
Identification and Clarification of VrCYCA1: A Key Genic Male Sterility-Related Gene in Mungbean by Multi-Omics Analysis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Genetic Analysis of Mungbean Male Sterile Mutants
2.3. Viability Test of Mature Pollen Grains and Phenotypic Investigation
2.4. Cytological Analysis
2.5. Whole-Genome Resequencing
2.6. SNP-Index and ED Methods to Identify the QTNs of Male Sterility
2.7. Candidate Gene Identification
2.8. RNA-Seq Analysis
2.9. Metabonomic Analysis
2.10. PPI (Protein-Protein Interaction)
3. Results
3.1. Phenotypic Analysis of GMS Mutants in Mungbean
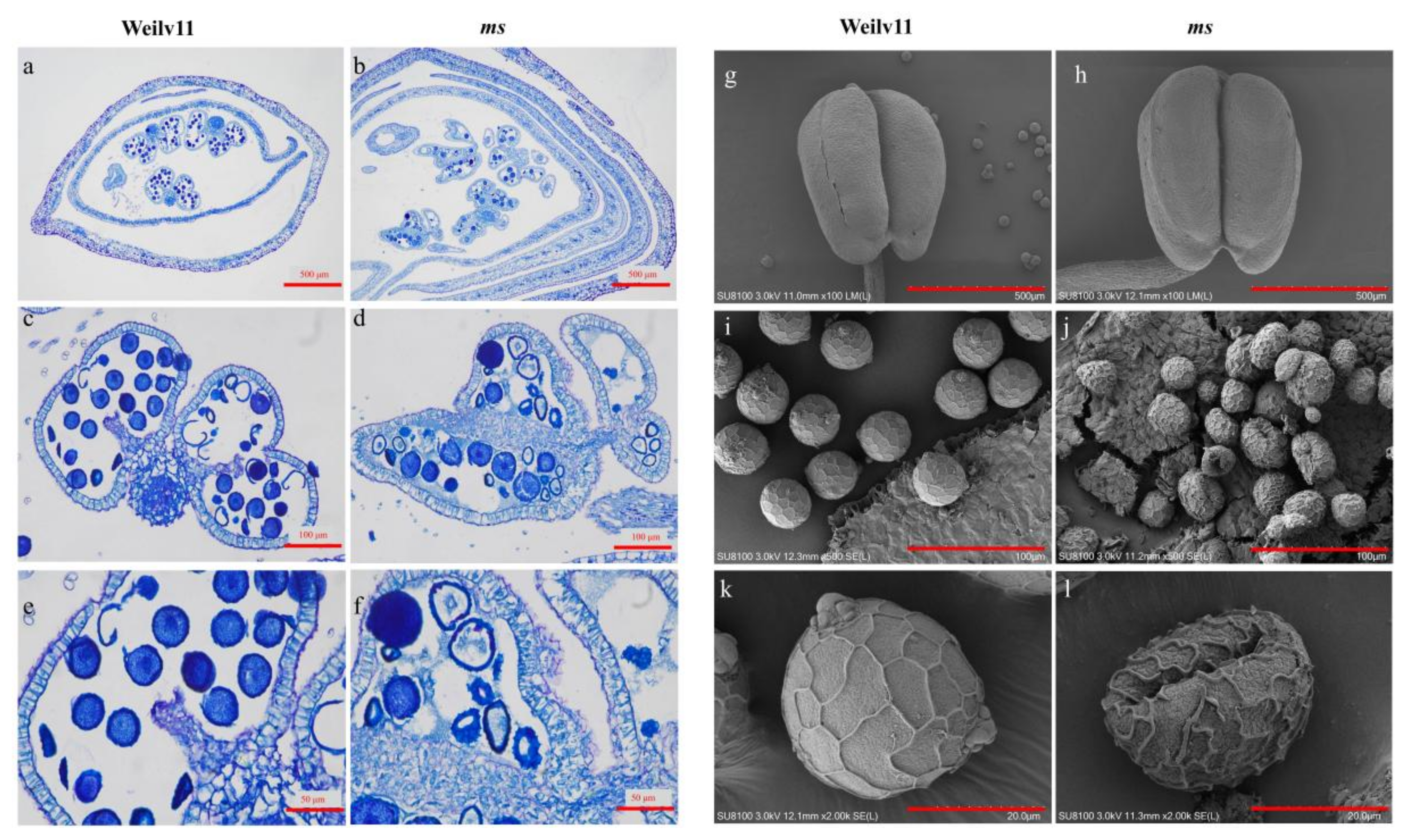
3.2. Histocytological Comparison of ms with Its Wild Type
3.3. Detection of Locus and Gene for GMS in Mungbean
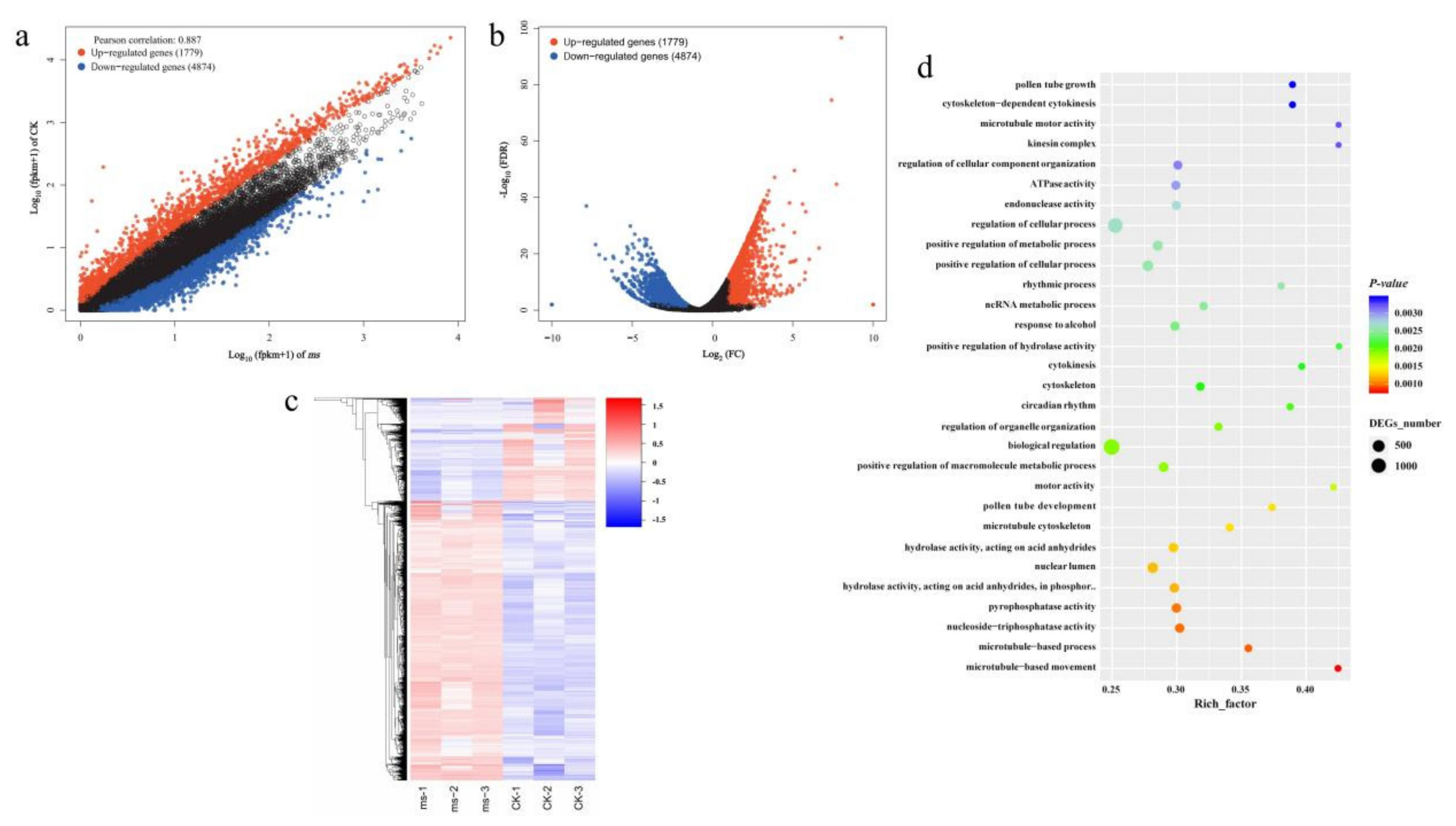
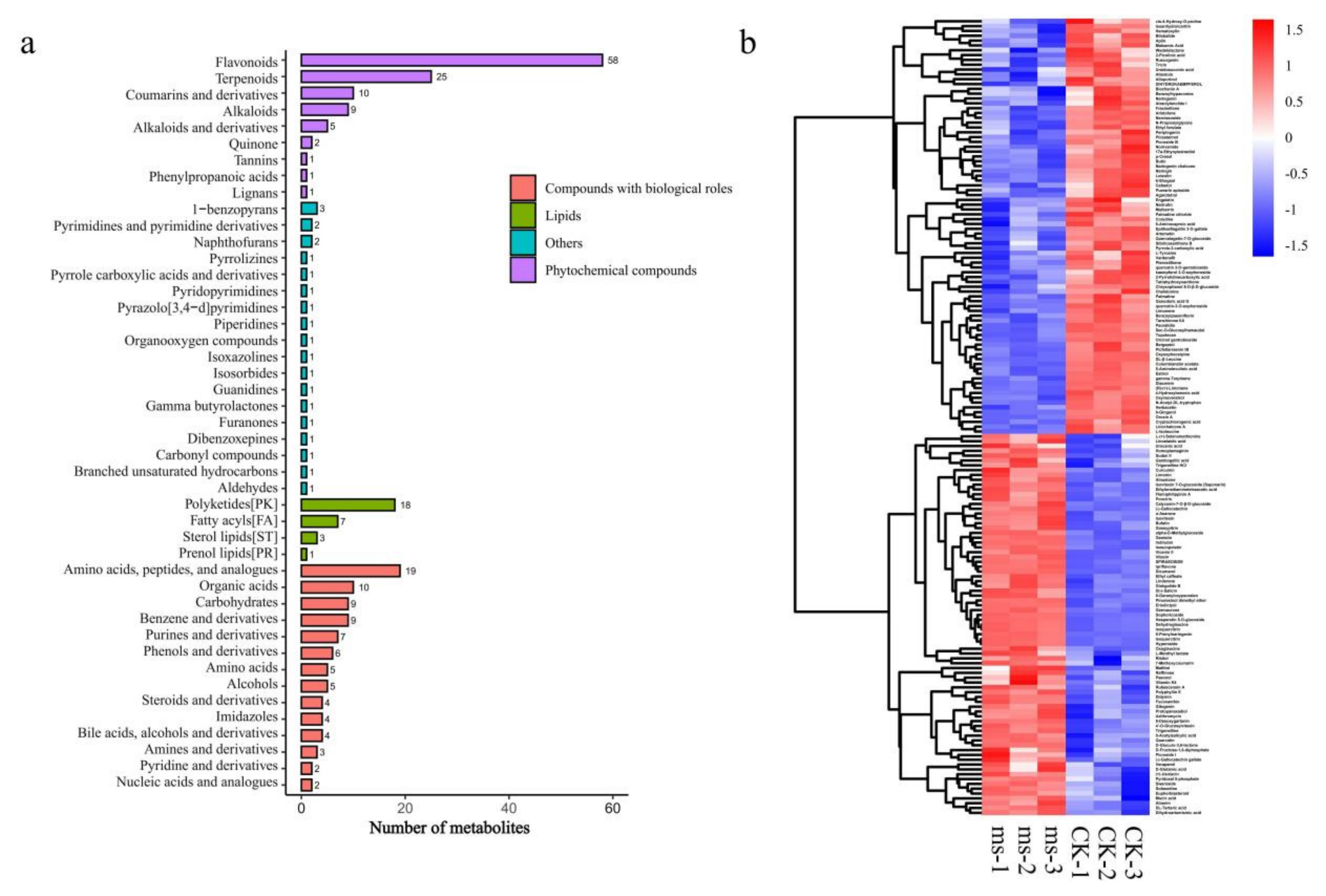
3.4. Identification of Differentially Expressed Genes and Metabolites in GMS Mutants
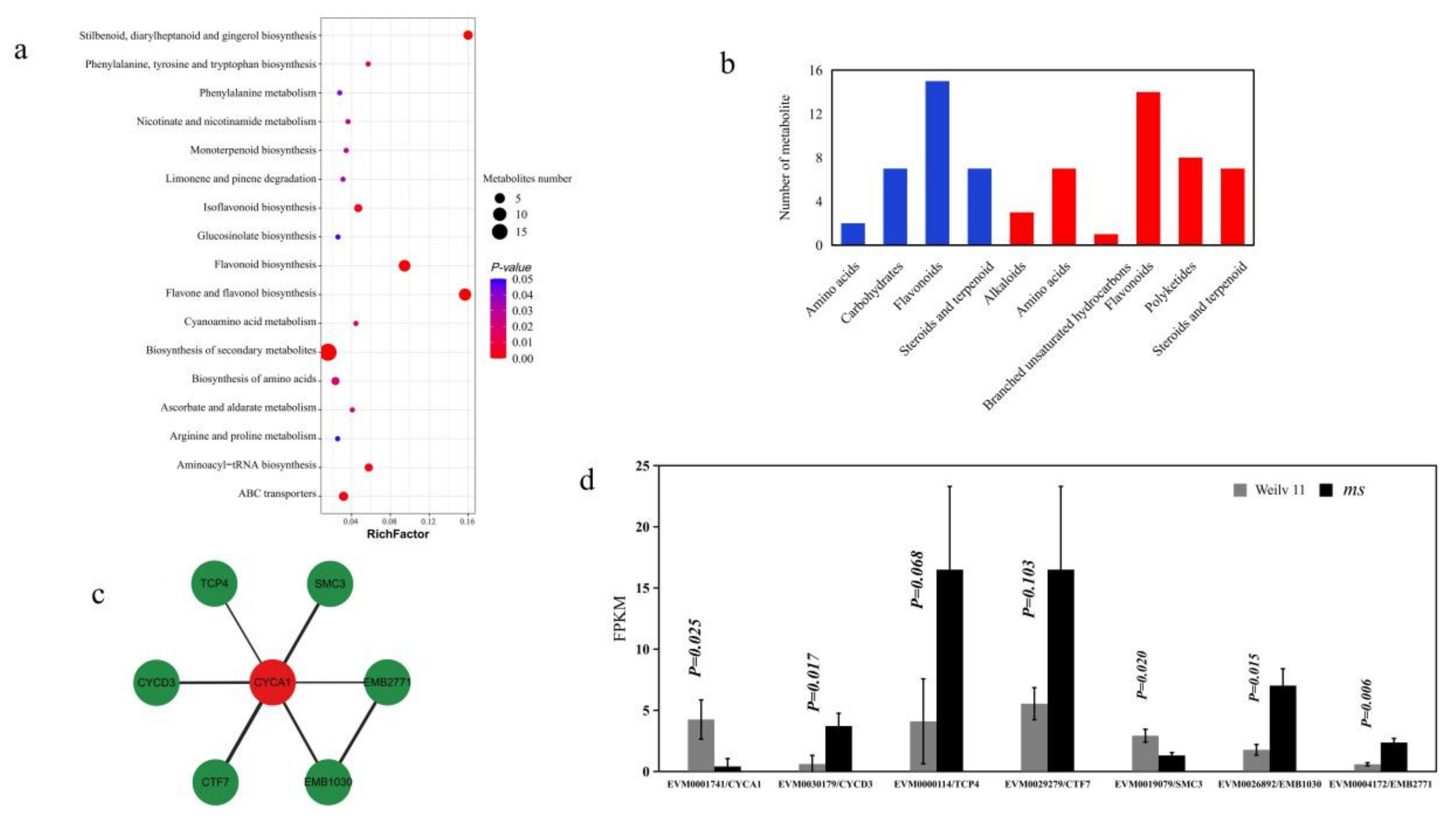
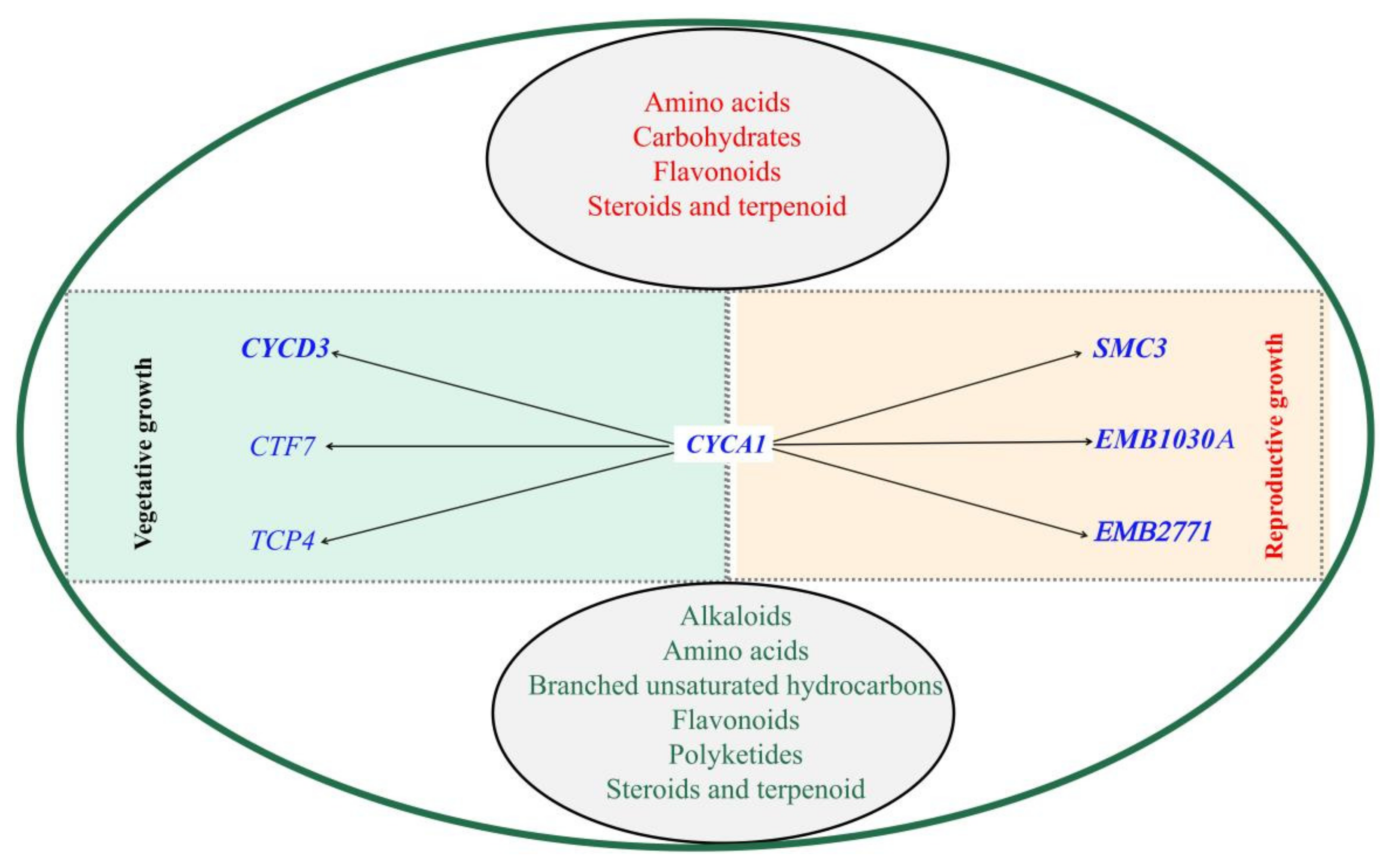
3.5. Genetic Relationships between VrCYCA1, Related Genes and Metabolites in Mungbean
4. Discussion
4.1. GMS in Mungbean
4.2. Significant Changes in VrCYCA1 Expression Led to GMS in Mungbean
4.3. Pollen Tube Growth, Biological Regulation and Pollen Tube Development Genes Were Altered in ms
4.4. Amino Acids Biosynthesis, Lipid Metabolism and Flavonoid Biosynthesis Pathways Were Altered in ms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BSA | Bulked segregate analysis |
| SNP | Single-nucleotide polymorphism |
| QTNs/QTL | Quantitative trait nucleotides/loci |
| EMS | Ethyl methane sulfonate |
| FPKM | Fragments reads per kilobases per million reads |
| PPI | Protein–protein interaction |
| GMS | Genic male sterility |
| LD | Linkage disequilibrium |
| DEGs | Differentially expressed genes |
| CYCA1 | Cyclin A1 |
References
- Chen, L.; Liu, Y.G. Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef] [PubMed]
- Yang, X. The Arabidopsis MALE MEIOCYTE DEATH1 gene encodes a PHD-finger protein that is required for male meiosis. Plant Cell 2003, 15, 281–1295. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.Y.; Wu, S.W.; Li, Z.W.; Dong, Z.Y.; An, X.L.; Ma, B.; Tian, Y.H.; Li, J.P. Maize genic male-sterility genes and their applications in hybrid breeding: Progress and perspectives. Mol. Plant 2019, 12, 321–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariizumi, T.; Hatakeyama, K.; Hinata, K.; Inatsugi, R.; Nishida, I.; Sato, S.; Kato, T.; Tabata, S.; Toriyama, K. Disruption of the novel plant protein NEF1 affects lipid accumulation in the plastids of the tapetum and exine formation of pollen, resulting in male sterility in Arabidopsis thaliana. Plant J. 2004, 39, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Zachgo, S. ROXY1 and ROXY2, two Arabidopsis glutaredoxin genes, are required for anther development. Plant J. 2008, 53, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.X.; Lu, J.Y.; Lou, Y.; Teng, X.D.; Gu, J.N.; Zhang, C.; Shi, Q.S.; Yang, Z.N.; Zhu, J. The transcription factors MS188 and AMS form a complex to activate the expression of CYP703A2 for sporopollenin biosynthesis in Arabidopsis thaliana. Plant J. 2016, 88, 936–946. [Google Scholar] [CrossRef]
- Mitterreiter, M.J.; Bosch, F.A.; Brylok, T.; Schwenkert, S. The ER luminal C-terminus of AtSec62 is critical for male fertility and plant growth in Arabidopsis thaliana. Plant J 2020, 101, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Ren, M.Y.; Han, W.J.; Zhang, Y.F.; Huang, M.J.; Wu, S.Y.; Huang, J.; Wang, Y.; Zhang, Z.; Yang, Z.N. Slow development allows redundant genes to restore the fertility of rpg1, a TGMS line in Arabidopsis. Plant J. 2021, 14, 1375–1385. [Google Scholar] [CrossRef]
- Li, X.; Gao, X.; Wei, Y.; Deng, L.; Ouyang, Y.; Chen, G.; Li, X.; Zhang, Q.; Wu, C. Rice APOPTOSIS INHIBITOR5 coupled with two DEAD-box adenosine 5′-triphosphate-dependent RNA helicases regulates tapetum degeneration. Plant Cell 2011, 23, 1416–1434. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.; Kim, S.R.; Zhao, G.; Yi, J.; Yoo, Y.; Jin, P.; Lee, S.W.; Jung, K.H.; Zhang, D.; An, G. Rice glycosyltransferase1 encodes a glycosyltransferase essential for pollen wall formation. Plant Physiol. 2013, 161, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Zhang, Y.; Sun, L.; Sinumporn, S.; Yang, Z.; Sun, B.; Xuan, D.; Li, Z.; Yu, P.; Wu, W.; et al. The Rice AAA-ATPase OsFIGNL1 Is Essential for Male Meiosis. Front. Plant Sci. 2017, 27, 1639. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Chen, Y.; Liu, L.; See, Y.H.B.; Mao, C.; Gan, Y.; Yu, H. OsFTIP7 determines auxin-mediated anther dehiscence in rice. Nat. Plants 2018, 4, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Long, T.; Wang, Y.; Tong, X.; Tang, J.; Li, J.; Wang, H.; Tang, L.; Li, Z.; Shu, Y.; et al. RMS2 Encoding a GDSL Lipase Mediates Lipid Homeostasis in Anthers to Determine Rice Male Fertility. Plant Physiol. 2020, 182, 2047–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernoud, V.; Laigle, G.; Rozier, F.; Meeley, R.B.; Perez, P.; Rogowsky, P.M. The HD-ZIP IV transcription factor OCL4 is necessary for trichome patterning and anther development in maize. Plant J. 2009, 59, 883–894. [Google Scholar] [CrossRef]
- Wang, C.J.; Nan, G.L.; Kelliher, T.; Timofejeva, L.; Vernoud, V.; Golubovskaya, I.N.; Harper, L.; Egger, R.; Walbot, V.; Cande, W.Z. Maize multiple archesporial cells 1 (mac1), an ortholog of rice TDL1A, modulates cell proliferation and identity in early anther development. Development 2012, 139, 2594–2603. [Google Scholar] [CrossRef] [Green Version]
- Nan, G.L.; Zhai, J.; Arikit, S.; Morrow, D.; Fernandes, J.; Mai, L.; Nguyen, N.; Meyers, B.C.; Walbot, V. MS23, a master basic helix-loop-helix factor, regulates the specification and development of the tapetum in maize. Development 2017, 144, 163–172. [Google Scholar] [CrossRef] [Green Version]
- An, X.; Dong, Z.; Tian, Y.; Xie, K.; Wu, S.; Zhu, T.; Zhang, D.; Zhou, Y.; Niu, C.; Ma, B.; et al. ZmMs30 encoding a novel GDSL lipase is essential for male fertility and valuable for hybrid breeding in maize. Mol. Plant 2019, 12, 343–359. [Google Scholar] [CrossRef] [Green Version]
- An, X.; Ma, B.; Duan, M.; Dong, Z.; Liu, R.; Yuan, D.; Hou, Q.; Wu, S.; Zhang, D.; Liu, D.; et al. Molecular regulation of ZmMs7 required for maize male fertility and development of a dominant male-sterility system in multiple species. Proc. Natl. Acad. Sci. USA 2020, 22, 23499–23509. [Google Scholar] [CrossRef]
- Huo, Y.; Pei, Y.; Tian, Y.; Zhang, Z.; Li, K.; Liu, J.; Xiao, S.; Chen, H.; Liu, J. IRREGULAR POLLENEXINE2 Encodes a GDSL Lipase Essential for Male Fertility in Maize. Plant Physiol. 2020, 184, 1438–1454. [Google Scholar] [CrossRef]
