Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review

 ,
,  , ,
, , 
Abstract
1. Introduction
2. LSDs Associated with Integral Lysosomal Membrane Proteins
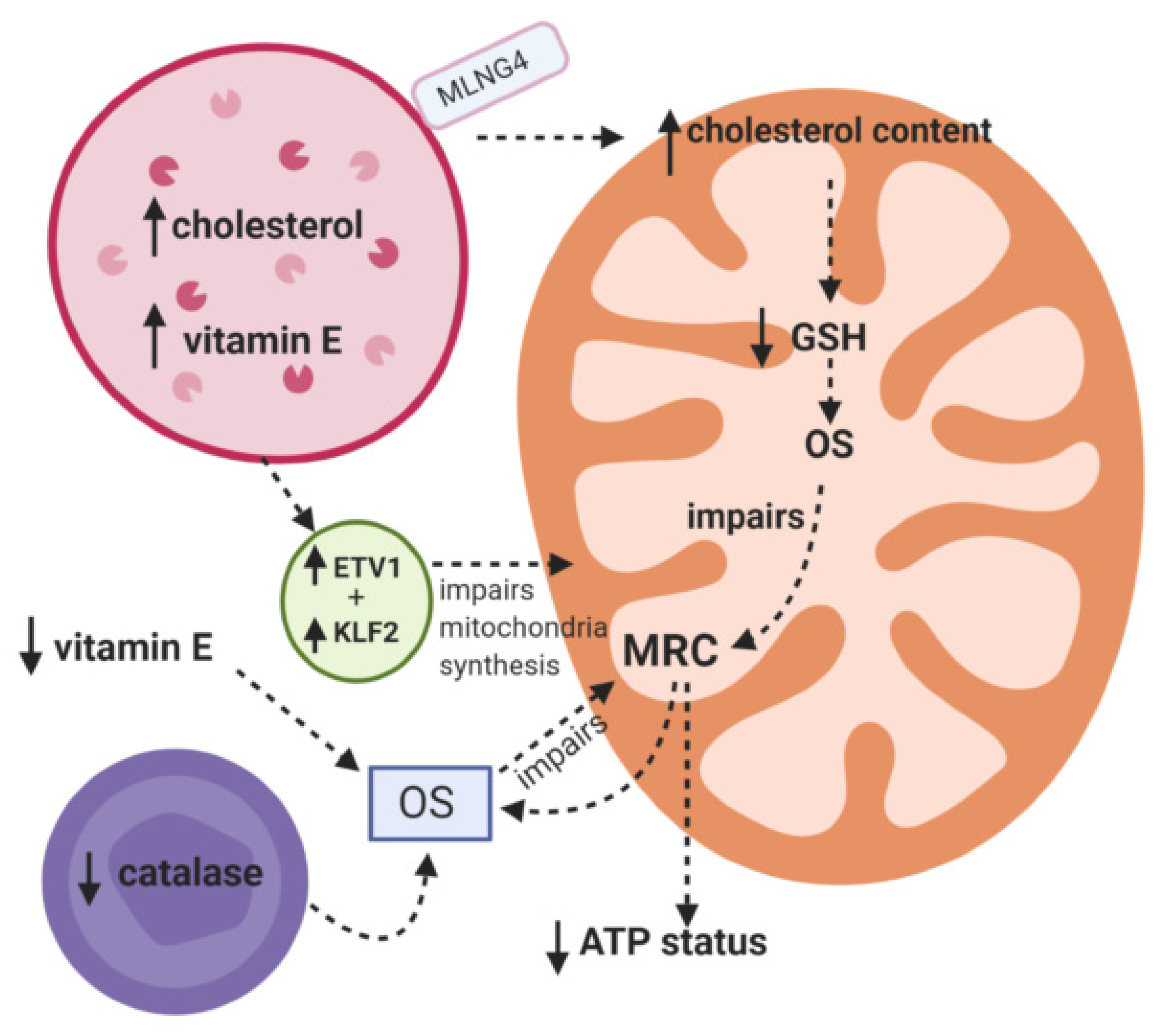
Niemann Pick C
3. LSDs Associated with Nonmembrane-Bound Lysosomal Hydrolases
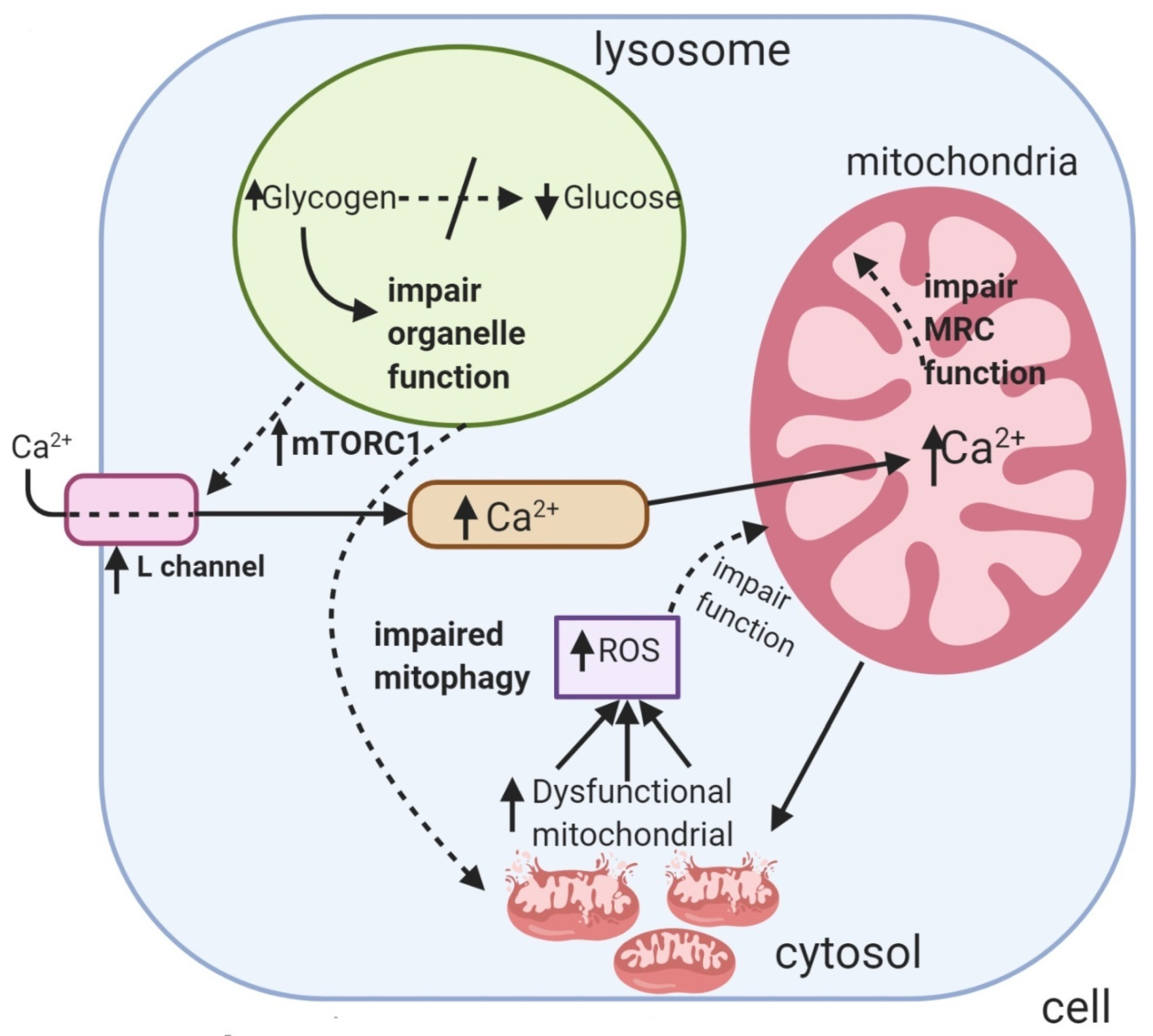
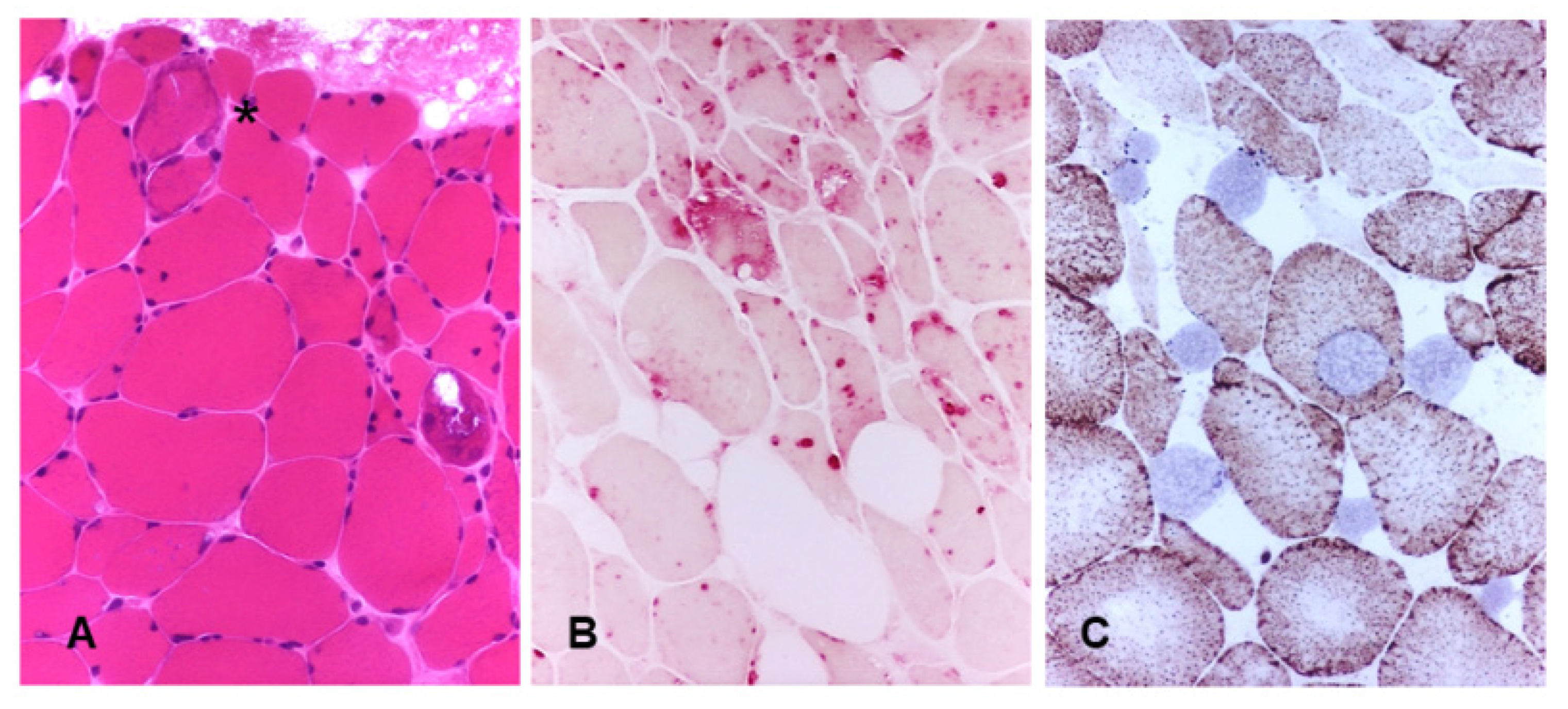
3.1. Pompe Disease
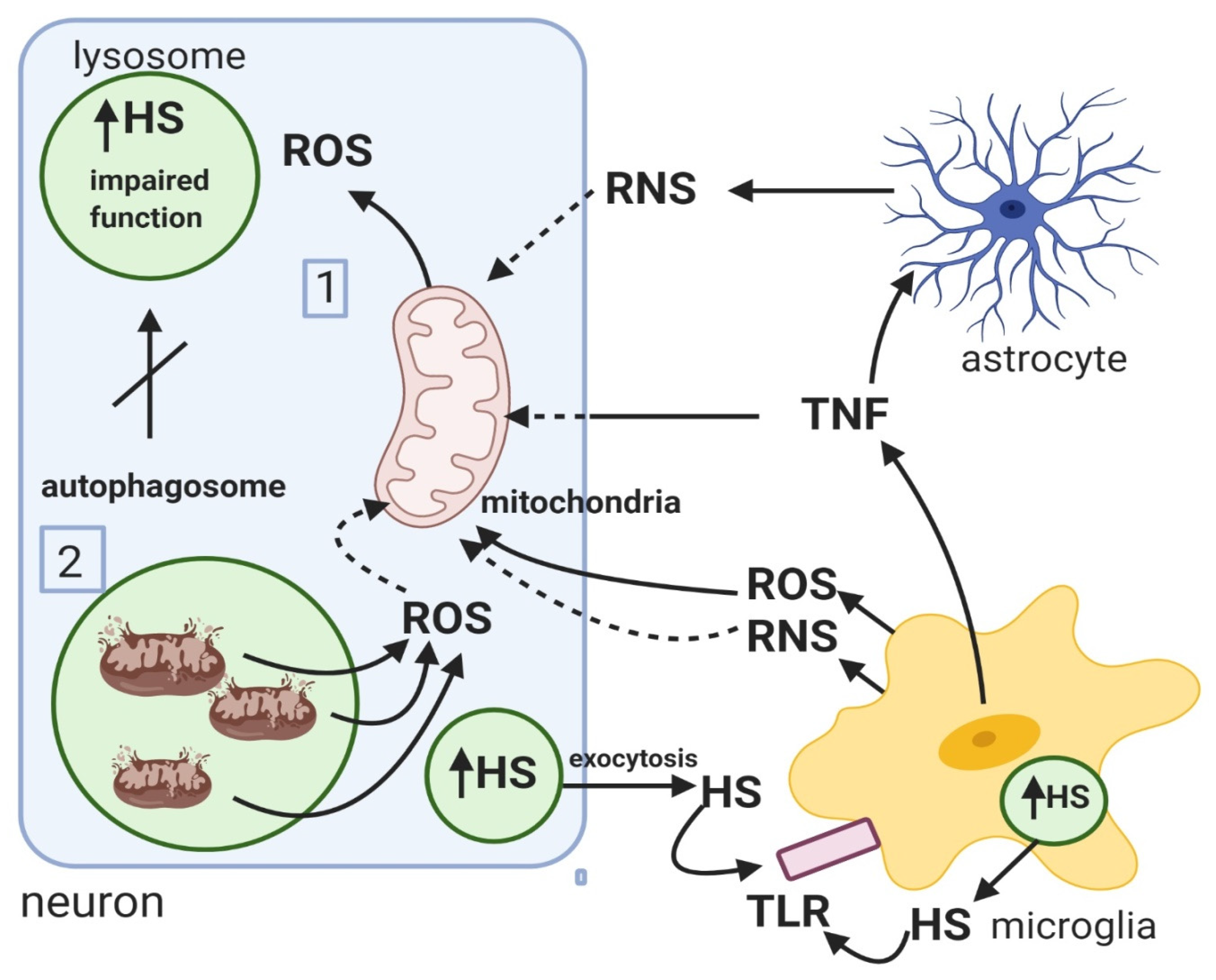
3.2. Mucopolysaccharidosis
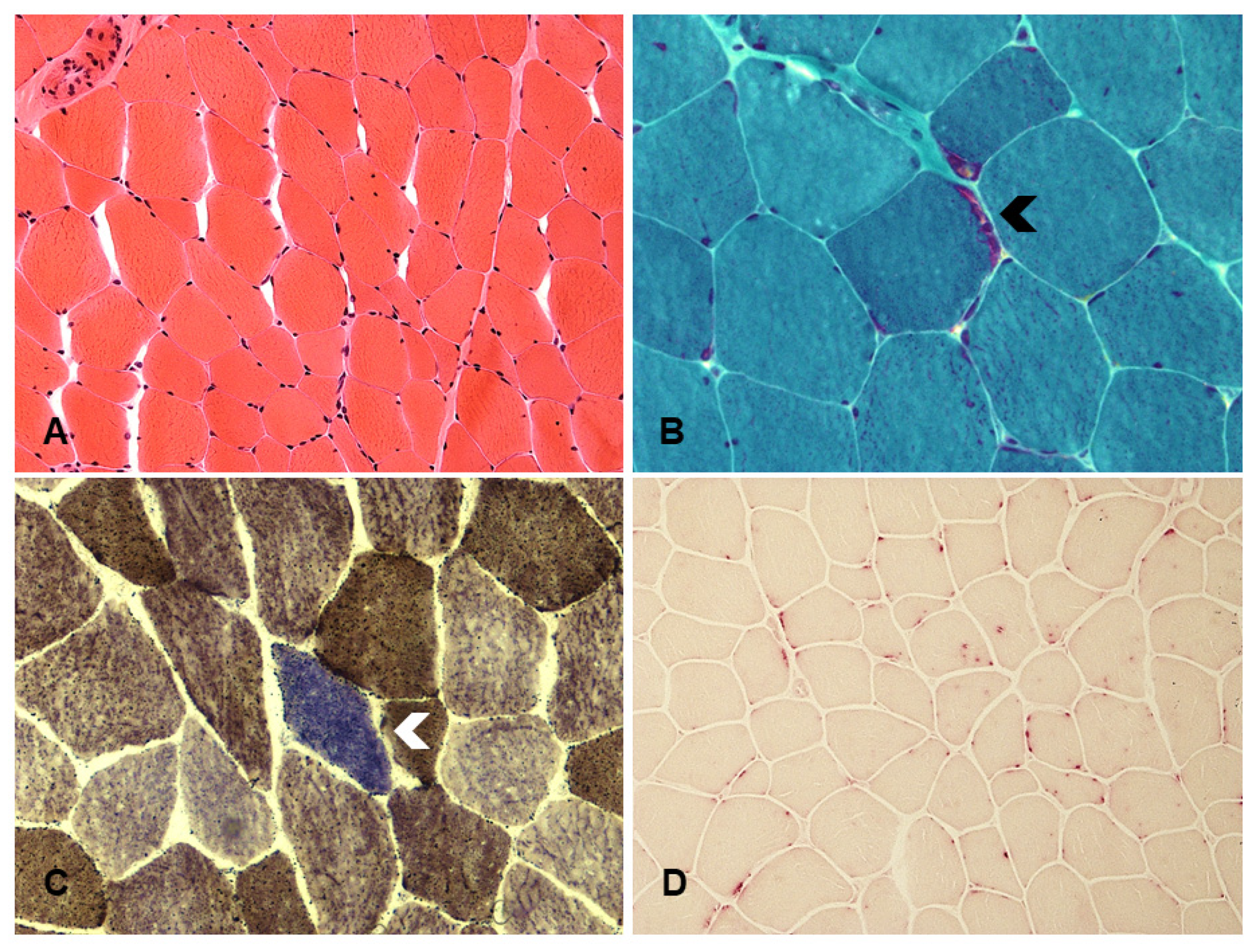
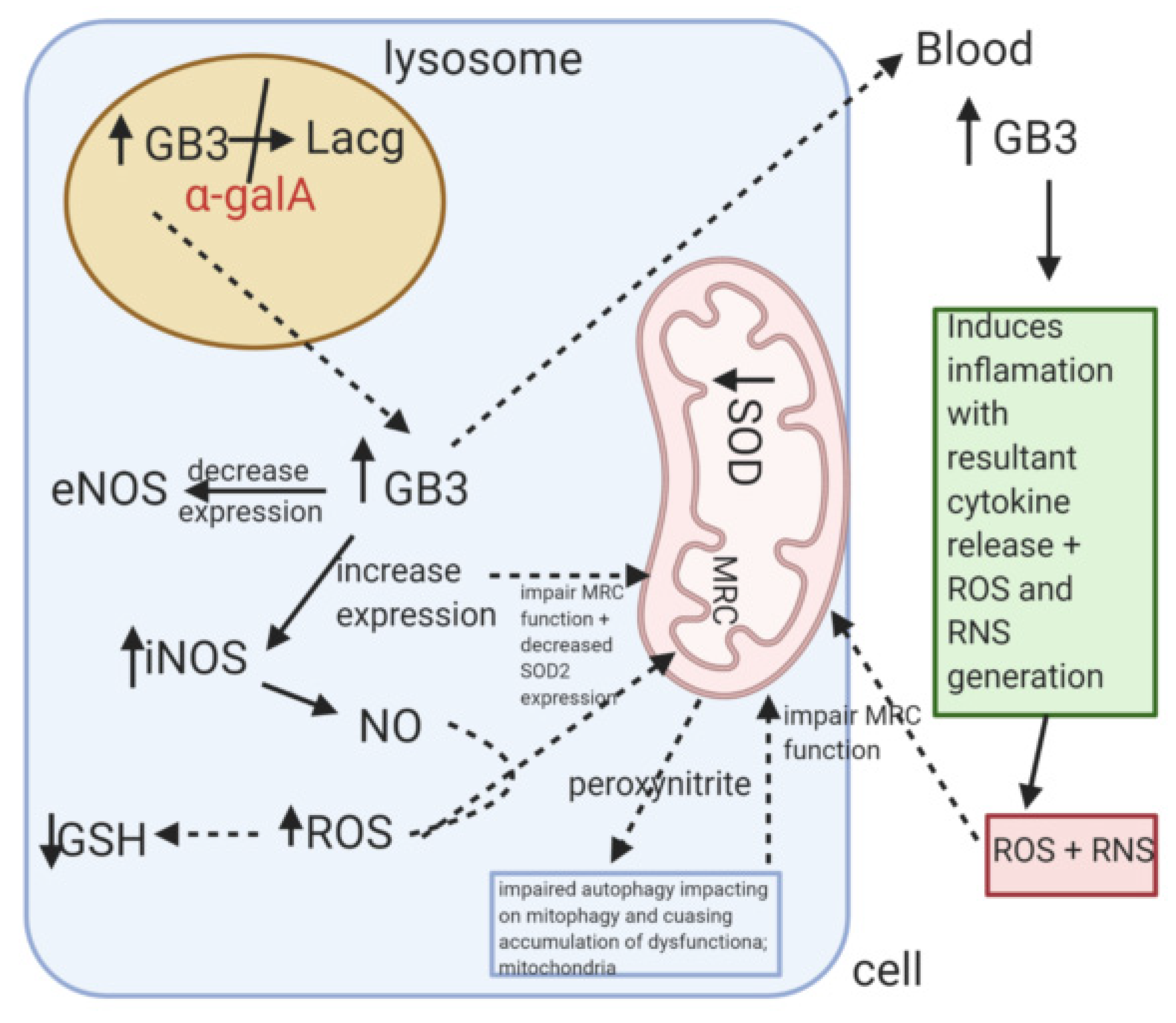
3.3. Fabry Disease
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CoQ10 | Co-enzyme Q10 |
| ERT | enzyme replacement therapy |
| LSD | lysosomal storage diseases |
| MRC | mitochondrial respiratory chain |
| NO | Nitric oxide |
| iNOS | inducible nitric oxide synthase |
| NPC | Niemann Pick C |
| OS | oxidative stress |
| ROS | redox oxidative stress |
| SCMAS | Subunit c of mitochondrial ATP synthase |
| SOD | Superoxide dismutase |
| V-ATPase | Lysosomal ATPase |
| TLR | Toll-like receptor |
References
- Platt, F.M.; Boland, B.; van der Spoel, A.C. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Parkinson-Lawrence, E.J.; Shandala, T.; Prodoehl, M.; Plew, R.; Borlace, G.N.; Brooks, D.A. Lysosomal storage disease: Revealing lysosomal function and physiology. Physiology 2010, 25, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E. Lysosomal disorders. Semin. Neonatol. 2002, 7, 75–83. [Google Scholar] [CrossRef]
- Audano, M.; Schneider, A.; Mitro, N. Mitochondria, lysosomes, and dysfunction: Their meaning in neurodegeneration. J. Neurochem. 2018, 147, 291–309. [Google Scholar] [CrossRef]
- Raben, N.; Wong, A.; Ralston, E.; Myerowitz, R. Autophagy and mitochondria in Pompe disease: Nothing is so new as what has long been forgotten. Am. J. Med. Gen. C Semin. Med. Gen. 2012, 160, 13–21. [Google Scholar] [CrossRef]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease—Links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef]
- Cleeter, M.W.; Chau, K.Y.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.W.; Hardy, J.; Mark Cooper, J.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef]
- Morén, C.; Juárez-Flores, D.L.; Chau, K.Y.; Gegg, M.; Garrabou, G.; González-Casacuberta, I.; Guitart-Mampel, M.; Tolosa, E.; Martí, M.J.; Cardellach, F.; et al. GBA mutation promotes early mitochondrial dysfunction in 3D neurosphere models. Aging 2019, 11, 10338–10355. [Google Scholar] [CrossRef]
