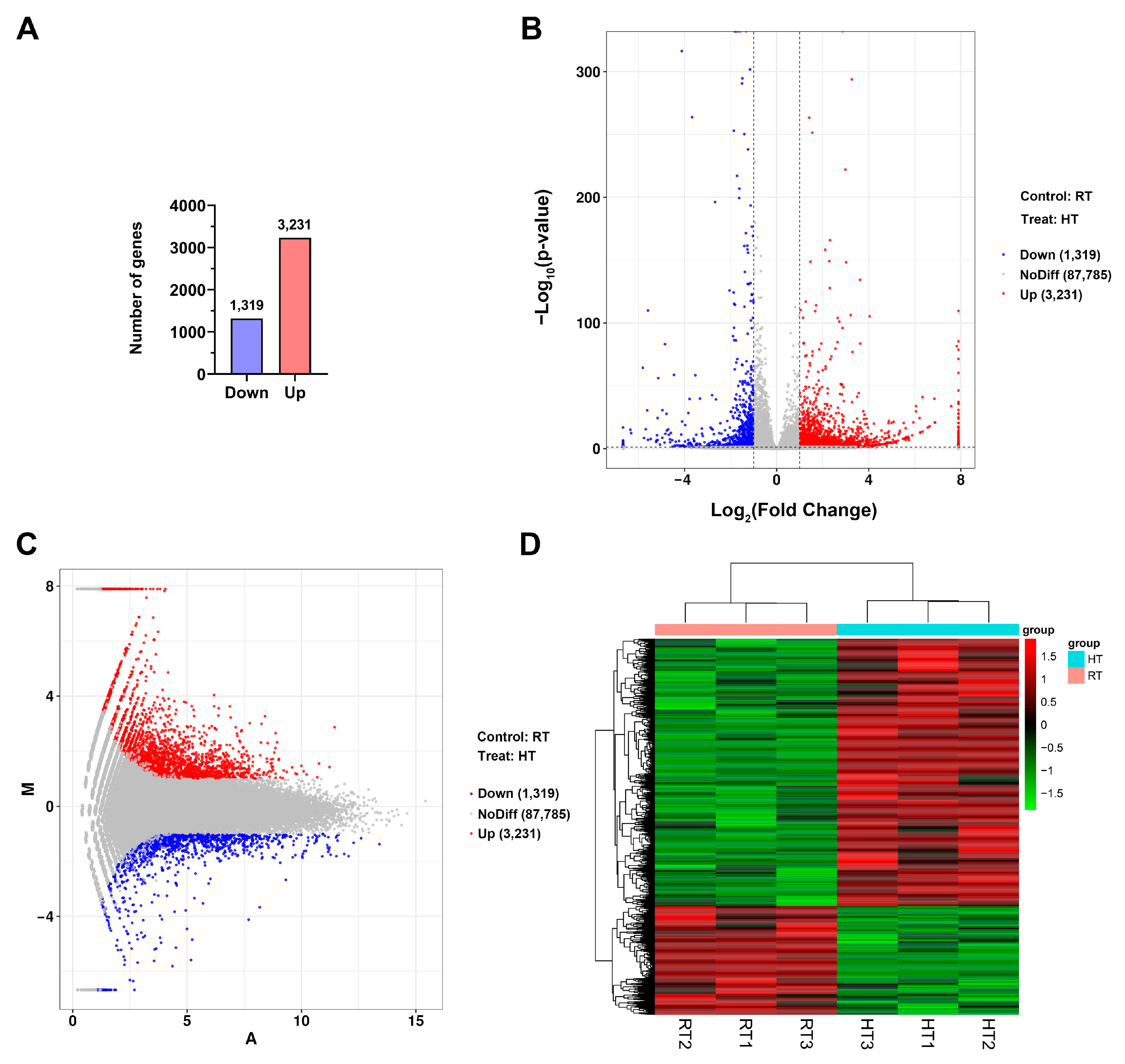
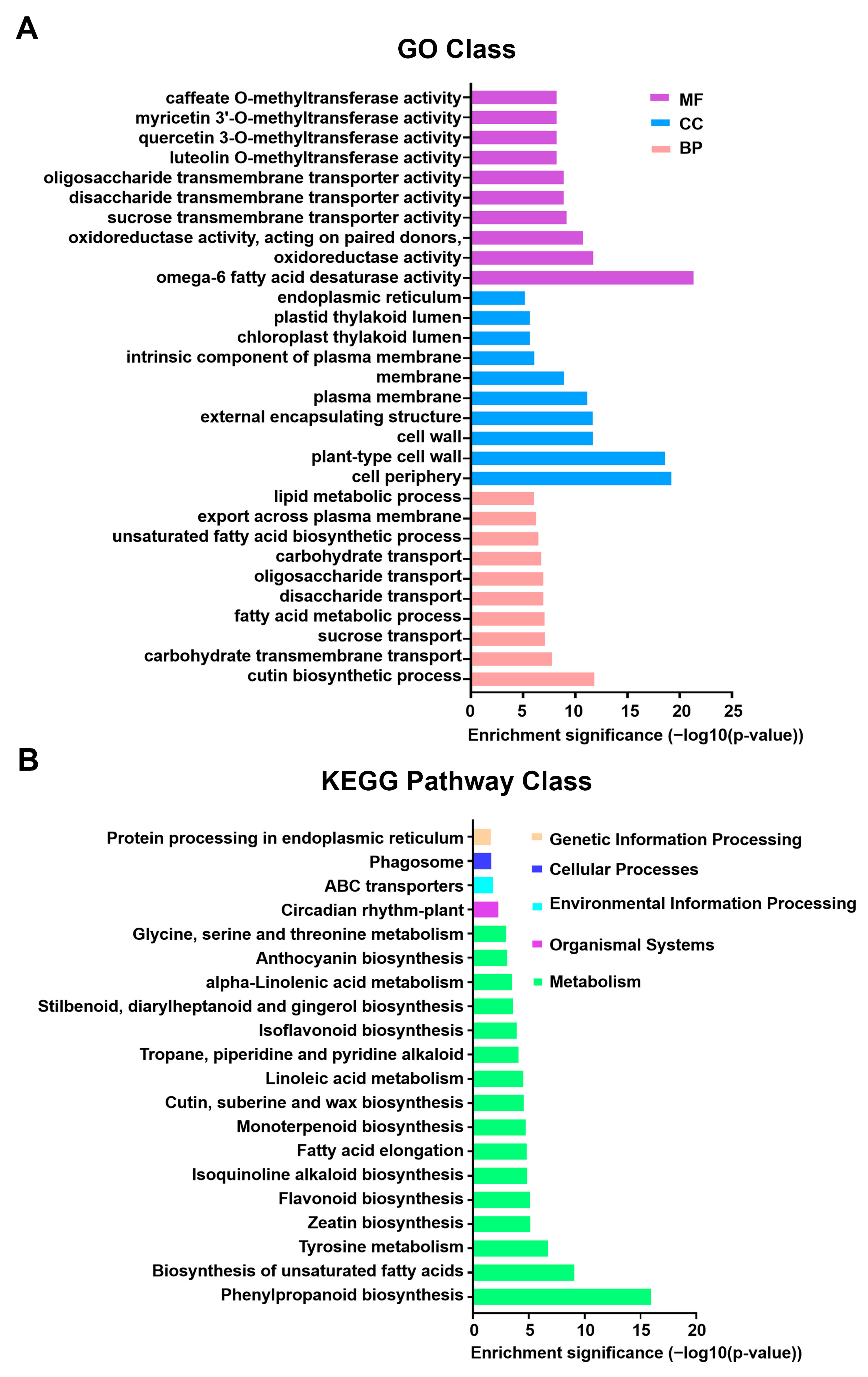
Gene annotation was conducted using data from the GO, SwissProt, KEGG, NR, eggNOG, eggNOG_Category, and SwissProtName databases. HTSeq was used to count reads mapped to each gene, representing the raw gene expression levels. FPKM normalization was applied to standardize gene expression across genes and samples for comparability. Differential gene expression analysis was performed using DESeq, with DEGs identified based on the criteria |log2FoldChange| > 1 and p < 0.05. Bidirectional clustering analysis of DEGs across different comparison groups and samples was conducted using the R package Pheatmap. Genes were clustered based on their expression profiles, while samples were grouped according to gene expression patterns. Euclidean distance was used to compute dissimilarities, and hierarchical clustering was performed using the complete linkage method. GO enrichment analysis was conducted using topGO, where DEGs associated with GO terms were evaluated based on their gene lists and counts per term. The hypergeometric distribution method was used to calculate p-values, with a significance threshold of p < 0.05. Significantly enriched GO terms were identified by comparing DEGs against the genomic background, providing insights into their major biological functions. KEGG enrichment analysis was assessed using the Rich factor, FDR value, and the number of genes mapped to each pathway.
2.5. Metabolite Extraction, Detection, and Statistical Analysis
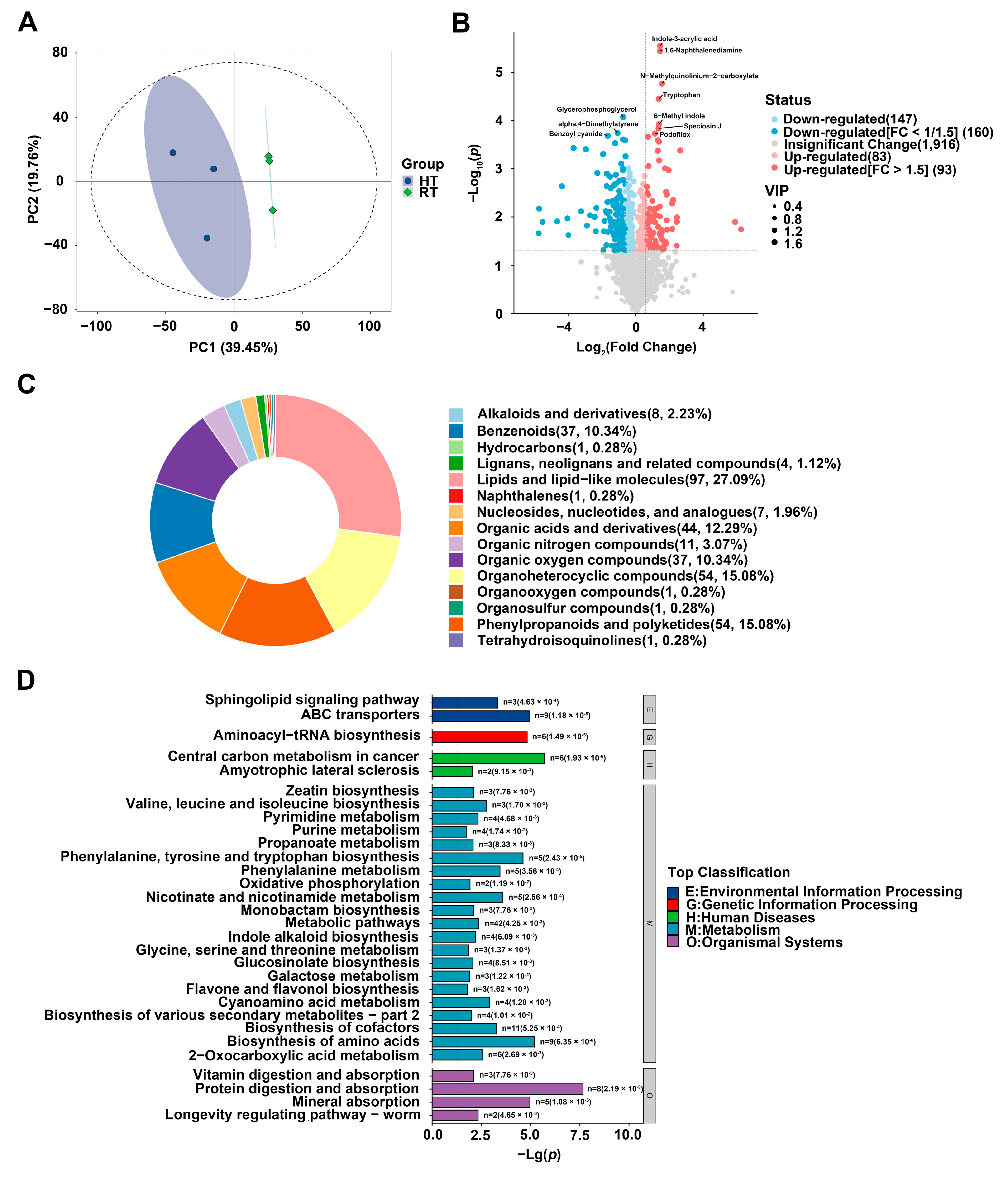
Freeze-dried samples were transferred to 2 mL centrifuge tubes with a 5 mm tungsten bead and homogenized at 65 Hz for 1 min using a grinding instrument. Next, 1 mL of pre-cooled extraction solvent (methanol/acetonitrile/water, 2:2:1, v/v/v) was added, followed by ultrasonic extraction in an ice bath for 1 h. The samples were then incubated at −20 °C for 1 h and centrifuged at 14,000 rpm for 20 min at 4 °C. The