- Zhang, H.W.; Wang, X.; Pan, Q.H.; Li, P.; Liu, Y.; Lu, X.; Zhong, W.; Li, M.; Han, L.; Li, J.; et al. QTG-Seq accelerates QTL fine mapping through QTL partitioning and whole-genome sequencing of bulked segregant samples. Mol. Plant 2019, 12, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Khouider, S.; Borges, F.; LeBlanc, C.; Ungru, A.; Schnittger, A.; Martienssen, R.; Colot, V.; Bouyer, D. Male fertility in Arabidopsis requires active DNA demethylation of genes that control pollen tube function. Nat. Commun. 2021, 18, 410. [Google Scholar] [CrossRef] [PubMed]
- Simko, I.; Haynes, K.G.; Jones, R.W. Assessment of linkage disequilibrium in potato genome with single nucleotide polymorphism markers. Genetics 2006, 173, 2237–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourad, R.; Sinoquet, C.; Leray, P. A hierarchical Bayesian network approach for linkage disequilibrium modeling and data-dimensionality reduction prior to genome-wide association studies. BMC Bioinform. 2011, 12, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubry, P.; de Bellis, F.; Avia, K.; Bouchet, S.; Pot, D.; Dufour, M.; Legnate, H.; Leroy, T. An initial assessment of linkage disequilibrium (LD) in coffee trees: LD patterns in groups of Coffea canephora Pierre using microsatellite analysis. BMC Genom. 2013, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Wang, P.; Xu, Y. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol. J. 2016, 14, 1941–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenger, J.W.; Schwartz, K.; Sherlock, G. Bulk segregant analysis by high-throughput sequencing reveals a novel xylose utilization gene from Saccharomyces cerevisiae. PLoS Genet. 2010, 6, e1000942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Chai, L.; Feng, B.; Liu, X.; Jiang, L.; Yuan, S.; Zhang, Z.; Li, H.; Zhang, J.; Fernando, D.; Xu, C.; et al. Fine Mapping of a Locus Underlying the Ectopic Blade-Like Outgrowths on Leaf and Screening Its Candidate Genes in Rapeseed (Brassica napus L.). Front. Plant Sci. 2021, 14, 616844. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, X.; Zhang, M.; Guo, L.; Qi, T.; Tang, H.; Zhu, H.; Wang, H.; Qiao, X.; Xing, C.; et al. Physical mapping and InDel marker development for the restorer gene Rf2 in cytoplasmic male sterile CMS-D8 cotton. BMC Genom. 2021, 6, 24. [Google Scholar] [CrossRef]
- Fekih, R.; Takagi, H.; Tamiru, M.; Abe, A.; Natsume, S.; Yaegashi, H.; Sharma, S.; Kanzaki, H.; Matsumura, H.; Saitoh, H.; et al. MutMap+: Genetic mapping and mutant identification without crossing in rice. PLoS ONE 2013, 10, e68529. [Google Scholar] [CrossRef]
