
Functional Characteristics and Cellulose Degradation Genes of the Microbial Community in Soils with Different Initial pH Values


Abstract
1. Introduction
2. Materials and Methods
2.1. Soil Sample
2.2. Soil Experimental Procedure
2.3. Soil Chemical Analysis
2.4. CO2 Flux Measurement
2.5. DNA Extraction, High-Throughput Sequencing, and qPCR Amplification
2.6. Statistical Analysis
3. Results and Discussion
3.1. Soil CO2 Emission and Soil Properties
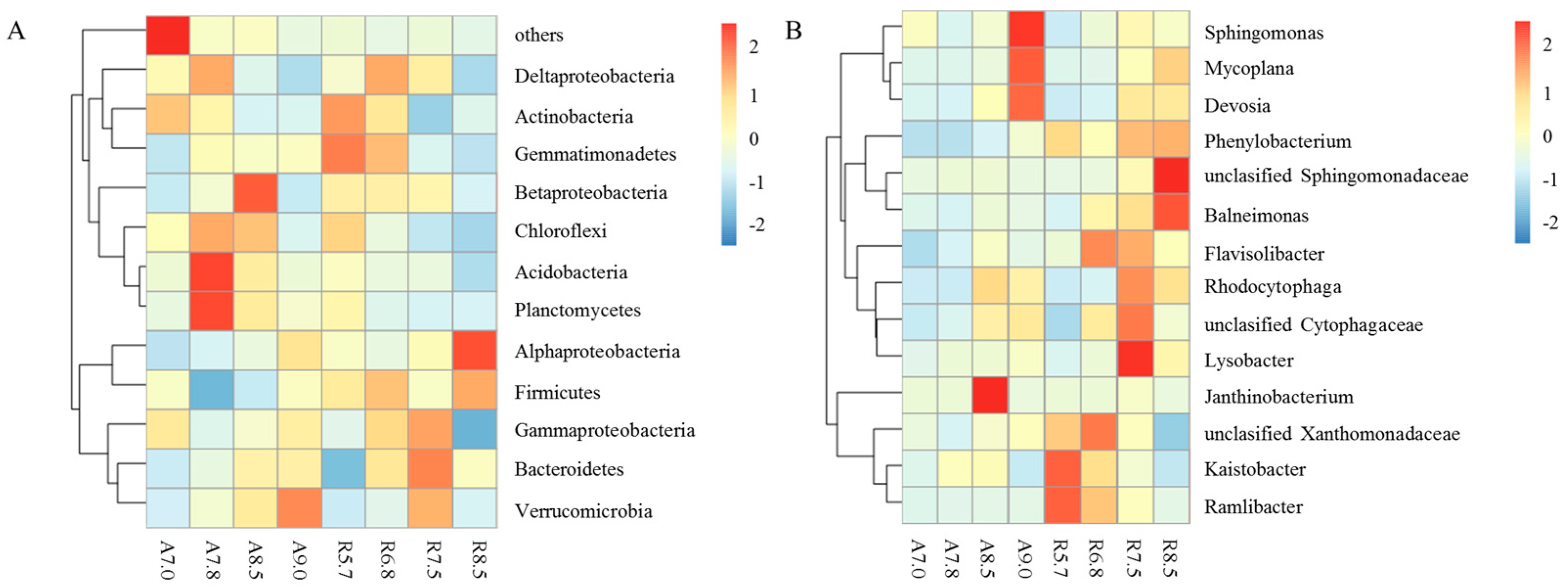
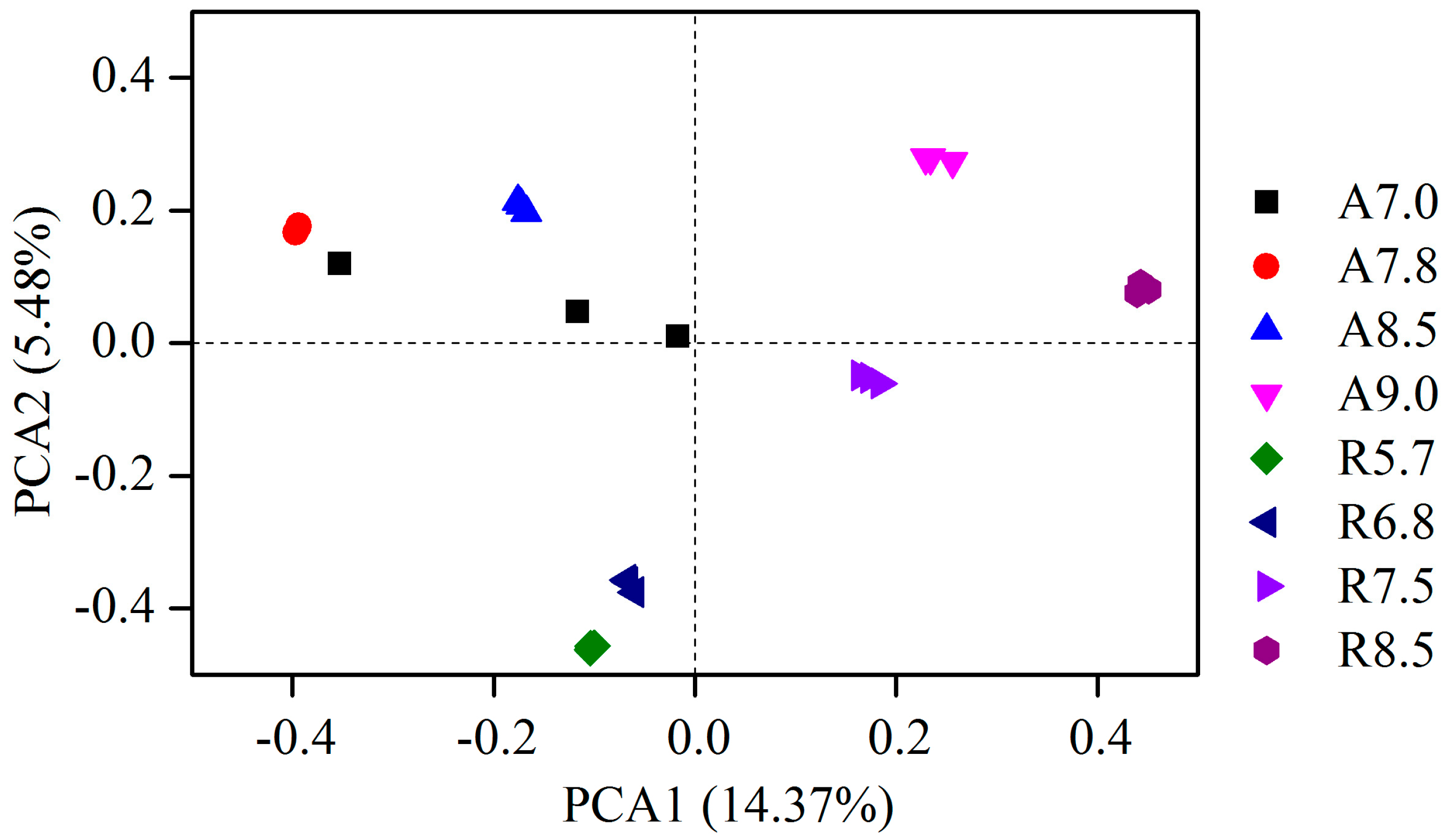
3.2. Bacterial Community Composition
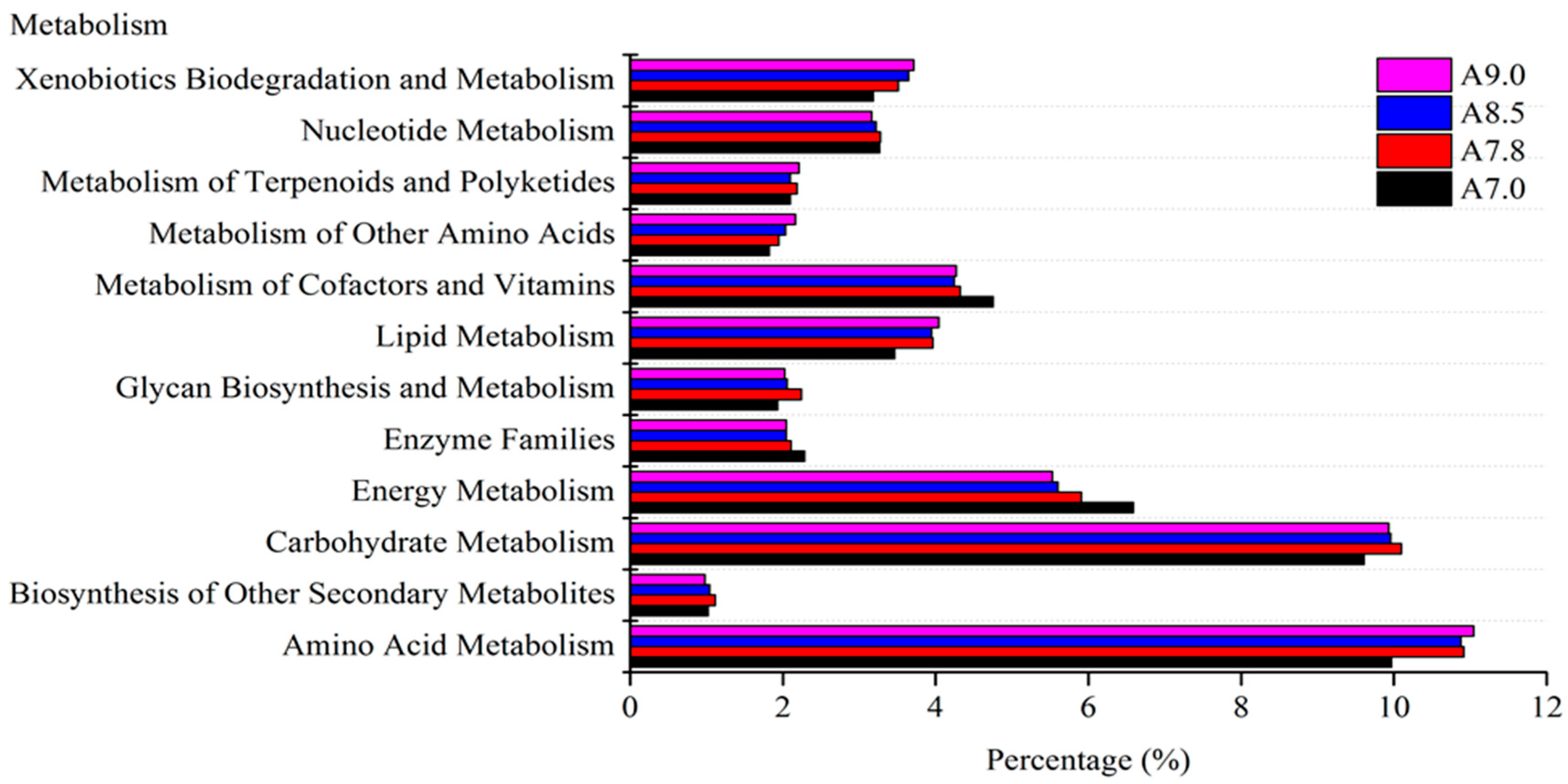
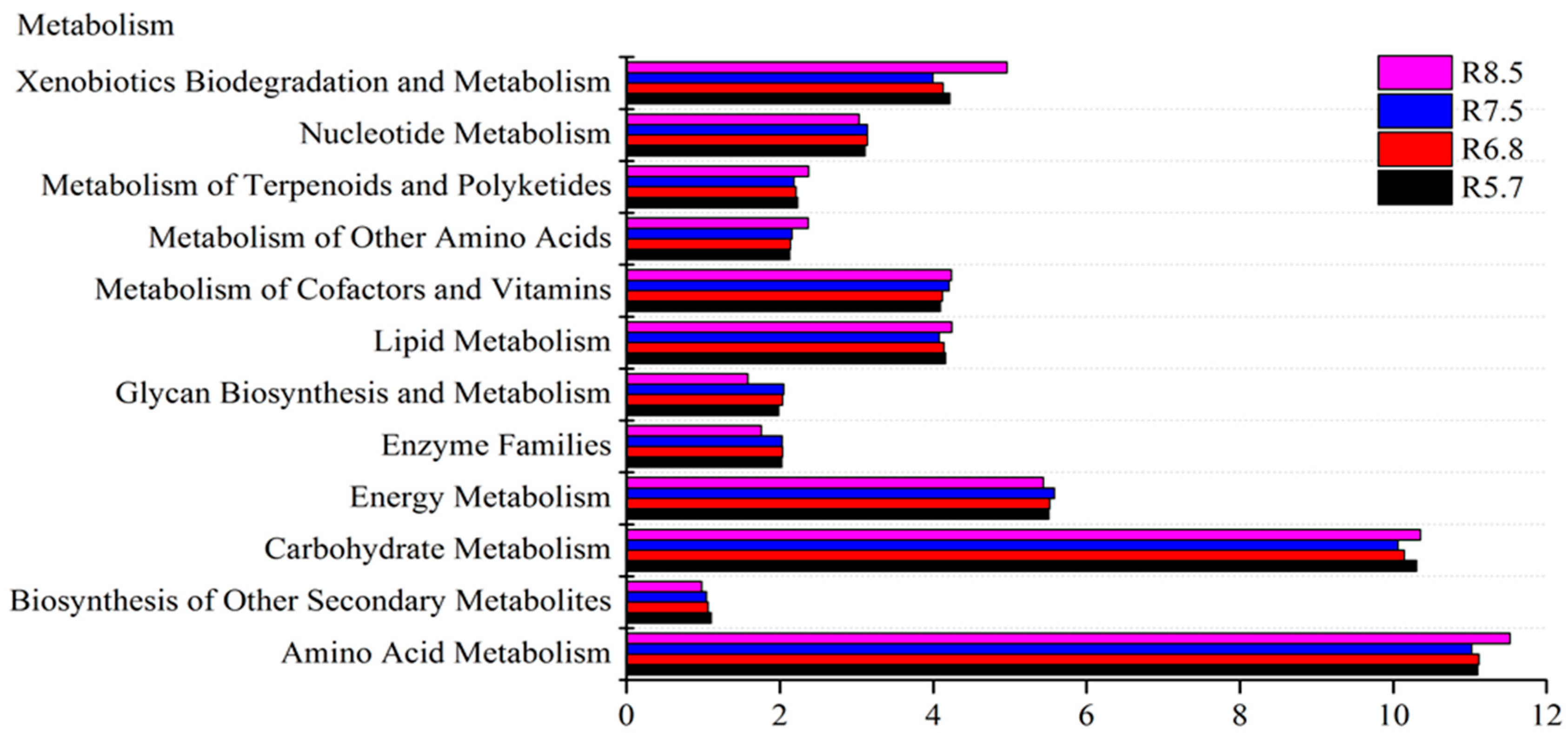
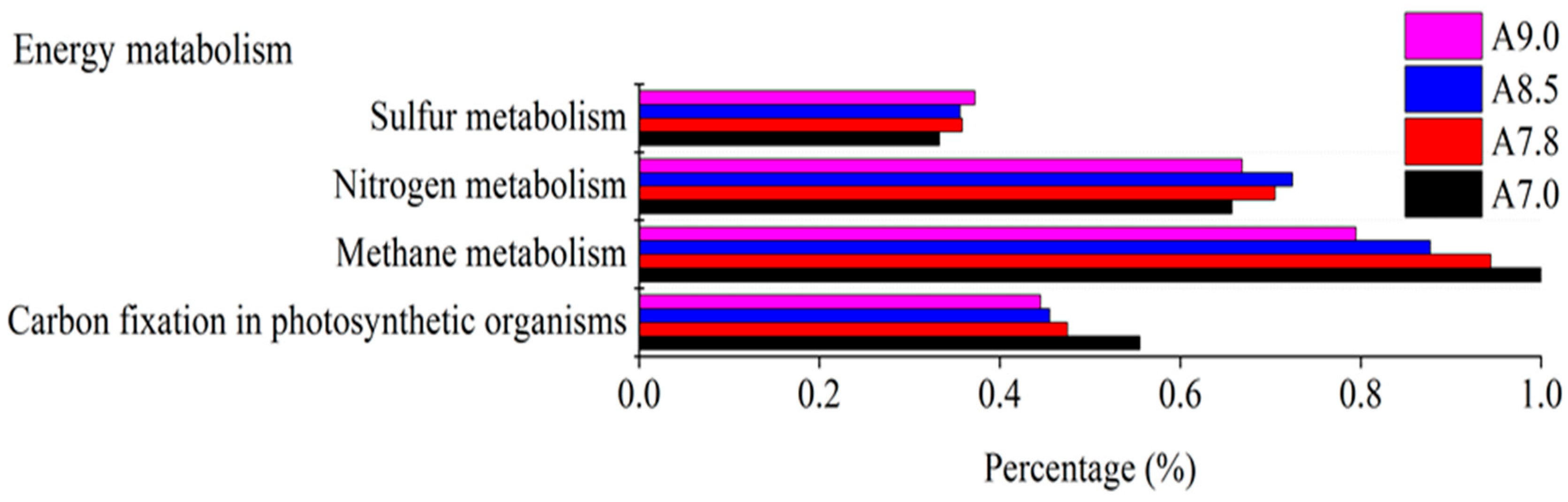
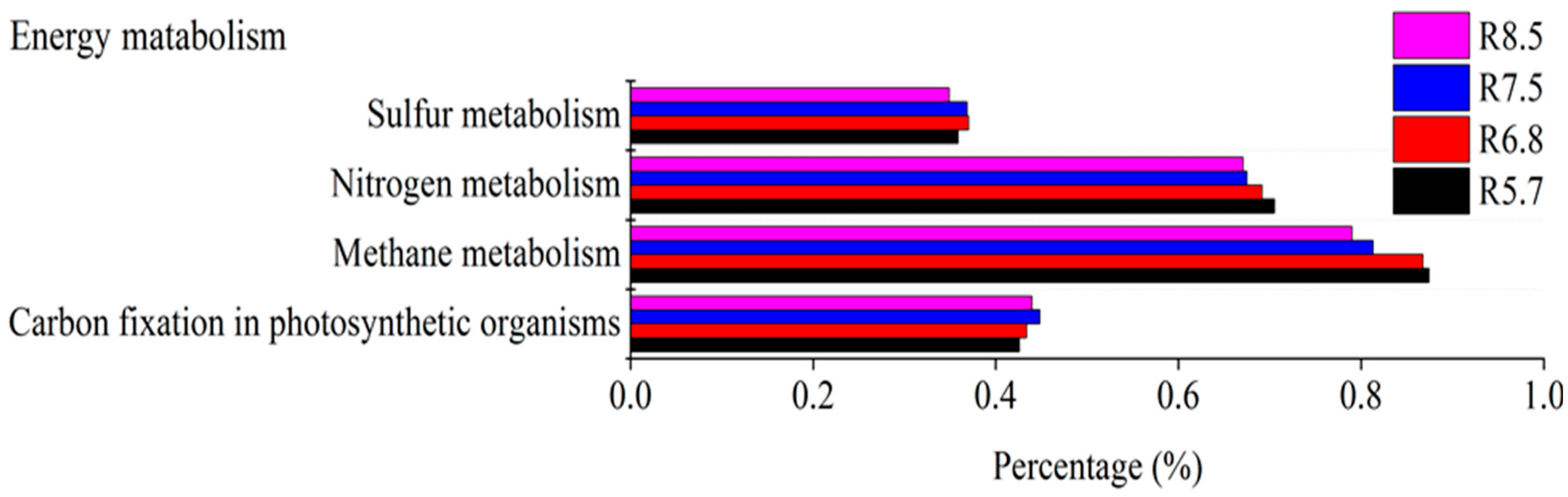
3.3. Bacterial Functional Prediction in Response to pH Gradient
3.4. GH48 and cbhI Gene Abundance Under Different pH Values
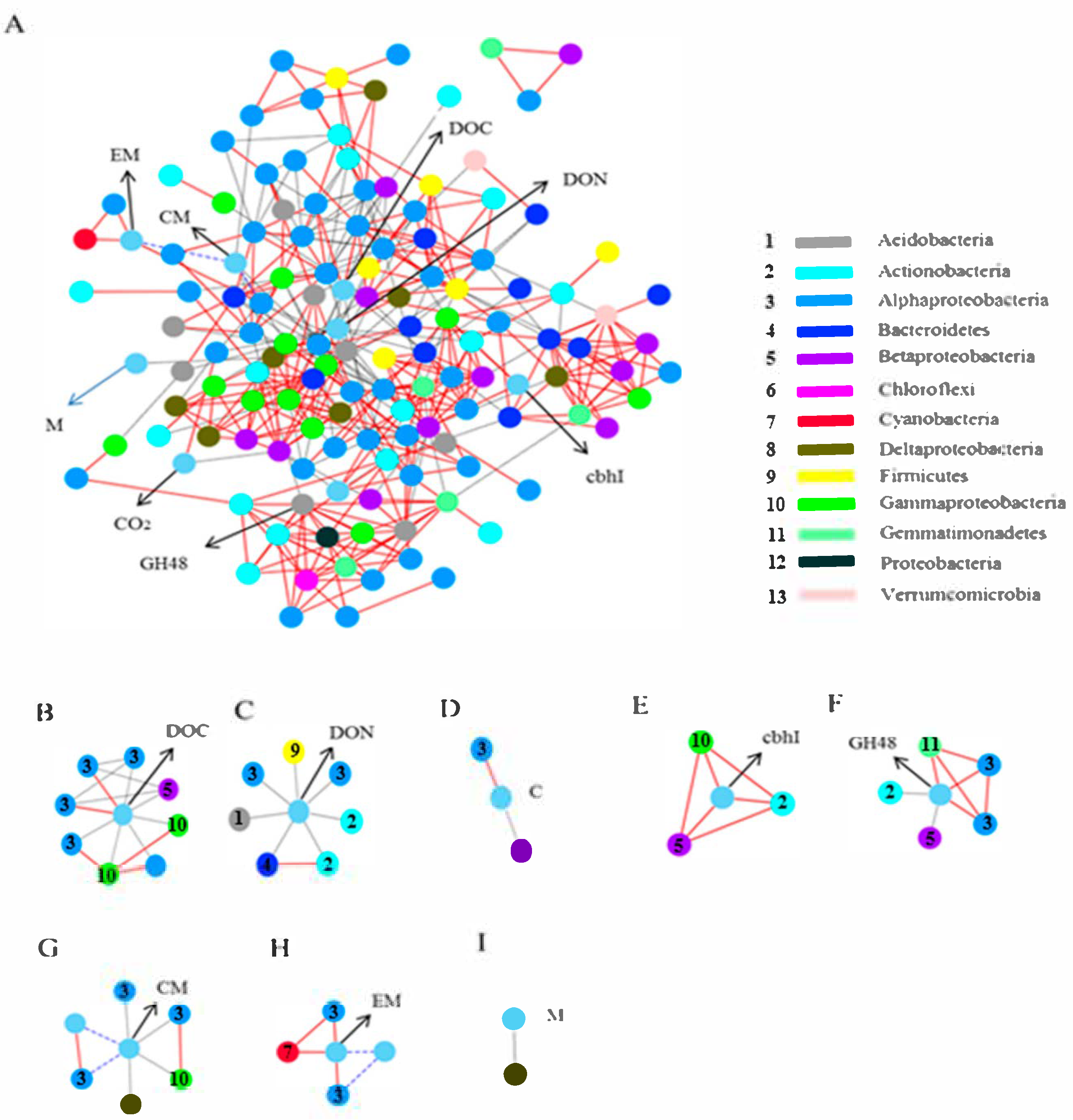
3.5. Network Associations Among Bacterial Community and Soil Chemical Characteristics, Functional Genes, and Microbial Metabolism
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, X.; Chen, Y.; Liu, Y.; Wang, S.; Jin, J.; Zhao, Y.; Yu, D. A framework combining CENTURY modeling and chronosequences sampling to estimate soil organic carbon stock in an agricultural region with large land use change. Agronomy 2023, 13, 1055. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, K.; Han, L.; Chen, Y.; Liu, J.; Xing, B. Biochar stability and impact on soil organic carbon mineralization depend on biochar processing, aging and soil clay content. Soil Biol. Biochem. 2022, 169, 108657. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, X.; Jing, Y.; Luo, J.; Guo, D.; Ma, Y. Dissolved N and C leaching losses mitigated by optimized fertilization management in intensive greenhouse system: Insights from DOM characteristics via EEM-PARAFAC. J. Soils Sediments 2023, 23, 657–671. [Google Scholar] [CrossRef]
- Jing, Y.; Zhang, Y.; Han, I.; Wang, P.; Mei, Q.; Huang, Y. Effects of different straw biochars on soil organic carbon, nitrogen, available phosphorus, and enzyme activity in paddy soil. Sci. Rep. 2020, 10, 8837. [Google Scholar] [CrossRef]
- Zhang, C.; Li, X.; Yan, H.; Ullah, I.; Zuo, Z.; Li, L.; Yu, J. Effects of irrigation quantity and biochar on soil physical properties, growth characteristics, yield and quality of greenhouse tomato. Agric. Water Manag. 2020, 241, 106263. [Google Scholar] [CrossRef]
- Feng, L.; Xu, Y.; Xiao, Y.; Song, J.; Li, D.; Zhang, Z.; Liu, C.; Liu, C.; Jiang, N.; Zhang, M.; et al. Effects of pre-drying treatments combined with explosion puffing drying on the physicochemical properties, antioxidant activities and flavor characteristics of apples. Food Chem. 2021, 338, 128015. [Google Scholar] [CrossRef]
- Tunio, M.H.; Gao, J.; Qureshi, W.A.; Sheikh, S.A.; Chen, J.; Chandio, F.A.; Lakhiar, I.A.; Solangi, K.A. Effects of droplet size and spray interval on root-to-shoot ratio, photosynthesis efficiency, and nutritional quality of aeroponically grown butterhead lettuce. Int. J. Agric. Biol. Eng. 2022, 15, 79–88. [Google Scholar]
- Mao, H.; Kumi, F.; Li, Q.; Han, L. Combining X-ray computed tomography with relevant techniques for analyzing soil–root dynamics—An overview. Acta Agric. Scand. Sect. B—Soil Plant Sci. 2016, 66, 1–19. [Google Scholar] [CrossRef]