- Heaton, R.A.; Heales, S.; Rahman, K.; Sexton, D.W.; Hargreaves, I. The effect of cellular coenzyme Q(10) deficiency on lysosomal acidification. J. Clin. Med. 2020, 9, 1923. [Google Scholar] [CrossRef]
- Montero, R.; Yubero, D.; Salgado, M.C.; González, M.J.; Campistol, J.; O’Callaghan, M.D.M.; Pineda, M.; Delgadillo, V.; Maynou, J.; Fernandez, G.; et al. Plasma coenzyme Q(10) status is impaired in selected genetic conditions. Sci. Rep. 2019, 9, 793. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T.; Duthel, S.; Rodriguez-Lafrasse, C.; Pentchev, P.; Carstea, E.D. Genetic heterogeneity in Niemann-Pick C disease: A study using somatic cell hybridization and linkage analysis. Am. J. Hum. Genet. 1996, 58, 118–125. [Google Scholar] [PubMed]
- Hargreaves, I.; Sheena, Y.; Land, J.; Heales, S. Glutathione deficiency in patients with mitochondrial disease: Implications for pathogenesis and treatment. J. Inherit. Metab. Dis. 2005, 28, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, M.C.; Balboa, E.; Alvarez, A.R.; Zanlungo, S. Oxidative stress: A pathogenic mechanism for Niemann-Pick type C disease. Oxid. Med. Cell. Longev. 2012, 2012, 205713. [Google Scholar] [CrossRef]