supernatant was vacuum-dried to remove solvents. Metabolomic profiling was performed using the UPLC-ESI-Q-Orbitrap-MS system (Nexera X2 UHPLC, Shimadzu, Japan) and the TripleTOF 6600 mass spectrometer (AB Sciex, Framingham, MA, USA). Liquid chromatography separation was conducted on an ACQUITY UPLC® HSS T3 column (2.1 × 100 mm, 1.8 μm, Waters, Milford, MA, USA) at a flow rate of 0.3 mL/min. The mobile phase comprised 0.1% formic acid in water (A) and pure acetonitrile (B). The elution gradient was as follows: 0–2 min, 0% B; 2–6 min, increased to 48% B; 6–10 min, increased to 100% B and held for 2 min; 10–10.1 min, decreased to 0% B; and 10.1–13 min, re-equilibration at 0% B. Mass spectrometry data were acquired in both positive and negative ionization modes. Raw MS data processing, including peak alignment, retention time correction, and peak area extraction, was performed using the MS-DIAL software (version 4.70). Metabolites were identified by cross-referencing public databases (HMDB, MassBank) and an in-house standard library, ensuring mass accuracy within 10 ppm and MS/MS deviation under 0.02 Da. Multivariate data analysis and modeling were conducted in R (version 4.0.3) using associated R libraries. Data were standardized using Pareto scaling, and models were constructed using principal component analysis (PCA), orthogonal PLS-DA (PLS-DA), and OPLS-DA. Statistically significant metabolites were identified via the OPLS-DA model based on VIP scores, followed by a two-tailed t-test on normalized data to determine key differential metabolites. Clustering analysis of identified differential metabolites was performed using R.
2.6. Gene Expression Analysis
Total RNA was extracted from chrysanthemum ray flower petals with a Quick RNA Isolation Kit (Huayueyang, Beijing, China), adhering to the manufacturer’s guidelines. First-strand cDNA was generated using HiScript II Q Select RT SuperMix for qPCR (Vazyme Biotech, Co., Ltd., Nanjing, China), following the provided protocol. Subsequently, the cDNA was adjusted to 400 ng μL