- Li, G.; Kim, S.; Han, S.H.; Chang, H.; Du, D.; Son, Y. Precipitation affects soil microbial and extracellular enzymatic responses to warming. Soil Biol. Biochem. 2018, 120, 212–221. [Google Scholar] [CrossRef]
- Li, P.; Li, Y.; Zheng, X.; Ding, L.; Ming, F.; Pan, A.; Lv, W.; Tang, X. Rice straw decomposition affects diversity and dynamics of soil fungal community, but not bacteria. J. Soils Sediments 2018, 18, 248–258. [Google Scholar] [CrossRef]
- Wang, C.; Wu, B.; Jiang, K.; Wei, M.; Wang, S. Effects of different concentrations and types of Cu and Pb on soil N-fixing bacterial communities in the wheat rhizosphere. Appl. Soil Ecol. 2019, 144, 51–59. [Google Scholar] [CrossRef]
- Shen, C.; Xiong, J.; Zhang, H.; Feng, Y.; Lin, X.; Li, X.; Liang, W.; Chu, H. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol. Biochem. 2013, 57, 204–211. [Google Scholar] [CrossRef]
- Yun, Y.; Wang, H.; Man, B.; Xiang, X.; Zhou, J.; Qiu, X.; Duan, Y.; Engel, A.S. The Relationship between pH and bacterial communities in a single karst ecosystem and its implication for soil acidification. Front. Microbiol. 2016, 7, 1955. [Google Scholar] [CrossRef] [PubMed]
- Rousk, J.; Brookes, P.C.; Bååth, E. Investigating the mechanisms for the opposing pH relationships of fungal and bacterial growth in soil. Soil Biol. Biochem. 2010, 42, 926–934. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, W.; Li, P.; Ten, B. Complementary Utilization of Nutrients between Phragmites Australis and Phalaris Arundinacea Based on Accumulative Dynamics of Soil Nutrient during Litter Decomposition. Commun. Soil Sci. Plant Anal. 2021, 52, 1622–1630. [Google Scholar] [CrossRef]
- Cayetano, R.D.A.; Park, J.; Kim, G.-B.; Jung, J.-H.; Kim, S.-H. Enhanced anaerobic digestion of waste-activated sludge via bioaugmentation strategy—Phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt2) analysis through hydrolytic enzymes and possible linkage to system performance. Bioresour. Technol. 2021, 332, 125014. [Google Scholar] [CrossRef]
- Abdualrahman, M.A.Y.; Ma, H.; Zhou, C.; Yagoub, A.E.A.; Hu, J.; Yang, X. Thermal and single frequency counter-current ultrasound pretreatments of sodium caseinate: Enzymolysis kinetics and thermodynamics, amino acids composition, molecular weight distribution and antioxidant peptides. J. Sci. Food Agric. 2016, 96, 4861–4873. [Google Scholar] [CrossRef]
- Vincze, É.-B.; Becze, A.; Laslo, É.; Mara, G. Beneficial Soil microbiomes and their potential role in plant growth and soil fertility. Agriculture 2024, 14, 152. [Google Scholar] [CrossRef]
- Zhang, Q.; Liang, G.; Guo, T.; He, P.; Wang, X.; Zhou, W. Evident variations of fungal and actinobacterial cellulolytic communities associated with different humified particle-size fractions in a long-term fertilizer experiment. Soil Biol. Biochem. 2017, 113, 1–13. [Google Scholar] [CrossRef]
- Min, X.; Xiao, L.; Li, Z.; Li, P.; Wang, F.; Liu, X.; Chen, S.; Wang, Z.; Pan, L. Effects of Incubation Temperature and Sludge Addition on Soil Organic Carbon and Nitrogen Mineralization Characteristics in Degraded Grassland Soil. Agronomy 2024, 14, 1590. [Google Scholar] [CrossRef]
