Understanding the Complexity of Sjögren’s Syndrome: Remarkable Progress in Elucidating NF-κB Mechanisms




Abstract
1. Introduction
2. Sjögren’s Syndrome
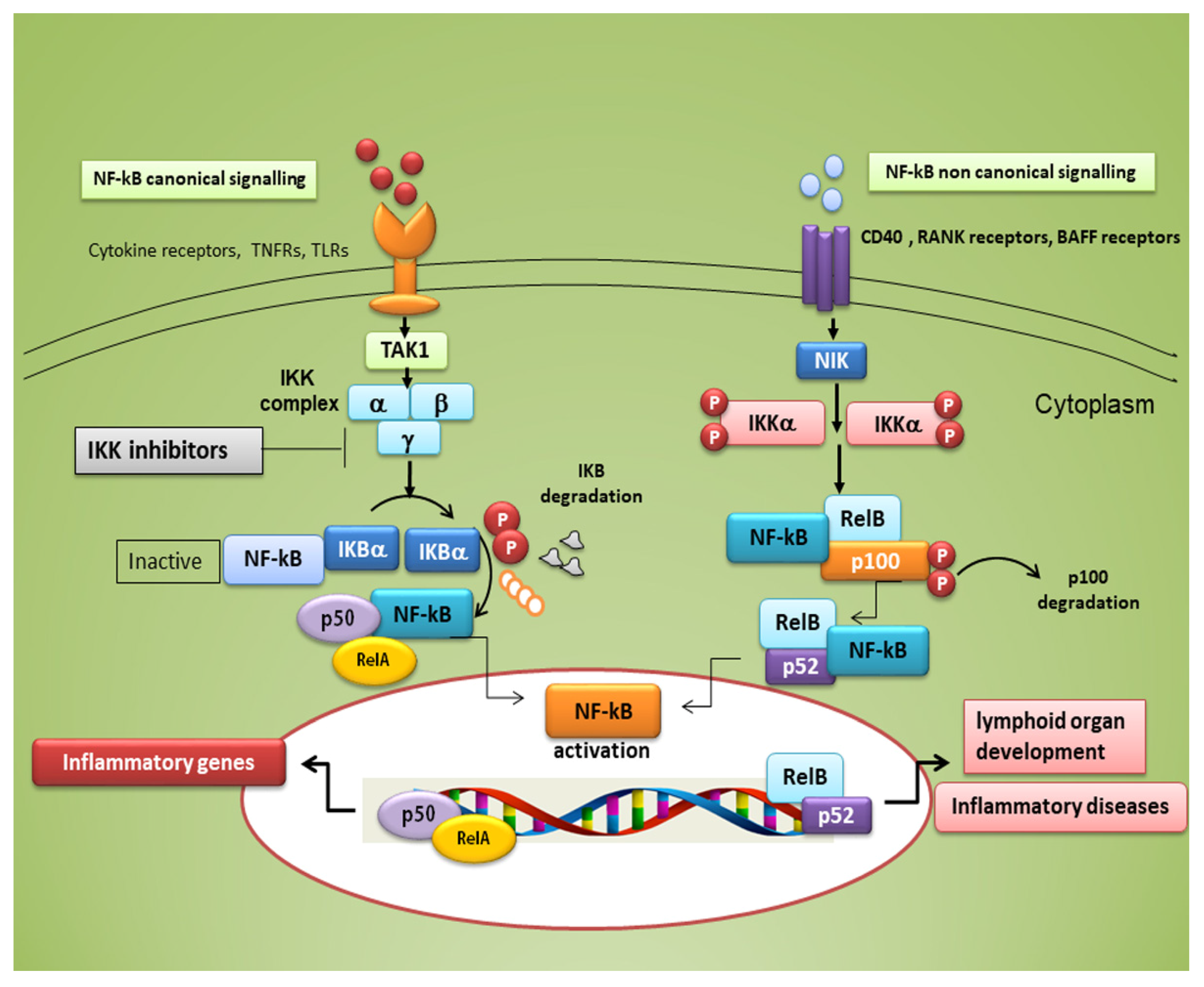
3. NF-κB Transcription Factors
4. Small-Molecule Inhibitors of NF-κB in Sjögren’s Syndrome
The Key Role of IκBα in NF-κB Modulation in pSS
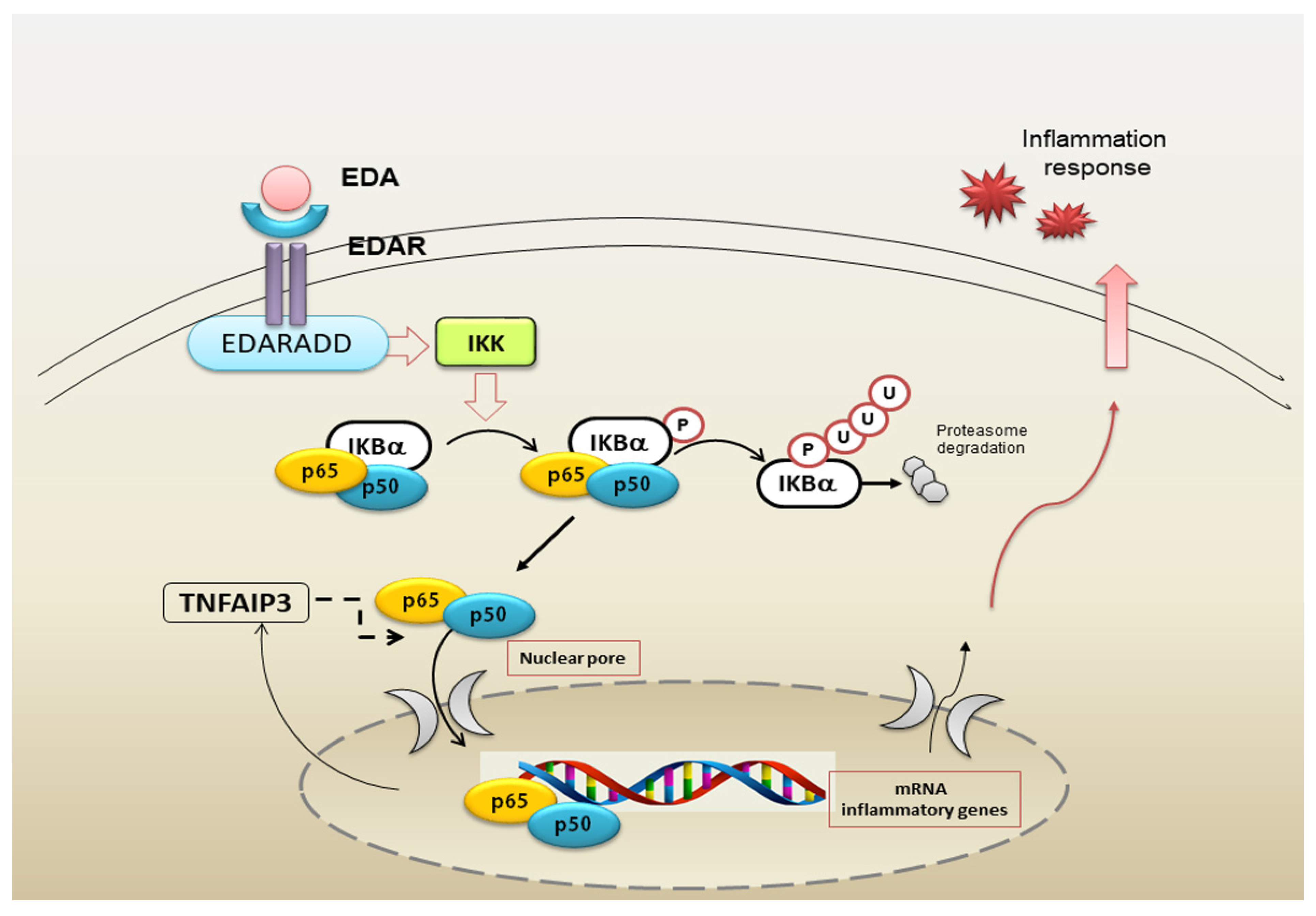
5. Impaired NF-κB Signalling Activated by EDA-A1/EDAR in pSS Salivary Glands
6. Toll-Like Receptor-Mediated NF-κB Activation in pSS
Role of TLRs in the NF-κB-Mediated Inflammatory State in pSS
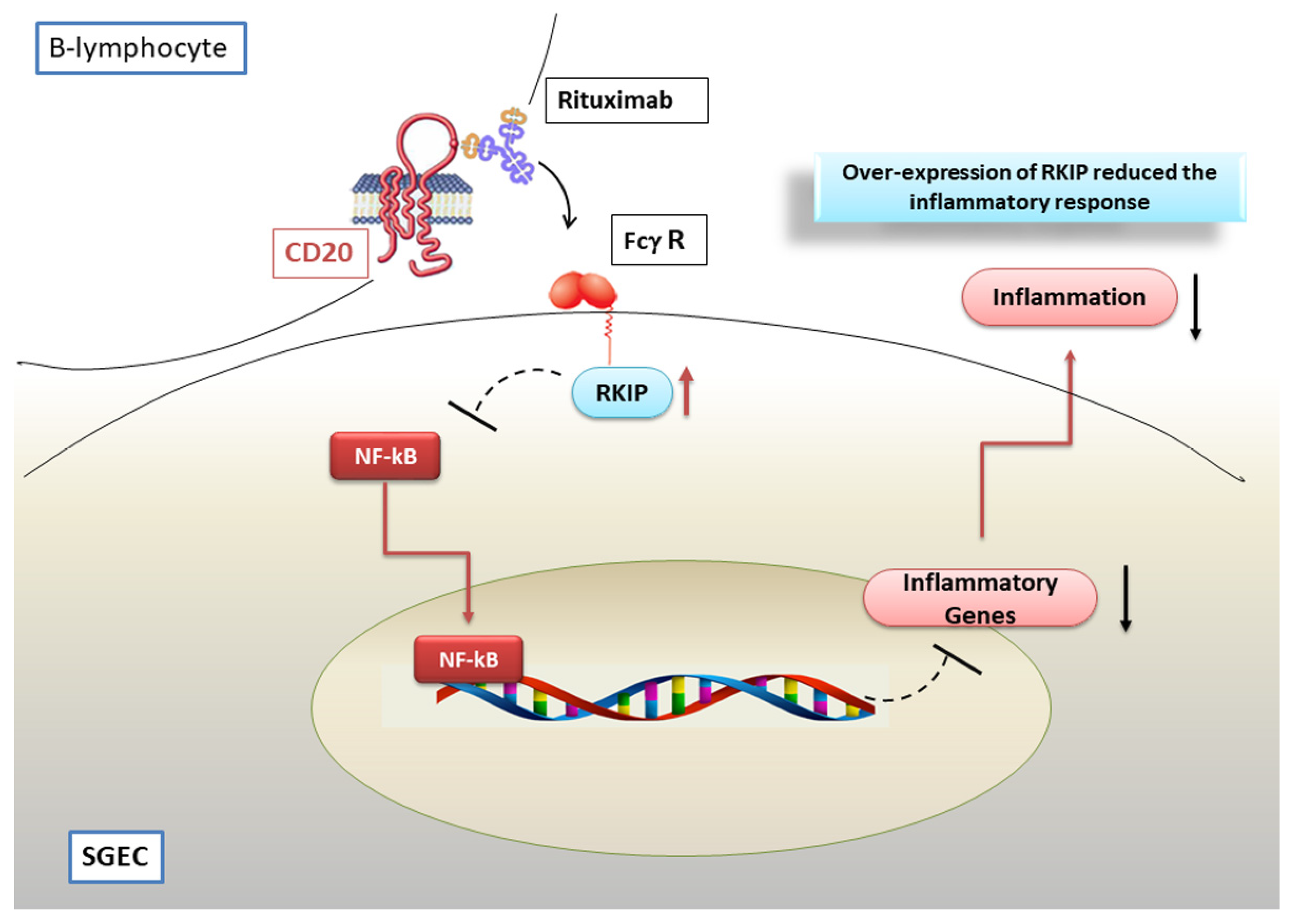
7. Modulation of NF-κB Activation by the Anti CD-20 Monoclonal Antibody Rituximab
Hypothetical Scenario Involving RTX as a Negative Regulator of the NF-κB Pathway in pSS
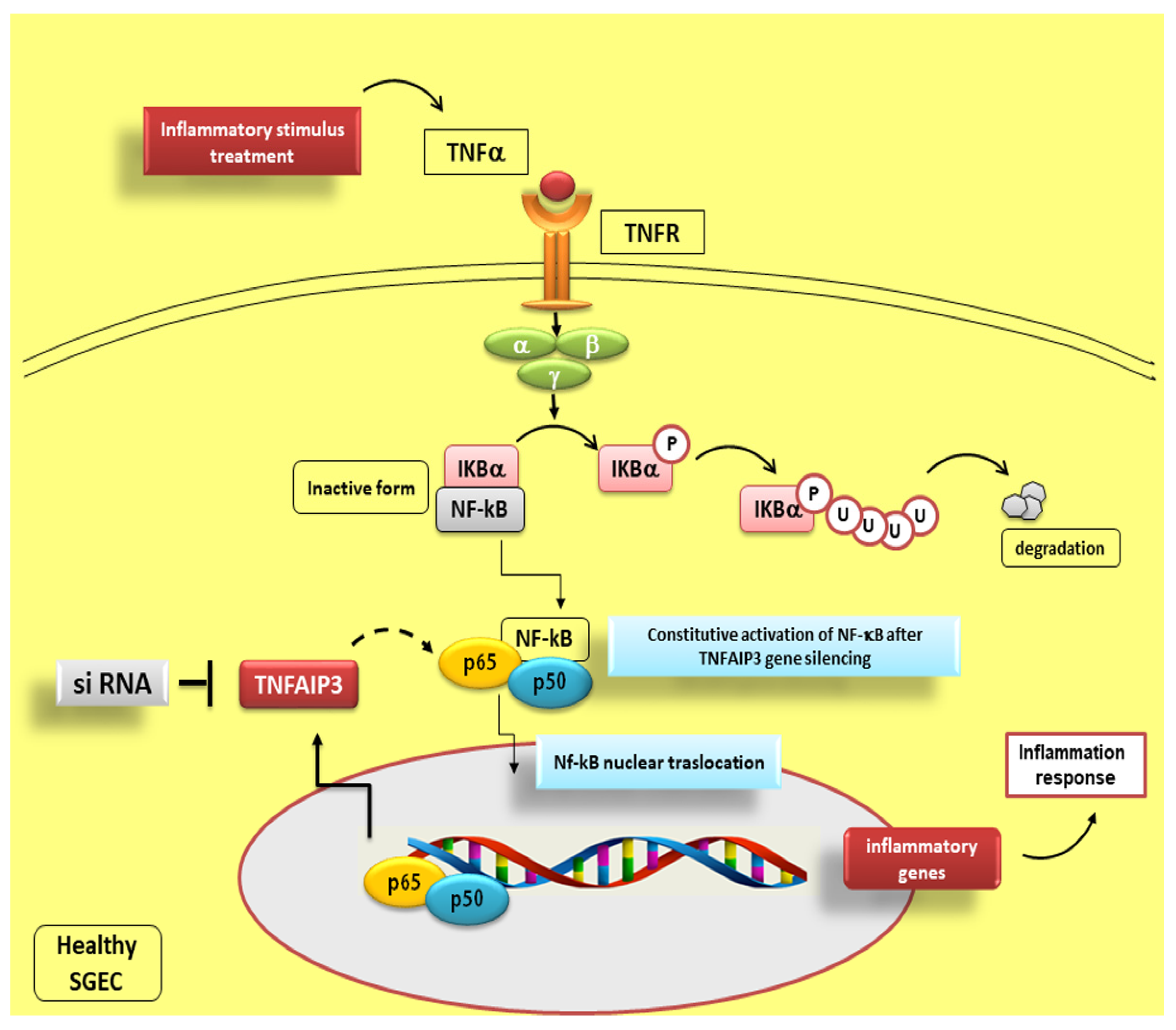
8. Fine Modulation of NF-κB Activity by TNFAIP3
Reduced TNFAIP3 Expression Levels in pSS Affect NF-κB Signalling
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Shaalan, A.; Liefers, S.; Coudenys, J.; Elewaut, D.; Proctor, G.B.; Bootsma, H.; Kroese, F.G.M.; Pringle, S. Dysregulation of NF-kB in glandular epithelial cells results in Sjögren’s-like features. PLoS ONE 2018, 13, e0200212. [Google Scholar] [CrossRef]
- Fox, P.C. Autoimmune Diseases and Sjögren’s Syndrome. Ann. N. Y. Acad. Sci. 2007, 1098, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lin, J.; Cao, H.; Xu, D.; Xu, B.; Xu, L.; Yue, L.; Sun, C.; Wu, G.; Qian, W. Local and Systemic IKKε and NF-κB Signaling Associated with Sjögren’s Syndrome Immunopathogenesis. J. Immunol. Res. 2015, 2015, 534648. [Google Scholar] [CrossRef]
- Cafaro, G.; Croia, C.; Argyropoulou, O.D.; Leone, M.C.; Orlandi, M.; Finamore, F.; Cecchettini, A.; Ferro, F.; Baldini, C.; Bartoloni, E. One year in review 2019: Sjögren’s syndrome. Clin. Exp. Rheumatol. 2019, 118, 3–15. [Google Scholar]
- Tzioufas, A.G.; Voulgarelis, M. Update on Sjögren’s syndrome autoimmune epithelitis: From classification to increased neoplasias. Best Pract. Res. Clin. Rheumatol. 2007, 21, 989–1010. [Google Scholar] [CrossRef]
- Papageorgiou, A.; Ziogas, D.C.; Mavragani, C.P.; Zintzaras, E.; Tzioufas, A.G.; Moutsopoulos, H.M.; Voulgarelis, M. Predicting the outcome of Sjogren’s syndrome-associated non-hodgkin’s lymphoma patients. PLoS ONE 2015, 10, e0116189. [Google Scholar] [CrossRef]
- Voulgarelis, M.; Tzioufas, A.G. Current aspects of pathogenesis in Sjögren’s syndrome. Ther. Adv. Musculoskelet. Dis. 2010, 2, 325–334. [Google Scholar] [CrossRef]
- Fox, P.C.; Speight, P.M. Current concepts of autoimmune exocrinopathy: Immunologic mechanisms in the salivary pathology of Sjögren’s syndrome. Crit. Rev. Oral Biol. Med. 1996, 7, 144–158. [Google Scholar] [CrossRef]
- Humphreys-Beher, M.G.; Peck, A.B.; Dang, H.; Talal, N. The role of apoptosis in the initiation of the autoimmune response in Sjögren’s syndrome. Clin. Exp. Immunol. 1999, 116, 383–387. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Kapsogeorgou, E.K. The role of epithelial cells in the pathogenesis of Sjögren’s syndrome. Clin. Rev. Allergy Immunol. 2007, 32, 225–230. [Google Scholar] [CrossRef]
- Moutsopoulos, H.M. Sjögren’s syndrome: Autoimmune epithelitis. Clin. Immunol. Immunopathol. 1994, 72, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Lisi, S.; Castellana, D.; Scagliusi, P.; D’Amore, M.; Caprio, S.; Scagliusi, A.; Acquafredda, A.A.; Panaro, M.A.; Mitolo, V. Autoantibodies from Sjögren’s syndrome induce activation of both the intrinsic and extrinsic apoptotic pathways in human salivary gland cell line A-253. J. Autoimmun. 2006, 27, 38–49. [Google Scholar] [CrossRef]