−1 using ddH
2O, and qRT-PCR was performed with TB Green Premix Ex Taq II (Tli RNaseH Plus) (Takara, Dalian, China) in a 20 μL reaction volume. CmEF1α and CmActin served as reference genes for normalization. Primers employed in this research are detailed in
Supplemental Table S1.
2.7. Statistical Analysis
All experiments were performed in triplicate, and bar graphs were generated using GraphPad Prism 7.05 (Chicago, IL, USA). One-way analysis of variance (ANOVA) was conducted using SPSS 19.0 to assess statistical significance.
4. Discussion
Chrysanthemums (
Chrysanthemum morifolium) typically bloom in the cool autumn season, but to meet high market demand, year-round production is necessary. However, petal fading due to high temperatures during summer cultivation significantly impacts their ornamental quality and commercial value, posing a major production challenge [
30,
31]. Understanding the mechanisms of flower color regulation under heat stress, identifying key regulatory genes, and developing heat-tolerant chrysanthemum varieties through molecular breeding are essential strategies to address this issue. Previous research has primarily focused on how heat stress suppresses anthocyanin biosynthesis, while anthocyanin degradation under heat stress remains largely unexplored. This knowledge gap limits a comprehensive understanding of anthocyanin metabolism in chrysanthemums and restricts the identification of superior genetic resources. In this study, we used the chrysanthemum cultivar ‘Nannong Fencui’ as the research material, subjected fully bloomed flowers—when anthocyanin accumulation peaked—to heat treatment, and conducted transcriptome sequencing to compare gene expression differences between heat-treated and control groups. Combined with metabolomic analysis of differential metabolites after heat treatment, we systematically examined the impact of heat stress on anthocyanin metabolism in fully bloomed chrysanthemums.