- Zang, X.; Liu, M.; Fan, Y.; Xu, J.; Xu, X.; Li, H. The structural and functional contributions of β-glucosidase-producing microbial communities to cellulose degradation in composting. Biotechnol. Biofuels 2018, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Qu, F.; Li, D.; Zhao, Y.; Li, X.; Niu, S.; Zhao, M.; Qi, H.; Wei, Z.; Song, C. Effect of Fenton pretreatment and bacterial inoculation on cellulose-degrading genes and fungal communities during rice straw composting. Sci. Total. Environ. 2022, 806, 151376. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Kirkby, C.A.; Schmutter, D.; Bissett, A.; Kirkegaard, J.A.; Richardson, A.E. Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biol. Biochem. 2016, 97, 188–198. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef]
- Yuan, Q.; Gao, Y.; Ma, G.; Wu, H.; Li, Q.; Zhang, Y.; Liu, S.; Jie, X.; Zhang, D.; Wang, D. The long-term effect of biochar amendment on soil biochemistry and phosphorus availability of calcareous soils. Agriculture 2025, 15, 458. [Google Scholar] [CrossRef]
- Li, H.; Zhou, B.; Zhuo, Z.; Wang, L.; Wang, Z.; Xie, C.; Jiang, F.; Lin, J.; Huang, Y.; Zhang, Y. Effects of Cover Measures on Soil Organic Nitrogen Fractions and Total Soluble Nitrogen Pools in Citrus Orchards of the Red Soil Hilly Region of Southern China. Agriculture 2024, 14, 1879. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, X.; Xu, B.; Yagoub, A.E.A.; Mustapha, A.T.; Zhou, C. Effect of intensive pulsed light on the activity, structure, physico-chemical properties and surface topography of polyphenol oxidase from mushroom. Innov. Food Sci. Emerg. Technol. 2021, 72, 102741. [Google Scholar] [CrossRef]
- Tian, Y.; Wu, L.; Lu, R.; Bao, H.; Zhou, Y.; Pang, M.; Brown, J.; Wang, J.; Wang, R.; Zhang, H. Virulent phage vB_CpeP_HN02 inhibits Clostridium perfringens on the surface of the chicken meat. Int. J. Food Microbiol. 2022, 363, 109514. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Liu, L.; Zhou, J.; Wan, Q.; Chen, J.; Cao, Y.; Zhang, L.; Feng, F.; Ning, Q.; et al. Bibliometric Analysis of Contemporary Research on the Amelioration of Saline Soils. Agronomy 2024, 14, 2935. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, Y.; Cayuela, M.L.; Sánchez-Monedero, M.A.; Wang, Q. Compost biochemical quality mediates nitrogen leaching loss in a greenhouse soil under vegetable cultivation. Geoderma 2020, 358, 113984. [Google Scholar] [CrossRef]
- Cai, C.; Wei, B.; Tian, Y.; Ma, R.; Chen, L.; Qiu, L.; Jin, Z. Structural changes of chemically modified rice starch by one-step reactive extrusion. Food Chem. 2019, 288, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Akhlaq, M.; Zhang, C.; Yan, H.; Ou, M.; Zhang, W.; Liang, S.; Ikram, R.M.A. Response of tomato growth to continuous elevated CO2 concentration under controlled environment. Int. J. Agric. Biol. Eng. 2022, 15, 51–59. [Google Scholar] [CrossRef]
- Sun, B.; Wang, X.; Wang, F.; Jiang, Y.; Zhang, X.-X. Assessing the relative effects of geographic location and soil type on microbial communities associated with straw decomposition. Appl. Environ. Microbiol. 2013, 79, 3327–3335. [Google Scholar] [CrossRef]
- Li, F.; Chen, L.; Zhang, J.; Yin, J.; Huang, S. Bacterial community structure after long-term organic and inorganic fertilization reveals important associations between soil nutrients and specific taxa involved in nutrient transformations. Front. Microbiol. 2017, 8, 187. [Google Scholar] [CrossRef]