- Sisto, M.; Lisi, S.; Lofrumento, D.; D’Amore, M.; Scagliusi, P.; Mitolo, V. Autoantibodies from Sjögren’s syndrome trigger apoptosis in salivary gland cell line. Ann. N. Y. Acad. Sci. 2007, 1108, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; Caprio, S.; Mitolo, V.; D’Amore, M. TNF blocker drugs modulate human TNF-α-converting enzyme pro-domain shedding induced by autoantibodies. Immunobiology 2010, 215, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; Ingravallo, G.; Mitolo, V.; D’Amore, M. Expression of pro-inflammatory TACE-TNF-α-amphiregulin axis in Sjögren’s syndrome salivary glands. Histochem. Cell Biol. 2010, 134, 345–353. [Google Scholar] [CrossRef]
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; Ingravallo, G.; Maiorano, E.; D’Amore, M. A failure of TNFAIP3 negative regulation maintains sustained NF-κB activation in Sjögren’s syndrome. Histochem. Cell Biol. 2011, 135, 615–625. [Google Scholar] [CrossRef]
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; D’Amore, M.; Frassanito, M.A.; Ribatti, D. Sjögren’s syndrome pathological neovascularization is regulated by VEGF-A-stimulated TACE-dependent crosstalk between VEGFR2 and NF-κB. Genes Immun. 2012, 13, 411–420. [Google Scholar] [CrossRef]
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; Ingravallo, G.; De Lucro, R.; D’Amore, M. Salivary gland expression level of IκBα regulatory protein in Sjögren’s syndrome. J. Mol. Histol. 2013, 44, 447–454. [Google Scholar] [CrossRef]
- Sisto, M.; Lisi, S.; D’Amore, M.; Lofrumento, D.D. The metalloproteinase ADAM17 and the epidermal growth factor receptor (EGFR) signaling drive the inflammatory epithelial response in Sjögren’s syndrome. Clin. Exp. Med. 2015, 15, 215–225. [Google Scholar] [CrossRef]
- Lisi, S.; Sisto, M.; Soleti, R.; Saponaro, C.; Scagliusi, P.; D’Amore, M.; Saccia, M.; Maffione, A.B.; Mitolo, V. Fcgamma receptors mediate internalization of anti-Ro and anti-La autoantibodies from Sjögren’s syndrome and apoptosis in human salivary gland cell line A-253. J. Oral Pathol. Med. 2007, 36, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Lisi, S.; Sisto, M.; Scagliusi, P.; Mitolo, V.; D’Amore, M. Siögren’s syndrome: Anti-Ro and anti-La autoantibodies trigger apoptotic mechanism in the human salivary gland cell line, A-253. Panminerva Med. 2007, 49, 103–108. [Google Scholar]
- Lisi, S.; D’Amore, M.; Scagliusi, P.; Mitolo, V.; Sisto, M. Anti-Ro/SSA autoantibody-mediated regulation of extracellular matrix fibulins in human epithelial cells of the salivary gland. Scand. J. Rheumatol. 2009, 38, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; Cucci, L.; Frassanito, M.A.; Mitolo, V.; D’Amore, M. Pro-inflammatory role of Anti-Ro/SSA autoantibodies through the activation of Furin-TACE-amphiregulin axis. J. Autoimmun. 2010, 35, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; D’Amore, M. Sjögren’s syndrome autoantibodies provoke changes in gene expression profiles of inflammatory cytokines triggering a pathway involving TACE/NF-κB. Lab. Investig. 2012, 92, 615–624. [Google Scholar] [CrossRef]