Heat stress-induced reductions in anthocyanin accumulation are widely observed across various plant species. For example, heat stress inhibits anthocyanin accumulation in apple (
Malus domestica) and cherry (
Prunus avium L.) fruits, leading to poor coloration [
32,
33]. Several key mechanisms underlie the impact of heat stress on anthocyanin accumulation. High temperatures promote the nuclear translocation of the E3 ubiquitin ligase COP1, which degrades anthocyanin biosynthesis activators such as HY5 [
34]. Under heat stress, phenylalanine metabolism shifts toward lignin and chlorogenic acid synthesis, reducing the availability of direct precursors for anthocyanin biosynthesis [
35]. The transcriptional expression of anthocyanin biosynthesis activators is suppressed under heat stress [
36], while the transcription of anthocyanin biosynthesis inhibitors is enhanced in response to heat stress [
17]. Additionally, PRX enzyme genes are upregulated under heat stress, leading to increased PRX enzyme levels and activity, which promote anthocyanin degradation [
9,
12]. Thus, heat stress affects anthocyanin metabolism through multiple interconnected regulatory mechanisms. In this study, we integrated transcriptomic and metabolomic approaches to comprehensively analyze the effects of heat stress on anthocyanin metabolism in chrysanthemum petals, considering both biosynthesis and degradation. Our research aims to unravel the regulatory network governing anthocyanin metabolism under heat stress, providing a foundation for further investigation of key regulatory pathways.
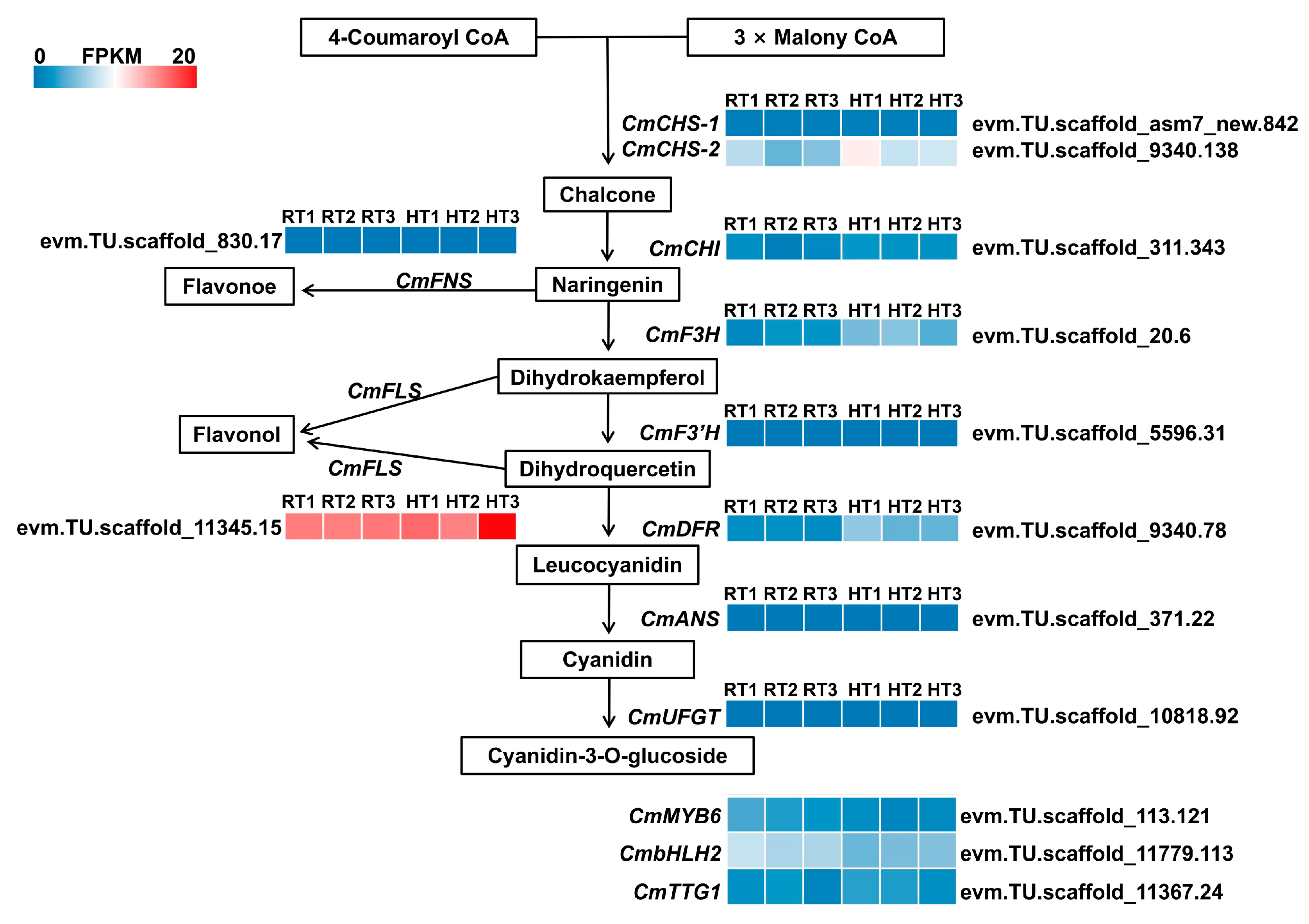
Transcriptomic analysis revealed that at full bloom, most structural genes involved in anthocyanin biosynthesis exhibited minimal expression levels, regardless of whether plants were exposed to normal or high temperatures (
Figure 7). This suggests that at this stage, anthocyanin accumulation in chrysanthemum petals has peaked, and the biosynthetic pathway is relatively inactive. Interestingly, the flavonol synthase gene
CmFLS maintained consistently high basal expression (FPKM values approximately 15–20) under both normal and high-temperature conditions, whereas the flavone synthase gene
CmFNS remained at low expression levels throughout (
Figure 7). These findings suggest that at full bloom, chrysanthemums may prioritize flavonol biosynthesis over anthocyanins or other flavonoids. This preferential metabolic branching could serve as a regulatory strategy to sustain flavonoid homeostasis after anthocyanin biosynthesis has concluded.