- Duan, N.; Gong, W.; Wu, S.; Wang, Z. Selection and Application of ssDNA Aptamers against Clenbuterol Hydrochloride Based on ssDNA Library Immobilized SELEX. J. Agric. Food Chem. 2017, 65, 1771–1777. [Google Scholar] [CrossRef]
- Lu, Y.Z.; Liu, P.F.; Montazar, A.; Paw, U.K.T.; Hu, Y.G. Soil water infiltration model for sprinkler irrigation control strategy: A case for tea plantation in Yangtze river region. Agriculture 2019, 9, 206. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, H.; Liu, J.; Yang, S.X. Control method of seedbed compactness based on fragment soil compaction dynamic characteristics. Soil Tillage Res. 2020, 198, 104551. [Google Scholar] [CrossRef]
- Edwards, I.P.; Upchurch, R.A.; Zak, D.R. Isolation of fungal cellobiohydrolase I genes from sporocarps and forest soils by PCR. Appl. Environ. Microbiol. 2008, 74, 3481–3489. [Google Scholar] [CrossRef]
- de Menezes, A.B.; Prendergast-Miller, M.T.; Poonpatana, P.; Farrell, M.; Bissett, A.; Macdonald, L.M.; Toscas, P.; Richardson, A.E.; Thrall, P.H. C/N Ratio drives soil actinobacterial cellobiohydrolase gene diversity. Appl. Environ. Microbiol. 2015, 81, 3016–3028. [Google Scholar] [CrossRef]
- Zeng, J.; Yuan, Q.; Xu, W.; Li, H.; Li, M.; Lei, X.; Wang, W.; Lin, Q.; Li, X.; Xu, R.; et al. Research on a Biofilter for a Typical Application Scenario in China: Treatment of Pesticide Residue Wastewater in Orchards. Agronomy 2024, 14, 934. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Hu, F.; Zhang, W.; Fang, D.; Dong, K.; Cao, J. Amino acid transporters of Brassica napus: Identification, evolution, expression and response to various stresses. Ind. Crop. Prod. 2023, 194, 116338. [Google Scholar] [CrossRef]
- Huang, Y.; Sun, Y.; Mehmood, A.; Lu, T.; Chen, X. Unraveling the temporal changes of Maillard reaction products and aroma profile in coffee leaves during hot-air drying. J. Food Compos. Anal. 2024, 128, 106055. [Google Scholar] [CrossRef]
- Reshef, D.N.; Reshef, Y.A.; Finucane, H.K.; Grossman, S.R.; McVean, G.; Turnbaugh, P.J.; Lander, E.S.; Mitzenmacher, M.; Sabeti, P.C. Detecting novel associations in large data sets. Science 2011, 334, 1518–1524. [Google Scholar] [CrossRef]
- Wang, K.; Zheng, X.; Su, Y.; Lu, Y.; Yang, Q.; Shi, Y.; Lanhuang, B.; Zhang, X.; Zhao, L.; Godana, E.A.; et al. A glycoside hydrolase superfamily gene plays a major role in Penicillium expansum growth and pathogenicity in apples. Postharvest Biol. Technol. 2023, 198, 112228. [Google Scholar] [CrossRef]
- Su, Z.; Li, Y.; Dong, Y.; Tang, Z.; Liang, Z. Simulation of rice threshing performance with concentric and non-concentric threshing gaps. Biosyst. Eng. 2020, 197, 270–284. [Google Scholar] [CrossRef]
- Jiang, N.J.; Wang, Y.J.; Chu, J.; Kawasaki, S.; Tang, C.S.; Cheng, L.; Du, Y.J.; Shashank, B.S.; Singh, D.N.; Han, X.L.; et al. Bio-mediated soil improvement: An introspection into processes, materials, characterization and applications. Soil Use Manage 2022, 38, 68–93. [Google Scholar] [CrossRef]
- Xu, J.; Tang, C.; Chen, Z. Chemical composition controls residue decomposition in soils differing in initial pH. Soil Biol. Biochem. 2006, 38, 544–552. [Google Scholar] [CrossRef]
- Zuo, Z.; Li, X.; Xu, C.; Yang, J.; Zhu, X.; Liu, S.; Song, F.; Liu, F.; Mao, H. Responses of barley Albina and Xantha mutants deficient in magnesium chelatase to soil salinity. Plant Soil Environ. 2017, 63, 348–354. [Google Scholar] [CrossRef]