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; D’Amore, M. Altered IκBα expression promotes NF-κB activation in monocytes from primary Sjögren’s syndrome patients. Pathology 2012, 44, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; D’Amore, M.; De Lucro, R.; Ribatti, D. A potential role of the GRO-α/CXCR2 system in Sjögren’s syndrome: Regulatory effects of pro-inflammatory cytokines. Histochem. Cell Biol. 2013, 139, 371–379. [Google Scholar] [CrossRef]
- Lisi, S.; Sisto, M.; D’Amore, M.; Lofrumento, D.D.; Ribatti, D. Emerging avenues linking inflammation, angiogenesis and Sjögren’s syndrome. Cytokine 2013, 61, 693–703. [Google Scholar] [CrossRef]
- Lisi, S.; Sisto, M.; Ribatti, D.; D’Amore, M.; De Lucro, R.; Frassanito, M.A.; Lorusso, L.; Vacca, A.; Lofrumento, D.D. Chronic inflammation enhances NGF-β/TrkA system expression via EGFR/MEK/ERK pathway activation in Sjögren’s syndrome. J. Mol. Med. 2014, 92, 523–537. [Google Scholar] [CrossRef]
- Pringle, S.; Wang, X.; Bootsma, H.; Spijkervet, F.K.L.; Vissink, A.; Kroese, F.G.M. Small-molecule inhibitors and the salivary gland epithelium in Sjögren’s syndrome. Expert Opin. Investig. Drugs. 2019, 28, 605–616. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef]
- Sun, S.C.; Ganchi, P.A.; Ballard, D.W.; Greene, W.C. NF-κB controls expression of inhibitor IκBα: Evidence for an inducible autoregulatory pathway. Science 1993, 259, 1912–1915. [Google Scholar] [CrossRef]
- Christman, J.W.; Sadikot, R.T.; Blackwell, T.S. The role of nuclear factor-kappa B in pulmonary diseases. Chest 2000, 117, 1482–1487. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Gaynor, R.B. Role of the NF-kB pathway in the pathogenesis of human disease states. Curr. Mol. Med. 2001, 1, 287–296. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Widera, D.; Kaltschmidt, C. Signaling via NF-kappaB in the nervous system. Biochim. Biophys. Acta 2005, 1745, 287–299. [Google Scholar] [CrossRef]
- Ledoux, A.C.; Perkins, N.D. NF-kappaB and the cell cycle. Biochem. Soc. Trans. 2014, 42, 76–81. [Google Scholar] [CrossRef]
- Han, Z.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. AP-1 and NF-kappa B regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity 1998, 28, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Makarov, S.S. NF-kappa B in rheumatoid arthritis: A pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. 2001, 3, 200–206. [Google Scholar] [CrossRef]
- Nakamura, H.; Kawakami, A.; Ida, H.; Koji, T.; Eguchi, K. EGF activates PI3K-Akt and NF-κB via distinct pathways in salivary epithelial cells in Sjögren’s syndrome. Rheumatol. Int. 2007, 28, 127–136. [Google Scholar] [CrossRef]
- Ping, L.; Ogawa, N.; Zhang, Y.; Sugai, S.; Masaki, Y.; Xiao, W. p38 mitogen-activated protein kinase and nuclear factor-kappaB facilitate CD40-mediated salivary epithelial cell death. J. Rheumatol. 2012, 39, 1256–1264. [Google Scholar] [CrossRef]
- Barnes, P.; Karin, M. Nuclear Factor-κB: A Pivotal Transcription Factor in Chronic Inflammatory Diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Dale, E.; Davis, M.; Faustman, D.L. A role for transcription factor NF-κB in autoimmunity: Possible interactions of genes, sex, and the immune response. Adv. Physiol. Educ. 2006, 30, 152–158. [Google Scholar] [CrossRef][Green Version]
- Thompson, N.; Isenberg, D.A.; Jury, E.C.; Ciurtin, C. Exploring BAFF: Its expression, receptors and contribution to the immunopathogenesis of Sjögren’s syndrome. Rheumatology 2016, 55, 1548–1555. [Google Scholar] [CrossRef]