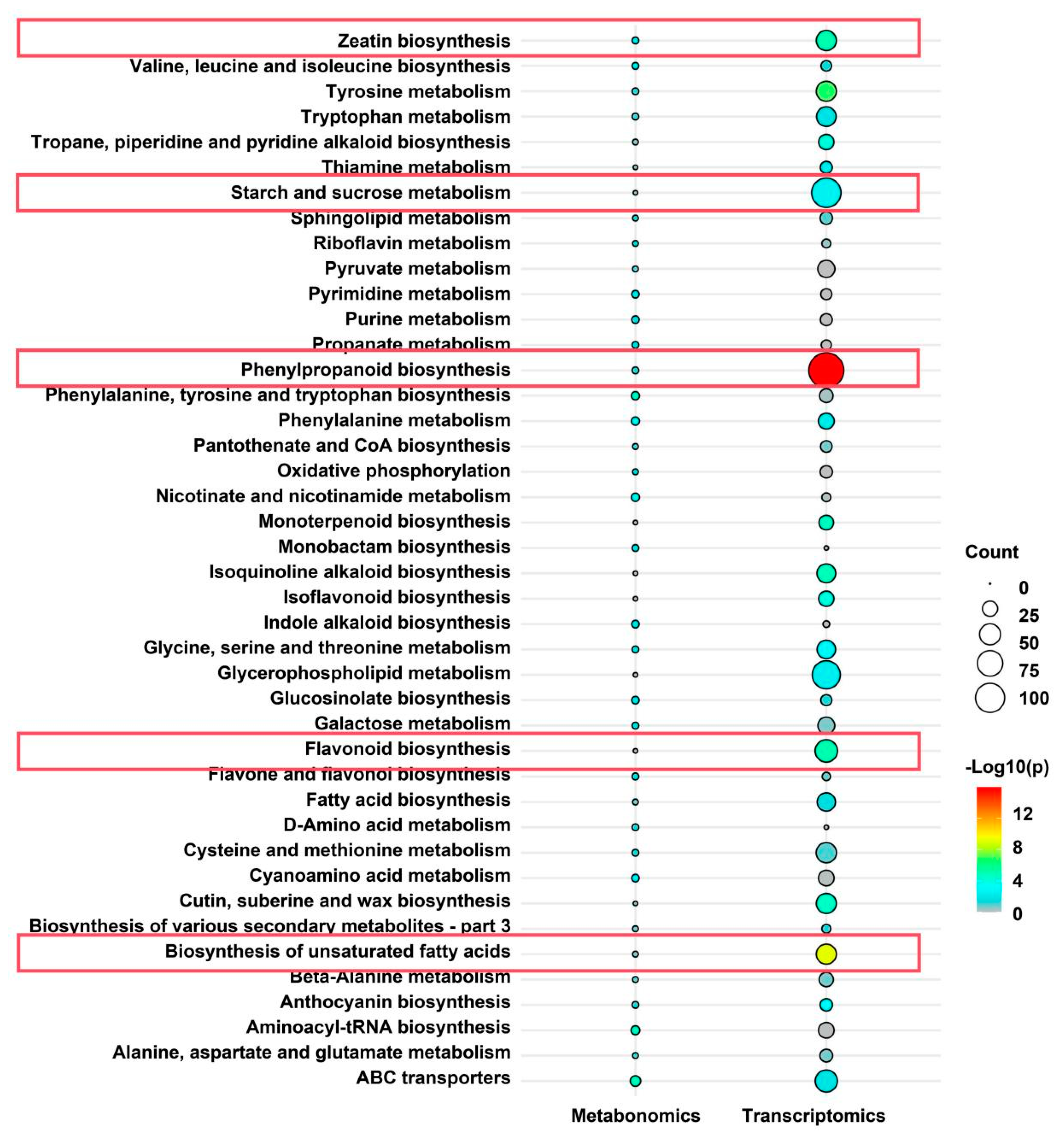
Metabolomic analysis provided additional insights into how heat stress affects anthocyanin metabolism-related compounds. KEGG metabolic pathway analysis revealed a significant enrichment of the phenylalanine metabolism pathway under heat stress, accompanied by a pronounced accumulation of phenylalanine in the heat-treated group (
Figure 5D and
Supplementary Table S2). As the primary precursor for flavonoid metabolism, phenylalanine accumulation may result from heat-induced inhibition of its normal flux into downstream pathways (e.g., anthocyanin biosynthesis), leading to precursor buildup. Beyond its effects on anthocyanin biosynthesis, heat stress also influences other secondary metabolites in the flavonoid metabolic pathway. Metabolomic analysis revealed a substantial decline in flavonol and flavone content following heat exposure (
Supplementary Table S2). However, transcriptomic analysis showed that
CmFLS and
CmFNS expression levels were only marginally affected by heat stress (
Figure 7). These findings suggest that heat stress likely reduces flavone and flavonol accumulation by modulating enzyme activity, altering metabolic flux, or promoting product degradation rather than directly suppressing biosynthetic gene expression. In conclusion, heat stress disrupts anthocyanin metabolism by altering precursor and branch metabolite levels, causing precursor accumulation while reducing the levels of end-products and branch compounds. This metabolic imbalance ultimately leads to decreased anthocyanin accumulation, negatively affecting petal pigmentation.