- Liu, E.; Terumasa, T. Effects of Applying Recycled Urban Green Waste Compost Made from Pruning Materials to Soil on the Growth of Plants. J. Soil Sci. Plant Nutr. 2022, 22, 1088–1097. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Goncalves, P.; Copeland, E.; Qi, S.-S.; Dai, Z.-C.; Li, G.-L.; Wang, C.-Y.; Du, D.-L.; Thomas, T. Invasion by the weed Conyza canadensis alters soil nutrient supply and shifts microbiota structure. Soil Biol. Biochem. 2020, 143, 107739. [Google Scholar] [CrossRef]
- Castro, H.F.; Classen, A.T.; Austin, E.E.; Norby, R.J.; Schadt, C.W. Soil microbial community responses to multiple experimental climate change drivers. Appl. Environ. Microbiol. 2010, 76, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Pawlett, M.; Hopkins, D.W.; Moffett, B.F.; Harris, J.A. The effect of earthworms and liming on soil microbial communities. Biol. Fertil. Soils 2009, 45, 361–369. [Google Scholar] [CrossRef]
- Bartram, A.K.; Jiang, X.; Lynch, M.D.; Masella, A.P.; Nicol, G.W.; Dushoff, J.; Neufeld, J.D. Exploring links between pH and bacterial community composition in soils from the Craibstone Experimental Farm. FEMS Microbiol. Ecol. 2014, 87, 403–415. [Google Scholar] [CrossRef]
- Hang, T.; Lu, N.; Takagaki, M.; Mao, H. Leaf area model based on thermal effectiveness and photosynthetically active radiation in lettuce grown in mini-plant factories under different light cycles. Sci. Hortic. 2019, 252, 113–120. [Google Scholar] [CrossRef]
- Lueders, T.; Kindler, R.; Miltner, A.; Friedrich, M.W.; Kaestner, M. Identification of bacterial micropredators distinctively active in a soil microbial food web. Appl. Environ. Microbiol. 2006, 72, 5342–5348. [Google Scholar] [CrossRef]
- Wakelin, S.A.; Colloff, M.J.; Kookana, R.S. Effect of wastewater treatment plant effluent on microbial function and community structure in the sediment of a freshwater stream with variable seasonal flow. Appl. Environ. Microbiol. 2008, 74, 2659–2668. [Google Scholar] [CrossRef]
- Lafortune, I.; Juteau, P.; Déziel, E.; Lépine, F.; Beaudet, R.; Villemur, R. Bacterial diversity of a consortium degrading high-molecular-weight polycyclic aromatic hydrocarbons in a two-liquid phase biosystem. Microb. Ecol. 2008, 57, 455–468. [Google Scholar] [CrossRef]
- Legrand, F.; Picot, A.; Cobo-Díaz, J.F.; Carof, M.; Chen, W.; Le Floch, G. Effect of tillage and static abiotic soil properties on microbial diversity. Appl. Soil Ecol. 2018, 132, 135–145. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Y.; Xia, D.; Jiang, X.; Fu, D.; Shen, L.; Wang, H.; Li, Q.B. Enhanced bioreduction of iron and arsenic in sediment by biochar amendment influencing microbial community composition and dissolved organic matter content and composition. J. Hazard. Mater. 2016, 311, 20–29. [Google Scholar] [CrossRef]