- Nordmark, G.; Wang, C.; Vasaitis, L.; Eriksson, P.; Theander, E.; Kvarnstrom, M.; Forsblad-d’Elia, H.; Jazebi, H.; Sjowall, C.; Reksten, T.R.; et al. Association of Genes in the NF-kappaB Pathway with Antibody-Positive Primary Sjogren’s Syndrome. Scand. J. Immunol. 2013, 78, 447–454. [Google Scholar] [CrossRef]
- Ou, T.T.; Lin, C.H.; Lin, Y.C.; Li, R.N.; Tsai, W.C.; Liu, H.W.; Yen, J.H. IkappaBalpha promoter polymorphisms in patients with primary Sjögren’s syndrome. J. Clin. Immunol. 2008, 28, 440–444. [Google Scholar] [CrossRef]
- Peng, B.; Ling, J.; Lee, A.J.; Wang, Z.; Chang, Z.; Jin, W.; Kang, Y.; Zhang, R.; Shim, D.; Wang, H.; et al. Defective feedback regulation of NF-kappaB underlies Sjogren’s syndrome in mice with mutated kappaB enhancers of the IkappaBalpha promoter. Proc. Natl. Acad. Sci. USA 2010, 107, 15193–15198. [Google Scholar] [CrossRef]
- Kwok, S.K.; Cho, M.L.; Her, Y.M.; Oh, H.J.; Park, M.K.; Lee, S.Y.; Woo, Y.J.; Ju, J.H.; Park, K.S.; Kim, H.Y.; et al. TLR2 ligation induces the production of IL-23/IL-17 via IL-6, STAT3 and NF-kB pathway in patients with primary Sjogren’s syndrome. Arthritis Res. Ther. 2012, 14, R64. [Google Scholar] [CrossRef]
- Sisto, M.; Lorusso, L.; Lisi, S. Interleukin-15 as a potential new target in Sjögren’s syndrome-associated inflammation. Pathology 2016, 48, 602–607. [Google Scholar] [CrossRef]
- Sisto, M.; Lorusso, L.; Lisi, S. TLR2 signals via NF-κB to drive IL-15 production in salivary gland epithelial cells derived from patients with primary Sjögren’s syndrome. Clin. Exp. Med. 2017, 17, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Chen, Z.; Hendriks, R.W.; Kool, M. A20/Tumor Necrosis Factor α-Induced Protein 3 in Immune Cells Controls Development of Autoinflammation and Autoimmunity: Lessons from Mouse Models. Front. Immunol. 2018, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Dawson, L.J.; Field, E.A.; Harmer, A.R.; Smith, P.M. Acetylcholine-evoked calcium mobilization and ion channel activation in human labial gland acinar cells from patients with primary Sjogren’s syndrome. Clin. Exp. Immunol. 2001, 124, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Lilienbaum, A.; Israël, A. From calcium to NF-kappa B signaling pathways in neurons. Mol. Cell. Biol. 2003, 23, 2680–2698. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Sig. Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Zhang, J.; Peng, B. NF-kappaB promotes iNOS and VEGF expression in salivary gland adenoid cystic carcinoma cells and enhances endothelial cell motility in vitro. Cell Prolif. 2009, 42, 150–161. [Google Scholar] [CrossRef]
- Miterski, B.; Böhringer, S.; Klein, W.; Sindern, E.; Haupts, M.; Schimrigk, S.; Epplen, J.T. Inhibitors in the NFkappaB cascade comprise prime candidate genes predisposing to multiple sclerosis, especially in selected combinations. Genes Immun. 2002, 3, 211–219. [Google Scholar] [CrossRef]
- Klein, W.; Tromm, A.; Folwaczny, C.; Hagedorn, M.; Duerig, N.; Epplen, J.T.; Schmiegel, W.H.; Griga, T. A polymorphism of the NFKBIA gene is associated with Crohn’s disease patients lacking a predisposing allele of the CARD15 gene. Int. J. Colorectal Dis. 2004, 19, 153–156. [Google Scholar] [CrossRef]
- Zubair, A.; Frieri, M. NF-κB and systemic lupus erythematosus: Examining the link. J. Nephrol. 2013, 26, 953–959. [Google Scholar] [CrossRef]