- Liu, J.Y.; Li, P.; Zhang, Y.W.; Zuo, J.F.; Li, G.; Han, X.; Dunwell, J.M.; Zhang, Y.M. Three-dimensional genetic networks among seed oil-related traits, metabolites and genes reveal the genetic foundations of oil synthesis in soybean. Plant J. 2020, 103, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Alam, M.K.; Xie, M.; Yang, L.; Liu, J.; Helal, M.; Huang, J.; Cheng, X.; Liu, Y.; Tong, C.; et al. Mapping of a major QTL controlling plant height using a high-density genetic map and QTL-seq methods based on whole-genome resequencing in Brassica napus. G3 2021, 11, jkab118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Jing, Y.; Luo, Z.; Gao, S.; Teng, W.; Zhan, Y.; Qiu, L.; Zheng, H.; Li, W.; Han, Y. GmST1, which encodes a sulfotransferase, confers resistance to soybean mosaic virus strains G2 and G3. Plant Cell Environ. 2021, 44, 2777–279210. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Hultman, J.; Waldrop, M.P.; Mackelprang, R.; David, M.M.; McFarland, J.; Blazewicz, S.J.; Harden, J.; Turetsky, M.R.; McGuire, A.D.; Shah, M.B.; et al. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 2015, 14, 208–212. [Google Scholar] [CrossRef]
- Jiang, L.; Yoshida, T.; Stiegert, S.; Jing, Y.; Alseekh, S.; Lenhard, M.; Pérez-Alfocea, F.; Fernie, A.R. Multi-omics approach reveals the contribution of KLU to leaf longevity and drought tolerance. Plant Physiol. 2021, 15, 352–368. [Google Scholar] [CrossRef]
- Lu, H.; Wang, Z.; Xu, C.; Li, L.; Yang, C. Multiomics analysis provides insights into alkali stress tolerance of sunflower (Helianthus annuus L.). Plant Physiol. Biochem. 2021, 166, 66–77. [Google Scholar] [CrossRef]
- Wan, X.; Wu, S.; Li, Z.; An, X.; Tian, Y. Lipid Metabolism: Critical Roles in Male Fertility and Other Aspects of Reproductive Development in Plants. Mol. Plant 2020, 6, 955–983. [Google Scholar] [CrossRef]
- Wang, L.; Lam, P.Y.; Lui, A.C.W.; Zhu, F.Y.; Chen, M.X.; Liu, H.; Zhang, J.; Lo, C. Flavonoids are indispensable for complete male fertility in rice. J. Exp. Bot. 2020, 6, 4715–4728. [Google Scholar] [CrossRef]
- Joanna, T. Confidence intervals for the power of student’s t-test. Stat. Probab. Lett. 2005, 73, 125–130. [Google Scholar]