- Jensen, M.; Michelsen, A.; Gashaw, M. Responses in plant, soil inorganic and microbial nutrient pools to experimental fire, ash and biomass addition in a woodland savanna. Oecologia 2001, 128, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Calviño, D.; Bååth, E. Growth response of the bacterial community to pH in soils differing in pH. FEMS Microbiol. Ecol. 2010, 73, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Meng, T.; Ma, Y. Sugars altered fungal community composition and caused high network complexity in a Fusarium wilt pathogen-infested soil. Biol. Fertil. Soils 2020, 56, 395–409. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, H.; Xu, C.; Zhong, Y.; Xu, X.; Yuan, J.; Wang, J.; Zhang, Y. Ten years of urea fertilization alter the pqqC-harbouring community and increase soil inorganic phosphorus mobilization. Eur. J. Soil Sci. 2024, 75, e13563. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Y.; Yang, J.; Fan, F.; Zhang, W.; Li, J.; Zhou, C.; Shi, G.; Tong, F.; Fan, G. Taxonomical and functional bacterial community selection in the rhizosphere of the rice genotypes with different nitrogen use efficiencies. Plant Soil 2022, 470, 111–125. [Google Scholar] [CrossRef]
- Solangi, K.A.; Siyal, A.A.; Wu, Y.; Abbasi, B.; Solangi, F.; Lakhiar, I.A.; Zhou, G. An Assessment of the spatial and temporal distribution of soil salinity in combination with field and satellite data: A case study in sujawal district. Agronomy 2019, 9, 869. [Google Scholar] [CrossRef]
- Zhong, R.; Wan, X.; Wang, D.; Zhao, C.; Liu, D.; Gao, L.; Wang, M.; Wu, C.; Nabavid, S.M.; Daglia, M.; et al. Polysaccharides from Marine Enteromorpha: Structure and function. Trends Food Sci. Technol. 2020, 99, 11–20. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, W.; Li, Z.; Zhang, N.; Wang, S.; Shi, J.; Zhai, X.; Zhang, J.; Shen, T. Preparation of a Dual-Functional Active Film Based on Bilayer Hydrogel and Red Cabbage Anthocyanin for Maintaining and Monitoring Pork Freshness. Foods 2023, 12, 4520. [Google Scholar] [CrossRef]
- Wen, C.; Zhang, J.; Duan, Y.; Zhang, H.; Ma, H. A Mini-Review on Brewer’s Spent Grain Protein: Isolation, Physicochemical Properties, Application of Protein, and Functional Properties of Hydrolysates. J. Food Sci. 2019, 84, 3330–3340. [Google Scholar] [CrossRef]
- Ouyang, X.-J.; Guo-Yi, Z.; Huang, Z.-L.; Ju-Xiu, L.; Zhang, D.-Q.; Jiong, L. Effect of simulated acid rain on potential carbon and nitrogen mineralization in forest soils. Pedosphere 2008, 18, 503–514. [Google Scholar] [CrossRef]
- Goleij, P.; Khandan, M.; Tabari, M.A.K.; Sanaye, P.M.; Alijanzadeh, D.; Soltani, A.; Hosseini, Z.; Larsen, D.S.; Khan, H.; Kumar, A.P.; et al. Unlocking the Potential: How Flavonoids Affect Angiogenesis, Oxidative Stress, Inflammation, Proliferation, Invasion, and Alter Receptor Interactions in Endometriosis. Food Sci. Nutr. 2025, 13, e4607. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Zhu, S.; Jiang, Z.; Ji, Y.; Wang, F.; Zhao, R.; Bie, T. Comparative mapping of powdery mildew resistance gene Pm21 and functional characterization of resistance-related genes in wheat. Theor. Appl. Genet. 2016, 129, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Apaliya, M.T.; Zhang, H.; Yang, Q.; Zheng, X.; Zhao, L.; Kwaw, E.; Mahunu, G.K. Hanseniaspora uvarum enhanced with trehalose induced defense-related enzyme activities and relative genes expression levels against Aspergillus tubingensis in table grapes. Postharvest Biol. Technol. 2017, 132, 162–170. [Google Scholar] [CrossRef]
- Pang, Y.; Li, H.; Tang, P.; Chen, C. Synchronization optimization of pipe diameter and operation frequency in a pressurized irrigation network based on the genetic algorithm. Agriculture 2022, 12, 673. [Google Scholar] [CrossRef]