- Gestermann, N.; Mekinian, A.; Comets, E.; Loiseau, P.; Puechal, X.; Hachulla, E.; Gottenberg, L.E.; Mariette, X.; Miceli-Richard, C. STAT4 is a confirmed genetic risk factor for Sjögren’s syndrome and could be involved in type 1 interferon pathway signaling. Genes Immun. 2010, 11, 432438. [Google Scholar]
- Palomino-Morales, R.J.; Diaz-Gallo, L.M.; Witte, T.; Anaya, J.M.; Martín, J. Influence of STAT4 polymorphism in primary Sjögren’s syndrome. J. Rheumatol. 2010, 37, 1016–1019. [Google Scholar] [CrossRef] [PubMed]
- Pispa, J.; Pummila, M.; Barker, P.A.; Thesleff, I.; Mikkola, M.L. Edar and Troy signalling pathways act redundantly to regulate initiation of hair follicle development. Hum. Mol. Genet. 2008, 17, 3380–3391. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, M.L. TNF superfamily in skin appendage development. Cytokine Growth Factor Rev. 2008, 19, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Melnick, M.; Jaskoll, T. Mouse submandibular gland morphogenesis: A paradigm for embryonic signal processing. Crit. Rev. Oral Biol. Med. 2000, 11, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Skaug, B.; Jiang, X.; Chen, Z.J. The role of ubiquitin in NF-kappaB regulatory pathways. Annu. Rev. Biochem. 2009, 78, 769–796. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Gaynor, R.B. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J. Clin. Investig. 2001, 107, 135–142. [Google Scholar] [CrossRef]
- Schmidt-Ullrich, R.; Aebischer, T.; Hulsken, J.; Birchmeier, W.; Klemm, U.; Scheidereit, C. Requirement of NF-kappaB/Rel for the development of hair follicles and other epidermal appendices. Development 2001, 128, 3843–3853. [Google Scholar]
- Courtney, J.M.; Blackburn, J.; Sharpe, P.T. The Ectodysplasin and NFkappaB signalling pathways in odontogenesis. Arch. Oral Biol. 2005, 50, 159–163. [Google Scholar] [CrossRef]
- Sisto, M.; Barca, A.; Lofrumento, D.D.; Lisi, S. Downstream activation of NF-κB in the EDA-A1/EDAR signalling in Sjögren’s syndrome and its regulation by the ubiquitin-editing enzyme A20. Clin. Exp. Immunol. 2016, 184, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, A.; Wahren-Herlenius, M. Update on the immunobiology of Sjögren’s syndrome. Curr. Opin. Rheumatol. 2015, 27, 468–475. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Brentano, F.; Kyburz, D.; Schorr, O.; Gay, R.; Gay, S. The role of Toll like receptor signaling in the pathogenesis of arthritis. Cell Immunol. 2005, 233, 90–96. [Google Scholar] [CrossRef]
- Kanczkowski, W.; Ziegler, C.G.; Zacharowski, K.; Bornstein, S.R. Toll-like receptors in endocrine disease and diabetes. NeuroImmunoModulation 2008, 15, 54–60. [Google Scholar] [CrossRef]
- Pisetsky, D.S. The role of innate immunity in the induction of autoimmunity. Autoimmun. Rev. 2008, 8, 69–72. [Google Scholar] [CrossRef]
- Wu, Y.W.; Tang, W.; Zuo, J.P. Toll-like receptors: Potential targets for lupus treatment. Acta Pharmacol. Sin. 2015, 36, 1395–1407. [Google Scholar] [CrossRef]
- Gooshe, M.; Aleyasin, A.R.; Abdolghaffari, A.H.; Rezaei, N. Toll like receptors: A new hope on the horizon to treat multiple sclerosis. Expert Rev. Clin. Immunol. 2014, 10, 1277–1279. [Google Scholar] [CrossRef]
- Sipos, F.; Furi, I.; Constantinovits, M.; Tulassay, Z.; Muzes, G. Contribution of TLR signaling to the pathogenesis of colitis-associated cancer in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 12713–12721. [Google Scholar] [CrossRef]