- Liu, J.Y.; Zhang, Y.W.; Han, X.; Zuo, J.F.; Zhang, Z.; Shang, H.; Song, Q.; Zhang, Y.M. An evolutionary population structure model reveals pleiotropic effects of GmPDAT for traits related to seed size and oil content in soybean. J. Exp. Bot. 2020, 71, 6988–7002. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.S.; Maxwell, P.W.; de Boer, S.H. Comparison of several methods for the extraction of DNA from potatoes and potato-derived products. J. Agric. Food Chem. 2005, 28, 9848–9859. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Li, Z.; Liu, Z.; Guo, Y.; Qiu, L.J. Next-Generation Sequencing from Bulked-Segregant Analysis Accelerates the Simultaneous Identification of Two Qualitative Genes in Soybean. Front. Plant Sci. 2017, 31, 919. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with burrows–wheeler transform. Bioinformatics 2010, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q.; Qiong, W.; Cheng, X.Z.; Wang, L.X.; Chen, X. High-quality genome assembly, annotation and evolutionary analysis of the mungbean (Vigna radiata) genome. Authorea Prepr. 2020. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative pcr and the 2-ddct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Cui, Q.; Lewis, I.A.; Hegeman, A.D.; Anderson, M.E.; Li, J.; Schulte, C.F.; Westler, W.M.; Eghbalnia, H.R.; Sussman, M.R.; Markley, J.L. Metabolite identification via the Madison Metabolomics Consortium Database. Nat. Biotechnol. 2008, 26, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Bulankova, P.; Riehs-Kearnan, N.; Nowack, M.K.; Schnittger, A.; Riha, K. Meiotic progression in Arabidopsis is governed by complex regulatory interactions between SMG7, TDM1, and the meiosis I-specific cyclin TAM. Plant Cell 2010, 22, 3791–3803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangiri, C.; Kaga, A.; Tomooka, N.; Vaughan, D.; Srinives, P. Genetic diversity of the mungbean (Vigna radiata, L) genepool on the basis of microsatellite analysis. Aust. J. Bot. 2007, 55, 837–847. [Google Scholar] [CrossRef]
- Chen, J.; Somta, P.; Chen, X.; Cui, X.; Yuan, X.; Srinives, P. Gene Mapping of a Mutant Mungbean (Vigna radiata L.) Using New Molecular Markers Suggests a Gene Encoding a YUC4-like Protein Regulates the Chasmogamous Flower Trait. Front. Plant Sci. 2016, 10, 830. [Google Scholar] [CrossRef] [Green Version]