- Fu, R.; Yanjanin, N.M.; Bianconi, S.; Pavan, W.J.; Porter, F.D. Oxidative stress in Niemann-Pick disease, type C. Mol. Genet. Metab. 2010, 101, 214–218. [Google Scholar] [CrossRef]
- Hargreaves, I.P. Ubiquinone: Cholesterol’s reclusive cousin. Ann. Clin. Biochem. 2003, 40, 207–218. [Google Scholar] [CrossRef]
- Ribas, G.S.; Pires, R.; Coelho, J.C.; Rodrigues, D.; Mescka, C.P.; Vanzin, C.S.; Biancini, G.B.; Negretto, G.; Wayhs, C.A.; Wajner, M.; et al. Oxidative stress in Niemann-Pick type C patients: A protective role of N-butyl-deoxynojirimycin therapy. Int. J. Dev. Neurosci. 2012, 30, 439–444. [Google Scholar] [CrossRef]
- Ulatowski, L.; Parker, R.; Davidson, C.; Yanjanin, N.; Kelley, T.J.; Corey, D.; Atkinson, J.; Porter, F.; Arai, H.; Walkley, S.U.; et al. Altered vitamin E status in Niemann-Pick type C disease. J. Lipid Res. 2011, 52, 1400–1410. [Google Scholar] [CrossRef]
- Schedin, S.; Sindelar, P.J.; Pentchev, P.; Brunk, U.; Dallner, G. Peroxisomal impairment in Niemann-Pick type C disease. J. Biol. Chem. 1997, 272, 6245–6251. [Google Scholar] [CrossRef]
- Charman, M.; Kennedy, B.E.; Osborne, N.; Karten, B. MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J. Lipid Res. 2010, 51, 1023–1034. [Google Scholar] [CrossRef]