- Shi, B.; Sreeram, V.; Zhao, D.; Duan, S.; Jiang, J. A wireless sensor network-based monitoring system for freshwater fishpond aquaculture. Biosyst. Eng. 2018, 172, 57–66. [Google Scholar] [CrossRef]
- Zhao, Z.; Jin, M.; Tian, C.; Yang, S.X. Prediction of seed distribution in rectangular vibrating tray using grey model and artificial neural network. Biosyst. Eng. 2018, 175, 194–205. [Google Scholar] [CrossRef]
- Wu, M.; Sun, J.; Lu, B.; Ge, X.; Zhou, X.; Zou, M. Application of deep brief network in transmission spectroscopy detection of pesticide residues in lettuce leaves. J. Food Process. Eng. 2019, 42, e13005. [Google Scholar] [CrossRef]
- Sauze, J.; Jones, S.P.; Wingate, L.; Wohl, S.; Ogée, J. The role of soil pH on soil carbonic anhydrase activity. Biogeosciences 2018, 15, 597–612. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, R.; Zhang, Y.; Wang, G.; Li, K. Impact of nutrient addition on diversity and fate of fecal bacteria. Sci. Total. Environ. 2018, 636, 717–726. [Google Scholar] [CrossRef]
- Dennis, P.G.; Newsham, K.K.; Rushton, S.P.; O’Donnell, A.G.; Hopkins, D.W. Soil bacterial diversity is positively associated with air temperature in the maritime Antarctic. Sci. Rep. 2019, 9, 2686. [Google Scholar] [CrossRef]
- Cao, B.; Zhang, Y.; Wang, Z.; Li, M.; Yang, F.; Jiang, D.; Jiang, Z. Insight Into the Variation of Bacterial Structure in Atrazine-Contaminated Soil Regulating by Potential Phytoremediator: Pennisetum americanum (L.) K. Schum. Front. Microbiol. 2018, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Teshita, A.; Khan, W.; Ullah, A.; Iqbal, B.; Ahmad, N. Soil Nematodes in Agroecosystems: Linking Cropping System’s Rhizosphere Ecology to Nematode Structure and Function. J. Soil Sci. Plant Nutr. 2024, 24, 6467–6482. [Google Scholar] [CrossRef]
- Zhu, L.; Hou, Z.; Hu, X.; Liu, X.; Dai, T.; Wang, X.; Zeng, C.; Wang, Y.; Wang, C.; Yang, S.; et al. Genomic and metabolic features of an unexpectedly predominant, thermophilic, assistant starter microorganism, Thermus thermophilus, in Chinese Inner Mongolian Cheese. Foods 2021, 10, 2962. [Google Scholar] [CrossRef]
- Hou, X.; Dai, C.; Tang, Y.; Xing, Z.; Mintah, B.K.; Dabbour, M.; Ding, Q.; He, R.; Ma, H. Thermophilic solid-state fermentation of rapeseed meal and analysis of microbial community diversity. LWT 2019, 116, 108520. [Google Scholar] [CrossRef]
- Wei, W.; Hu, X.; Hou, Z.; Wang, Y.; Zhu, L. Microbial community structure and diversity in different types of non-bovine milk. Curr. Opin. Food Sci. 2021, 40, 51–57. [Google Scholar] [CrossRef]
- Zhao, Q.; Shi, Y.; Ngea, G.L.N.; Zhang, X.; Yang, Q.; Zhang, Q.; Xu, X.; Zhang, H. Changes of the microbial community in kiwifruit during storage after postharvest application of Wickerhamomyces anomalus. Food Chem. 2023, 404, 134593. [Google Scholar] [CrossRef]
- Li, H.; Li, P.; Li, J.; Jiang, Y.; Huang, X. Influence of micro/nano aeration on the diversity of the microbial community in drip irrigation to reduce emitter clogging. Biosyst. Eng. 2023, 235, 116–130. [Google Scholar] [CrossRef]
- Zheng, Z.; He, Y.; He, Y.; Zhan, J.; Shi, C.; Xu, Y.; Wang, X.; Wang, J.; Zhang, C. Micro-nano bubble water subsurface drip irrigation affects strawberry yield and quality by modulation of microbial communities. Agric. Water Manag. 2025, 307, 109228. [Google Scholar] [CrossRef]