- Kiran, K.; Ansari, S.A.; Srivastava, R.; Lodhi, N.; Chaturvedi, C.P.; Sawant, S.V.; Tuli, R. The TATA-box sequence in the basal promoter contributes to determining light-dependent gene expression in plants. Plant Physiol. 2006, 142, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Grosschedl, R.; Wasylyk, B.; Chambon, P.; Birnstiel, M.L. Point mutation in the TATA box curtails expression of sea urchin H2A histone gene in vivo. Nature 1981, 12, 178–180. [Google Scholar] [CrossRef]
- D’Erfurth, I.; Cromer, L.; Jolivet, S.; Girard, C.; Horlow, C.; Sun, Y.; To, J.P.; Berchowitz, L.E.; Copenhaver, G.P.; Mercier, R. The cyclin-A CYCA1;2/TAM is required for the meiosis I to meiosis II transition and cooperates with OSD1 for the prophase to first meiotic division transition. PLoS Genet. 2010, 17, e1000989. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, M. Loss-of-function mutants and overexpression lines of the Arabidopsis cyclin CYCA1;2/Tardy Asynchronous Meiosis exhibit different defects in prophase-i meiocytes but produce the same meiotic products. PLoS ONE 2014, 17, e113348. [Google Scholar] [CrossRef]
- Karidas, P.; Challa, K.R.; Nath, U. The tarani mutation alters surface curvature in Arabidopsis leaves by perturbing the patterns of surface expansion and cell division. J. Exp. Bot. 2015, 66, 2107–2122. [Google Scholar] [CrossRef] [Green Version]
- Koyama, T.; Sato, F.; Ohme-Takagi, M. Roles of miR319 and TCP Transcription Factors in Leaf Development. Plant Physiol. 2017, 175, 874–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, K.; Bolaños-Villegas, P.; Mitra, S.; Yang, X.; Homan, G.; Jauh, G.Y.; Makaroff, C.A. The Opposing Actions of Arabidopsis CHROMOSOME TRANSMISSION FIDELITY7 and WINGS APART-LIKE1 and 2 Differ in Mitotic and Meiotic Cells. Plant Cell 2016, 28, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; McElver, J.; Tzafrir, I.; Joosen, R.; Wittich, P.; Patton, D.; van Lammeren, A.A.; Meinke, D. Condensin and cohesin knockouts in Arabidopsis exhibit a titan seed phenotype. Plant J. 2002, 29, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Errampalli, D.; Patton, D.; Castle, L.; Mickelson, L.; Hansen, K.; Schnall, J.; Feldmann, K.; Meinke, D. Embryonic Lethals and T-DNA Insertional Mutagenesis in Arabidopsis. Plant Cell 1991, 3, 149–157. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, Y.; Gu, H.; Kang, D.; Chen, Z.L.; Liu, J.; Qu, L.J. The Arabidopsis anaphase-promoting complex/cyclosome subunit 1 is critical for both female gametogenesis and embryogenesis(F). J. Integr. Plant Biol. 2013, 55, 64–74. [Google Scholar] [CrossRef]