- Yu, W.; Gong, J.-S.; Ko, M.; Garver, W.S.; Yanagisawa, K.; Michikawa, M. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J. Biol. Chem. 2005, 280, 11731–11739. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.E.; Murphy, E.J.; Mitchell, D.C.; Golovko, M.Y.; Scaglia, F.; Barceló-Coblijn, G.C.; Nussbaum, R.L. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol. Cell Biol. 2005, 25, 10190–10201. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Kaplowitz, N. Hepatic mitochondrial glutathione: Transport and role in disease and toxicity. Toxicol. Appl. Pharm. 2005, 204, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Matías, N.; Baulies, A.; Nuñez, S.; Alarcon-Vila, C.; Martinez, L.; Nuño, N.; Fernandez, A.; Caballeria, J.; Levade, T.; et al. Mitochondrial GSH replenishment as a potential therapeutic approach for Niemann-Pick type C disease. Redox Biol. 2017, 11, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Balboa, E.; Castro, J.; Pinochet, M.J.; Cancino, G.I.; Matías, N.; Sáez, P.J.; Martínez, A.; Álvarez, A.R.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; et al. MLN64 induces mitochondrial dysfunction associated with increased mitochondrial cholesterol content. Redox Biol. 2017, 12, 274–284. [Google Scholar] [CrossRef]