In plants, anthocyanin biosynthesis is primarily regulated by the MBW complex (MYB-bHLH-WD40), with MYB and bHLH transcription factors serving as key transcriptional regulators. Transcriptomic analysis revealed that the expression of genes encoding MBW complex members in chrysanthemums (
CmMYB6,
CmbHLH2, and
CmTTG1) remained low at the full-bloom stage. Specifically, the gene encoding the key regulatory factor
CmMYB6 (
evm.TU.scaffold_113.121) had a relatively low expression level under normal temperature conditions (FPKM~2.76), which further declined to approximately 1.92 after high-temperature treatment. Meanwhile,
CmbHLH2 (
evm.TU.scaffold_11779.113) and
CmTTG1 (
evm.TU.scaffold_11367.24) exhibited extremely low expression levels under both temperature conditions (
Figure 7). These findings indicate that at full bloom, the transcriptional activators responsible for anthocyanin biosynthesis in chrysanthemums are minimally active, and high temperatures further suppress anthocyanin production by downregulating
CmMYB6 expression. This observation is consistent with findings in other plant species, where high temperatures commonly repress MYB transcription factors in the MBW complex, leading to reduced anthocyanin accumulation [
37].
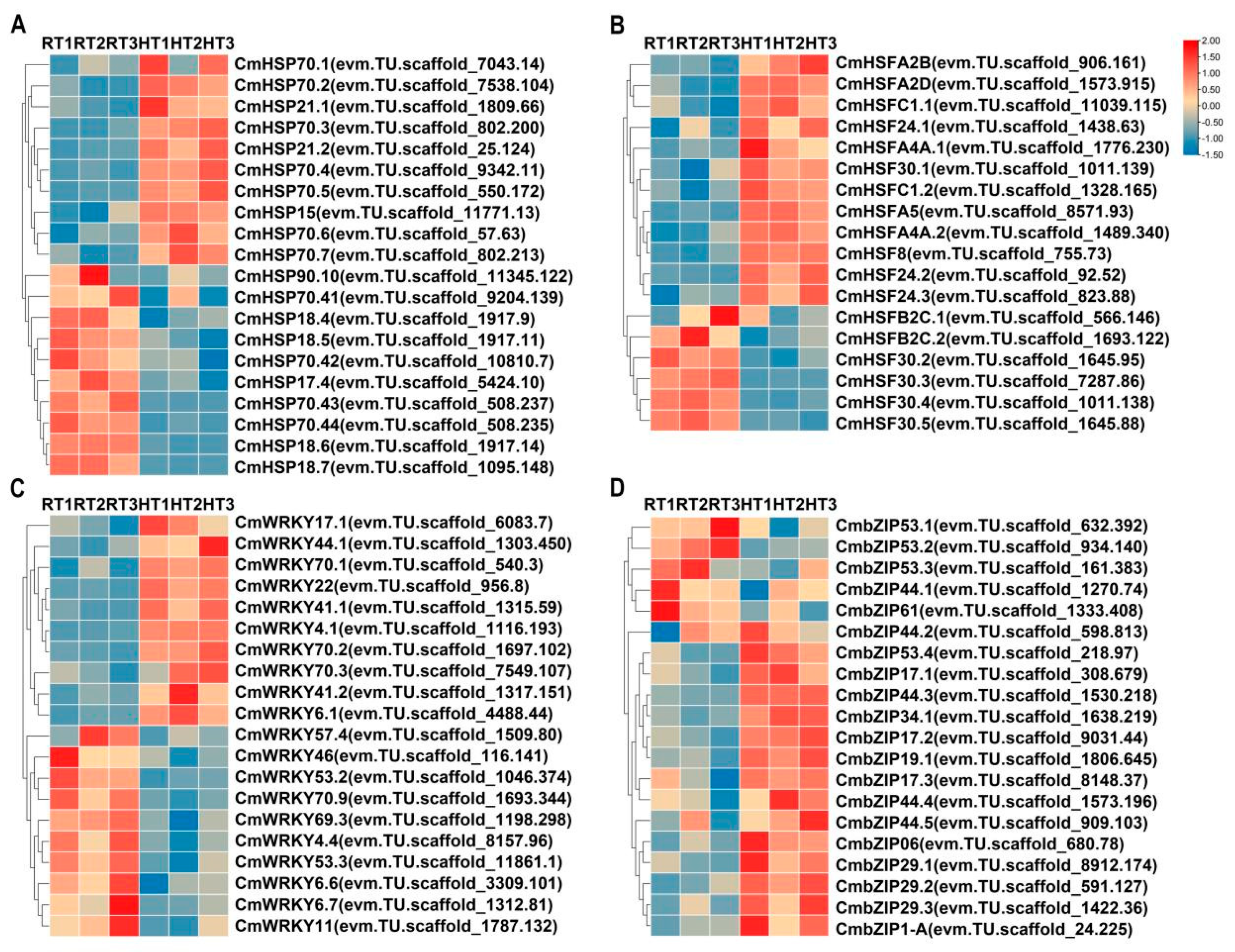
Transcriptomic analysis revealed that, in addition to the MBW complex, heat stress induced substantial changes in other transcription factor families in chrysanthemums. The heat shock transcription factor (HSF) family plays a central role in plant thermotolerance by activating protective genes such as heat shock proteins (HSPs) [
38,
39,
40]. Our study identified 12
CmHSF genes that were significantly upregulated in response to heat stress. Additionally, heat stress reprogrammed the transcriptional profiles of numerous WRKY and bZIP transcription factors. Among them, 42
CmWRKY genes were upregulated, while 21 were downregulated; similarly, 23
CmbZIP genes were upregulated, whereas 5 were downregulated under heat stress. WRKY transcription factors are key regulators of plant adaptation to heat stress. They enhance thermotolerance, partly by binding to W-box elements in target gene promoters and modulating reactive oxygen species (ROS), phytohormone signaling, and MAPK-mediated pathways [
41,
42,
43]. Moreover, bZIP transcription factors respond to heat stress, often being upregulated under the regulation of abscisic acid (ABA) and other plant hormones. This activation leads to the expression of downstream genes that enhance heat resistance [
44]. The widespread transcriptional changes in WRKY and bZIP transcription factors suggest that heat stress extensively remodels the transcription factor network in chrysanthemums, altering metabolic and defense-related gene expression. This large-scale transcriptional reprogramming not only regulates heat stress defense mechanisms but may also indirectly impact anthocyanin metabolism.