- Birchler, T.; Seibl, R.; Buchner, K.; Loeliger, S.; Seger, R.; Hossle, J.P.; Aguzzi, A.; Lauener, R.P. Human Toll-like receptor 2 mediates induction of the antimicrobial peptide human beta-defensin 2 in response to bacterial lipoprotein. Eur. J. Immunol. 2001, 31, 3131–3137. [Google Scholar] [CrossRef]
- Hertz, C.J.; Wu, Q.; Porter, E.M.; Zhang, Y.J.; Weismüller, K.H.; Godowski, P.J.; Ganz, T.; Randell, S.H.; Modlin, R.L. Activation of Toll-like receptor 2 on human tracheobronchial epithelial cells induces the antimicrobial peptide human beta defensin-2. J. Immunol. 2003, 171, 6820–6826. [Google Scholar] [CrossRef]
- Spachidou, M.P.; Bourazopoulou, E.; Maratheftis, C.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Tzioufas, A.G.; Manoussakis, M.N. Expression of functional Toll-like receptors by salivary gland epithelial cells: Increased mRNA expression in cells derived from patients with primary Sjogren’s syndrome. Clin. Exp. Immunol. 2007, 147, 497–503. [Google Scholar] [CrossRef]
- Liu, Y.; Yin, H.; Zhao, M.; Lu, Q. TLR2 and TLR4 in Autoimmune Diseases: A Comprehensive Review. Clin. Rev. Allerg. Immunol. 2014, 47, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Kiripolsky, J.; Kramer, J.M. Current and Emerging Evidence for Toll-Like Receptor Activation in Sjögren’s Syndrome. J. Immunol. Res. 2018, 2018, 1246818. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Franziska Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Kirschning, C.J.; Häcker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, Z.; Yu, C.; Yang, C. Expression of Toll-like receptors 7, 8, and 9 in primary Sjogren’s syndrome. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2010, 109, 844–850. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J. Clin. Investig. 2015, 125, 2228–2233. [Google Scholar] [CrossRef]
- Shimizu, T.; Nakamura, H.; Takatani, A.; Umeda, V.; Horai, Y.; Kurushima, S.; Michitsuji, T.; Nakashima, Y.; Kawakami, A. Activation of Toll-like receptor 7 signaling in labial salivary glands of primary Sjögren’s syndrome patients. Clin. Exp. Immunol. 2019, 196, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Flynn, C.M.; Garbers, Y.; Lokau, J.; Wesch, D.; Schulte, D.M.; Laudes, M.; Lieb, W.; Aparicio-Siegmund, S.; Garbers, C. Activation of Toll Like Receptor 2 (TLR2) induces interleukin-6 trans-signalling. Sci. Rep. 2019, 9, 7306. [Google Scholar] [CrossRef] [PubMed]
- Nadler, L.M.; Ritz, J.; Hardy, R.; Pesando, J.M.; Schlossman, S.F.; Stashenko, P. A unique cell surface antigen identifying lymphoid malignancies of B cell origin. J. Clin. Investig. 1981, 67, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Cragg, M.S.; Walshe, C.A.; Ivanov, A.O.; Glennie, M.J. The biology of CD20 and its potential as a target for mAb therapy In: B cell trophic factors and B cell antagonism in autoimmune disease. Curr. Dir. Autoimmun. 2005, 8, 140–174. [Google Scholar]
- Walshe, C.A.; Beers, S.A.; French, R.R.; Chan, C.H.T.; Johnson, P.W.; Packham, G.K.; Glennie, M.J.; Cragg, M.S. Induction of cytosolic calcium flux by CD20 is dependent upon B Cell antigen receptor signaling. J. Biol. Chem. 2008, 283, 16971–16984. [Google Scholar] [CrossRef]
- Leandro, M.J. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res. Ther. 2013, 15, S3. [Google Scholar] [CrossRef]
- Rehnberg, M.; Amu, S.; Tarkowski, A.; Bokarewa, M.J.; Brisslert, M. Short- and long-term effects of anti-CD20 treatment on B cell ontogeny in bone marrow of patients with rheumatoid arthritis. Arthritis Res. Ther. 2009, 11, R123. [Google Scholar] [CrossRef]