- Yambire, K.F.; Fernandez-Mosquera, L.; Steinfeld, R.; Mühle, C.; Ikonen, E.; Milosevic, I.; Raimundo, N. Mitochondrial biogenesis is transcriptionally repressed in lysosomal lipid storage diseases. eLife 2019, 8. [Google Scholar] [CrossRef]
- Pascarella, A.; Terracciano, C.; Farina, O.; Lombardi, L.; Esposito, T.; Napolitano, F.; Franzese, G.; Panella, G.; Tuccillo, F.; la Marca, G. Vacuolated PAS-positive lymphocytes as an hallmark of Pompe disease and other myopathies related to impaired autophagy. J. Cell. Physiol. 2018, 233, 5829–5837. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef]
- Lim, J.-A.; Kakhlon, O.; Li, L.; Myerowitz, R.; Raben, N. Pompe disease: Shared and unshared features of lysosomal storage disorders. Rare Dis. 2015, 3, e1068978. [Google Scholar] [CrossRef]
- Fukuda, T.; Ahearn, M.; Roberts, A.; Mattaliano, R.J.; Zaal, K.; Ralston, E.; Plotz, P.H.; Raben, N. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol. Ther. 2006, 14, 831–839. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Fanin, M.; Masiero, E.; Angelini, C.; Sandri, M. The role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII). Cell Death Differ. 2012, 19, 1698–1708. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Fanin, M.; Tasca, E.; Angelini, C.; Sandri, M. Impaired autophagy affects acid α-glucosidase processing and enzyme replacement therapy efficacy in late-onset glycogen storage disease type II. Neuropathol. Appl. Neurobiol. 2015, 41, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Shea, L.; Raben, N. Autophagy in skeletal muscle: Implications for Pompe disease. Int. J. Clin. Pharmacol. Ther. 2009, 47, S42. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Schreiner, C.; Baum, R.; Takikita, S.; Xu, S.; Xie, T.; Myerowitz, R.; Komatsu, M.; Van Der Meulen, J.H.; Nagaraju, K. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder—Murine Pompe disease. Autophagy 2010, 6, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-A.; Li, L.; Kakhlon, O.; Myerowitz, R.; Raben, N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 2015, 11, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; de Chadarevian, J.P.; Melvin, J.J.; Grover, W.D.; Salganicoff, L.; Kaye, E.M. Mitochondrial activity in Pompe’s disease. Pediatric Neurol. 2000, 23, 54–57. [Google Scholar] [CrossRef]
- Sarnat, H.B.; Roth, S.I.; Carroll, J.E.; Brown, B.I.; Dungan, W.T. Lipid storage myopathy in infantile Pompe’s disease. Arch. Neurol. 1982, 39, 180–183. [Google Scholar] [CrossRef]