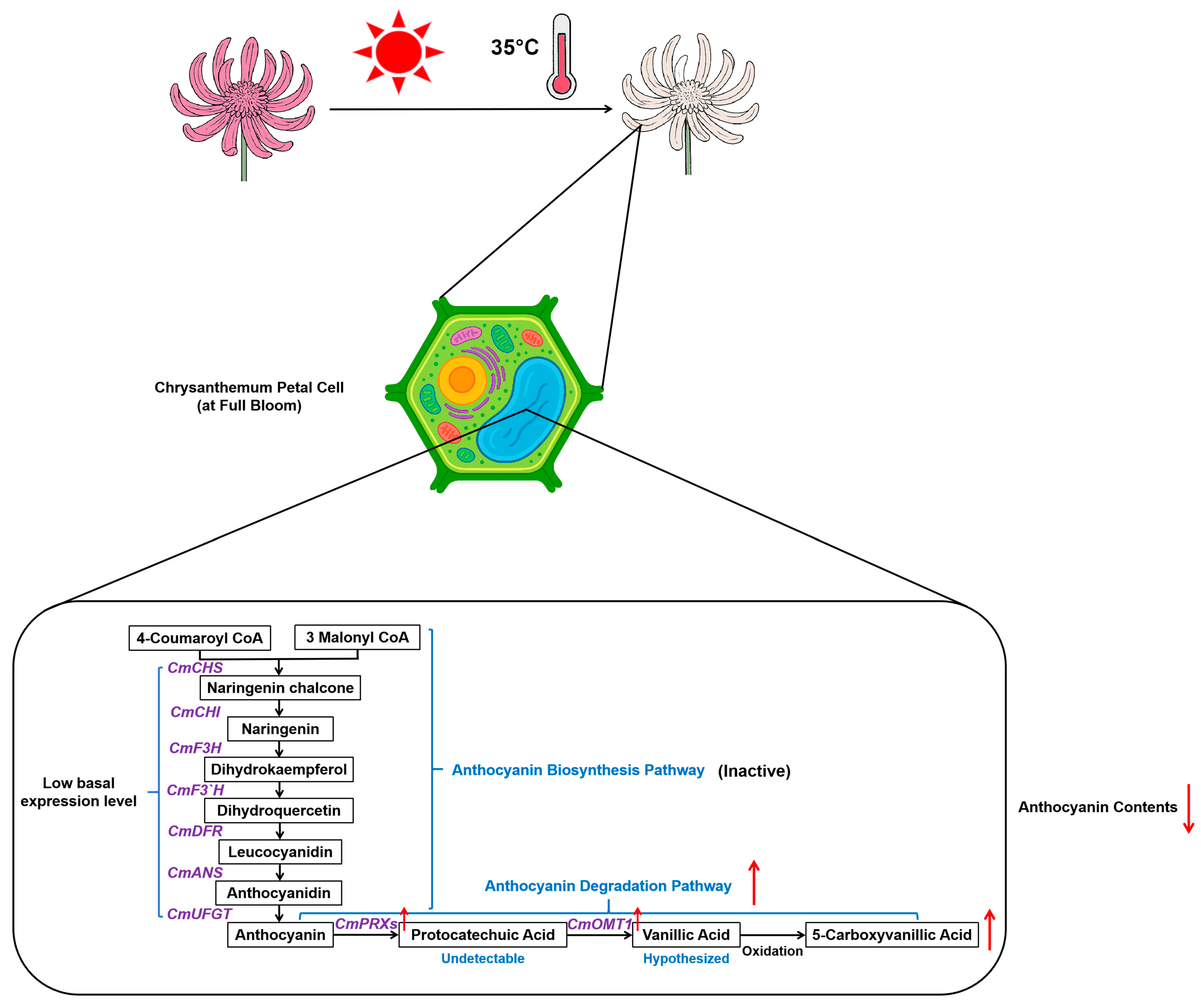
Since anthocyanin biosynthesis is relatively inactive at full bloom, the decline in anthocyanin accumulation and petal fading under heat stress is more likely attributable to anthocyanin degradation. By integrating metabolomic and transcriptomic analyses, our study investigates the potential enzymatic pathways involved in anthocyanin degradation in chrysanthemums under heat stress. Class III peroxidases (
PRXs) have been reported to facilitate anthocyanin degradation into protocatechuic acid in plants [
12]. However, our metabolomic data revealed no substantial accumulation of protocatechuic acid under heat stress, suggesting that it may undergo further metabolic conversion. According to a pioneering work, protocatechuic acid in plants can undergo O-methylation to form vanillic acid or hydroxylation to produce gallic acid [
29]. Notably, our metabolomic analysis revealed a significant accumulation of 5-carboxyvanillic acid, an oxidized derivative of vanillic acid, in response to heat treatment, whereas gallic acid was undetectable under both conditions (
Supplementary Table S2). The observed accumulation of 5-carboxyvanillic acid suggests a possible pathway in which anthocyanins are first degraded into protocatechuic acid by PRX, subsequently methylated into vanillic acid by O-methyltransferase (OMT), and then further oxidized into 5-carboxyvanillic acid. Supporting this, several key enzyme genes associated with anthocyanin degradation were markedly upregulated in response to heat stress. Among them, a class III peroxidase gene,
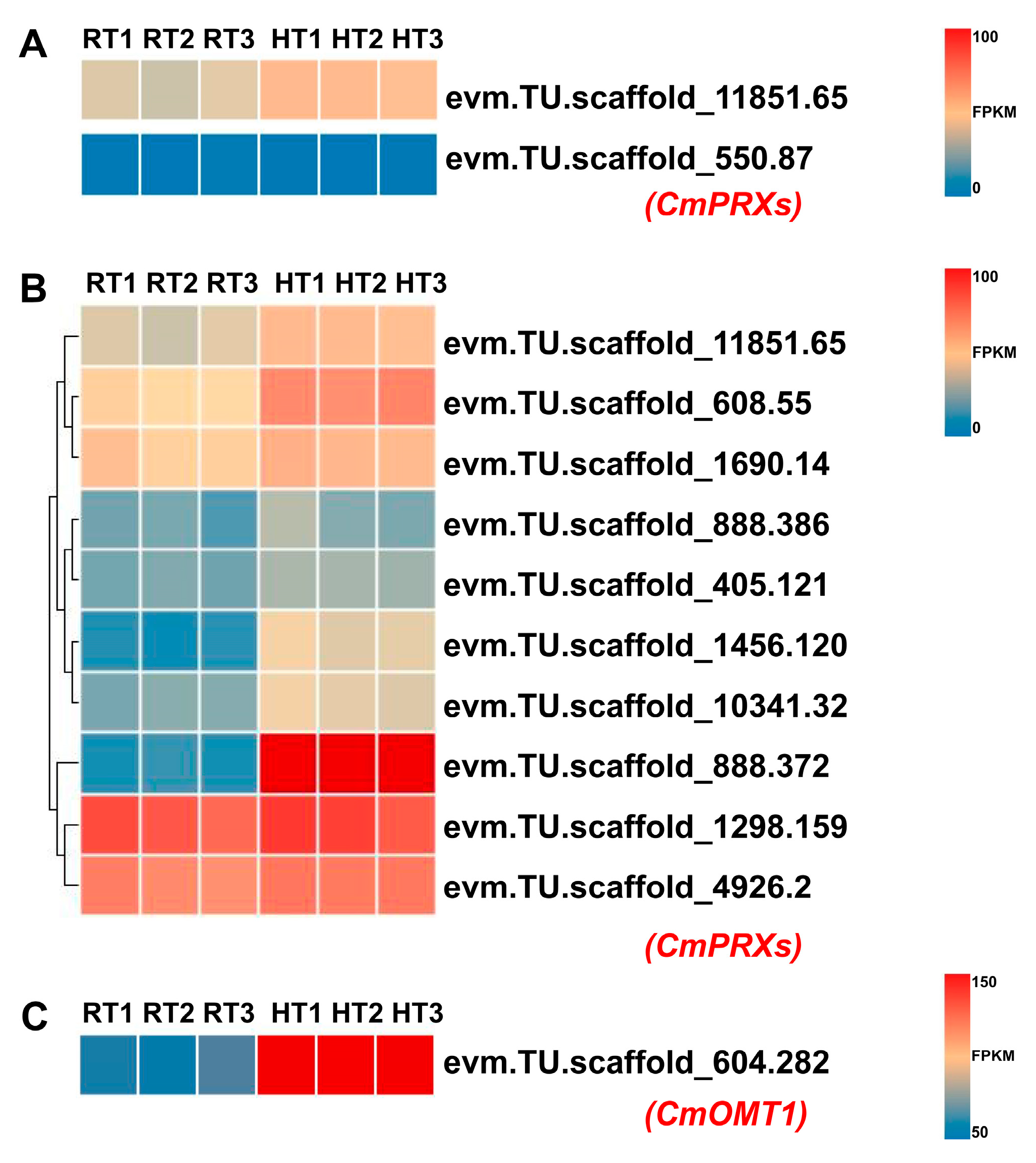
CmPRX (
evm.TU.scaffold_11851.65), homologous to grape
VviPRX31—known to participate in anthocyanin degradation—showed slight induction. Additional heat-responsive
CmPRX genes (
evm.TU.scaffold_1456.120,