- Qin, Y.; Yu, H.; Chen, K.; Cui, R.; Cao, J.; Wang, Z.; Zhang, Z.-H.; Soteyome, T. Effects of chitosan/eugenol-loaded IRMOF-3 nanoparticles composite films on reactive oxygen species metabolism and microbial community dynamics in postharvest strawberries. Food Biosci. 2025, 63, 105652. [Google Scholar] [CrossRef]
- Yergeau, E.; Kang, S.; He, Z.; Zhou, J.; Kowalchuk, G.A. Functional microarray analysis of nitrogen and carbon cycling genes across an Antarctic latitudinal transect. ISME J. 2007, 1, 163–179. [Google Scholar] [CrossRef]
- Bodenhausen, N.; Horton, M.W.; Bergelson, J. Bacterial Communities associated with the leaves and the roots of Arabidopsis thaliana. PLoS ONE 2013, 8, e56329. [Google Scholar] [CrossRef] [PubMed]
- Yeager, C.M.; Gallegos-Graves, L.V.; Dunbar, J.; Hesse, C.N.; Daligault, H.; Kuske, C.R. Polysaccharide degradation capability of actinomycetales soil isolates from a semiarid grassland of the colorado plateau. Appl. Environ. Microbiol. 2017, 83, e03020-16. [Google Scholar] [CrossRef]
- Wei, H.; Wang, L.; Hassan, M.; Xie, B. Succession of the functional microbial communities and the metabolic functions in maize straw composting process. Bioresour. Technol. 2018, 256, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Pan, T.; Li, Y.; Chen, S.; Li, G. Development of analytical method associating near-infrared spectroscopy with one-dimensional convolution neural network: A case study. J. Food Meas. Charact. 2021, 15, 2963–2973. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Q.; Li, X.; Obadi, M.; Jiang, S.; Li, S.; Xu, B. Effects of dough resting time on the development of gluten network in different sheeting directions and the textural properties of noodle dough. LWT 2021, 141, 110920. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkaline Soil | Red Soil | |||
|---|---|---|---|---|
| r | p | r | p | |
| Dominant Phyla | ||||
| Alphaproteobacteria | 0.76 | <0.0001 | 0.58 | 0.003 |
| Bacteroidetes | 0.69 | 0.001 | 0.9 | <0.0001 |
| Actinobacteria | 0.78 | 0.0001 | 0.63 | 0.001 |
| Acidobacteria | 0.63 | 0.005 | 0.83 | <0.0001 |
| Functional Genes | ||||
| GH48 | 0.145 | 0.121 | 0.601 | 0.002 |
| cbhI | −0.095 | 0.839 | 0.543 | 0.006 |
| Soil Properties | ||||
| DOC | 0.91 | <0.0001 | 0.95 | <0.0001 |
| DON | 0.95 | <0.0001 | 0.95 | <0.0001 |
| CO2 flux | 0.864 | <0.0001 | 0.714 | 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Xu, B.; Wang, Q. Functional Characteristics and Cellulose Degradation Genes of the Microbial Community in Soils with Different Initial pH Values. Agriculture 2025, 15, 1068. https://doi.org/10.3390/agriculture15101068
Jiang L, Xu B, Wang Q. Functional Characteristics and Cellulose Degradation Genes of the Microbial Community in Soils with Different Initial pH Values. Agriculture. 2025; 15(10):1068. https://doi.org/10.3390/agriculture15101068
Chicago/Turabian StyleJiang, Li, Boyan Xu, and Qi Wang. 2025. "Functional Characteristics and Cellulose Degradation Genes of the Microbial Community in Soils with Different Initial pH Values" Agriculture 15, no. 10: 1068. https://doi.org/10.3390/agriculture15101068
APA StyleJiang, L., Xu, B., & Wang, Q. (2025). Functional Characteristics and Cellulose Degradation Genes of the Microbial Community in Soils with Different Initial pH Values. Agriculture, 15(10), 1068. https://doi.org/10.3390/agriculture15101068