- Polyak, M.J.; Li, H.; Shariat, N.; Deans, J.P. CD20 homo-oligomers physically associate with the B cell antigen receptor. J. Biol. Chem. 2008, 283, 18545–18552. [Google Scholar] [CrossRef]
- Devauchelle-Pensec, V.; Pennec, Y.; Morvan, J.; Pers, J.O.; Daridon, C.; Jousse-Joulin, S.; Roudaut, A.; Jamin, C.; Renaudineau, Y.; Roué, I.Q.; et al. Improvement of Sjögren’s syndrome after two infusions of rituximab (anti-CD20). Arthritis Rheum. 2007, 57, 310–317. [Google Scholar] [CrossRef]
- Meijer, J.M.; Meiners, P.M.; Vissink, A.; Spijkervet, F.K.L.; Abdulahad, W.; Kamminga, N.; Brouwer, E.; Kallenberg, C.G.M.; Bootsma, H. Effectiveness of rituximab treatment in primary Sjögren’s syndrome: A randomized, double-blind, placebo controlled trial. Arthritis Rheum. 2010, 62, 960–968. [Google Scholar] [CrossRef]
- Abdulahad, W.H.; Meijer, J.M.; Kroese, F.G.; Meiners, P.M.; Vissink, A.; Spijkervet, F.K.; Kallenberg, C.G.; Bootsma, H. B cell reconstitution and T helper cell balance after rituximab treatment of active primary Sjogren’s syndrome: A double-blind, placebo-controlled study. Arthritis Rheum. 2011, 63, 1116–1123. [Google Scholar] [CrossRef]
- Edwards, J.C.; Szczepanski, L.; Szechinski, J.; Filipowicz-Sosnowska, A.; Emery, P.; Close, D.R.; Stevens, R.M.; Shaw, T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N. Engl. J. Med. 2004, 350, 2572–2581. [Google Scholar] [CrossRef]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Merkel, P.A.; Seo, P.; Spiera, R.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; Clair, E.W.S.; et al. Rituximab versus cyclophosphamide for ANCA associated vasculitis. N. Engl. J. Med. 2010, 363, 221–232. [Google Scholar] [CrossRef]
- Sisto, M.; Lisi, S.; D’Amore, M.; Lofrumento, D.D. Rituximab-mediated Raf kinase inhibitor protein induction modulates NF-κB in Sjögren syndrome. Immunology 2014, 143, 42–51. [Google Scholar] [CrossRef]
- Lisi, S.; Sisto, M.; D’Amore, M.; Lofrumento, D.D. Co-culture system of human salivary gland epithelial cells and immune cells from primary Sjögren’s syndrome patients: An in vitro approach to study the effects of Rituximab on the activation of the Raf-1/ERK1/2 pathway. Int. Immunol. 2015, 27, 183–194. [Google Scholar] [CrossRef][Green Version]
- Jazirehi, A.R.; Huerta-Yepez, S.; Cheng, G.; Bonavida, B. Rituximab (chimeric anti-CD20 monoclonal antibody) inhibits the constitutive nuclear factor-jB signalling pathway in non-Hodgkin’s lymphoma B-cell lines: Role in sensitization to chemotherapeutic drug induced apoptosis. Cancer Res. 2005, 65, 264–276. [Google Scholar]
- Zeng, L.; Imamoto, A.; Rosner, M.R. Raf kinase inhibitory protein (RKIP): A physiological regulator and future therapeutic target. Expert Opin. Ther. Targets 2008, 12, 1275–1287. [Google Scholar] [CrossRef]
- Al-Mulla, F.; Bitar, M.S.; Taqi, Z.; Yeung, K.C. RKIP: Much more than Raf kinase inhibitory protein. J. Cell Physiol. 2013, 228, 1688–1702. [Google Scholar] [CrossRef]
- Casey, E.; Bournazos, S.; Mo, G.; Mondello, P.; Tan, K.S.; Ravetch, J.V.; Scheinberg, D.A. A new mouse expressing human Fcγ receptors to better predict therapeutic efficacy of human anti-cancer antibodies. Leukemia 2018, 32, 547–549. [Google Scholar] [CrossRef]
- Zhao, J.; Wenzel, S. Interactions of RKIP with Inflammatory Signaling Pathways critical reviews in oncogenesis. Crit. Rev. Oncog. 2014, 19, 497–504. [Google Scholar] [CrossRef]