- Hu, Y.; Wu, Q.; Liu, S.; Wei, L.; Chen, X.; Yan, Z.; Yu, J.; Zeng, L.; Ding, Y. Study of rice pollen grains by multispectral imaging microscopy. Microsc. Res. Tech. 2005, 15, 335–346. [Google Scholar] [CrossRef]
- Mattioli, R.; Biancucci, M.; Lonoce, C.; Costantino, P.; Trovato, M. Proline is required for male gametophyte development in Arabidopsis. BMC Plant Biol. 2012, 12, 236. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Chen, Z.; Li, H.; Sun, Y.; Wang, L.; Zeng, H.; He, Y. Lipid metabolism is involved in male fertility regulation of the photoperiod- and thermo sensitive genic male sterile rice line Peiai 64S. Plant Sci. 2020, 299, 110581. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Observed Number | Theoretical Number | χ2 (3:1) |
|---|---|---|---|
| Fertile plants | 177 | 165 | 3.491 |
| Sterile plants | 43 | 55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Lin, Y.; Chen, J.; Xue, C.; Wu, R.; Yan, Q.; Chen, X.; Yuan, X. Identification and Clarification of VrCYCA1: A Key Genic Male Sterility-Related Gene in Mungbean by Multi-Omics Analysis. Agriculture 2022, 12, 686. https://doi.org/10.3390/agriculture12050686
Liu J, Lin Y, Chen J, Xue C, Wu R, Yan Q, Chen X, Yuan X. Identification and Clarification of VrCYCA1: A Key Genic Male Sterility-Related Gene in Mungbean by Multi-Omics Analysis. Agriculture. 2022; 12(5):686. https://doi.org/10.3390/agriculture12050686
Chicago/Turabian StyleLiu, Jinyang, Yun Lin, Jinbin Chen, Chenchen Xue, Ranran Wu, Qiang Yan, Xin Chen, and Xingxing Yuan. 2022. "Identification and Clarification of VrCYCA1: A Key Genic Male Sterility-Related Gene in Mungbean by Multi-Omics Analysis" Agriculture 12, no. 5: 686. https://doi.org/10.3390/agriculture12050686
APA StyleLiu, J., Lin, Y., Chen, J., Xue, C., Wu, R., Yan, Q., Chen, X., & Yuan, X. (2022). Identification and Clarification of VrCYCA1: A Key Genic Male Sterility-Related Gene in Mungbean by Multi-Omics Analysis. Agriculture, 12(5), 686. https://doi.org/10.3390/agriculture12050686