evm.TU.scaffold_10341.32, and
evm.TU.scaffold_888.372) also exhibited elevated expression, with
evm.TU.scaffold_888.372 showing the highest upregulation among all candidates. Furthermore,
CmOMT1 (
evm.TU.scaffold_604.282), an O-methyltransferase gene likely involved in the downstream methylation of anthocyanin degradation products, was significantly upregulated (
Figure 8). Combined with our finding that anthocyanin levels decline under heat stress without transcriptional repression of biosynthetic genes at the full-bloom stage, these results provide direct transcriptional evidence supporting our central hypothesis: heat stress induces petal fading by activating specific enzymatic degradation pathways of anthocyanins.
Looking ahead, several avenues warrant further investigation based on our findings. Functional validation of the key candidate genes identified, particularly the highly heat-responsive CmPRX (e.g., evm.TU.scaffold_888.372) and CmOMT1 (evm.TU.scaffold_604.282), using techniques like CRISPR/Cas9 or VIGS in chrysanthemum is crucial to confirm their direct roles in heat-induced anthocyanin degradation. Elucidating the upstream signaling pathways and transcriptional regulators that mediate the heat induction of these degradation genes represents another important research direction. Furthermore, assessing the conservation of this PRX-OMT1 mediated degradation mechanism across diverse chrysanthemum cultivars and other ornamental species would be valuable. Ultimately, translating this knowledge into practical applications, such as developing molecular markers for breeding programs or designing strategies to mitigate anthocyanin degradation, could significantly benefit the horticulture industry by improving flower color stability under challenging high-temperature conditions.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}