- Bedford, L.; Lowe, J.; Dick, L.; Mayer, R.J.; Brownell, J.E. Ubiquitin-like protein conjugation and the ubiquitin–proteasome system as drug targets. Nat. Rev. Drug Discov. 2011, 10, 29–46. [Google Scholar] [CrossRef]
- Plenge, R.M.; Cotsapas, C.; Davies, L.; Price, A.L.; de Bakker, P.I.; Maller, J.; Pe’er, I.; Burtt, N.P.; Blumenstiel, B.; DeFelice, M.; et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat. Genet. 2007, 39, 1477–1482. [Google Scholar] [CrossRef]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.J.; et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappa B pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef]
- Graham, R.R.; Cotsapas, C.; Davies, L.; Hackett, R.; Lessard, C.J.; Leon, J.M.; Burtt, N.P.; Guiducci, C.; Parkin, M.; Gates, C.; et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 1059–1061. [Google Scholar] [CrossRef] [PubMed]
- Fung, E.Y.; Smyth, D.J.; Howson, J.M.; Cooper, J.D.; Walker, N.M.; Stevens, H.; Wicker, L.S.; Todd, J.A. Analysis of 17 autoimmune disease associated variants in type 1 diabetes identifi es 6q23/TNFAIP3 as a susceptibility. Genes Immun. 2009, 10, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Musone, S.L.; Taylor, K.E.; Lu, T.T.; Nititham, J.; Ferreira, R.C.; Ortmann, W.; Shifrin, N.; Petri, M.A.; Kamboh, M.I.; Manzi, S.; et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Boone, D.L.; Chai, S.; Libby, S.L.; Chien, M.; Lodolce, J.P.; Ma, A. Failure to regulate TNF-induced NF-kappa B and cell death responses in A20-deficient mice. Science 2000, 289, 2350–2354. [Google Scholar] [CrossRef]
- Song, X.T.; Evel-Kabler, K.; Shen, L.; Rollins, L.; Huang, X.F.; Chen, S.Y. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat. Med. 2008, 14, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Heyninck, K.; Beyaert, R. A20 inhibits NF-kappa B activation by dual ubiquitin-editing functions. Trends Biochem. Sci. 2005, 30, 1–4. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Small Inhibitors of NF-kB in pSS | Study | References |
|---|---|---|
| Iguratimod | Clinical study | [30] |
| Syk-inihibitor-Gs-9876 | Clinical study | [30] |
| IKKε | Pre-clinical study | [4,30] |
| IκBα | Pre-clinical study | [19,26,47,48] |
| anti-Ro/SSA autoantibodies | Pre-clinical study | [14,16,24,25] |
| TNFAIP3 | Pre-clinical study | [17] |
| Calcium mobilization | Pre-clinical study | [53,54] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sisto, M.; Ribatti, D.; Lisi, S. Understanding the Complexity of Sjögren’s Syndrome: Remarkable Progress in Elucidating NF-κB Mechanisms. J. Clin. Med. 2020, 9, 2821. https://doi.org/10.3390/jcm9092821
Sisto M, Ribatti D, Lisi S. Understanding the Complexity of Sjögren’s Syndrome: Remarkable Progress in Elucidating NF-κB Mechanisms. Journal of Clinical Medicine. 2020; 9(9):2821. https://doi.org/10.3390/jcm9092821
Chicago/Turabian StyleSisto, Margherita, Domenico Ribatti, and Sabrina Lisi. 2020. "Understanding the Complexity of Sjögren’s Syndrome: Remarkable Progress in Elucidating NF-κB Mechanisms" Journal of Clinical Medicine 9, no. 9: 2821. https://doi.org/10.3390/jcm9092821
APA StyleSisto, M., Ribatti, D., & Lisi, S. (2020). Understanding the Complexity of Sjögren’s Syndrome: Remarkable Progress in Elucidating NF-κB Mechanisms. Journal of Clinical Medicine, 9(9), 2821. https://doi.org/10.3390/jcm9092821