- Verity, M.A. Infantile Pompe’s disease, lipid storage, and partial carnitine deficiency. Muscle Nerve 1991, 14, 435–440. [Google Scholar] [CrossRef]
- Sato, Y.; Kobayashi, H.; Higuchi, T.; Shimada, Y.; Ida, H.; Ohashi, T. Metabolomic profiling of pompe disease-induced pluripotent stem cell-derived cardiomyocytes reveals that oxidative stress is associated with cardiac and skeletal muscle pathology. Stem Cells Transl. Med. 2017, 6, 31–39. [Google Scholar] [CrossRef]
- Engel, A.G.; Dale, A. Autophagic glycogenosis of late onset with mitochondrial abnormalities: Light and electron microscopic observations. Mayo Clin. Proc. 1968, 43, 233–279. [Google Scholar]
- Fernández, R.; Fernández, J.M.; Cervera, C.; Teijeira, S.; Teijeiro, A.; Domínguez, C.; Navarro, C. Adult glycogenosis II with paracrystalline mitochondrial inclusions and Hirano bodies in skeletal muscle. Neuromuscul. Disord. 1999, 9, 136–143. [Google Scholar] [CrossRef]
- Lewandowska, E.; Wierzba-Bobrowicz, T.; Rola, R.; Modzelewska, J.; Stepien, T.; Lugowska, A.; Pasennik, E.; Ryglewicz, D. Pathology of skeletal muscle cells in adult-onset glycogenosis type II (Pompe disease): Ultrastructural study. Folia Neuropathol. 2008, 46, 123–133. [Google Scholar]
- Huang, H.-P.; Chen, P.-H.; Hwu, W.-L.; Chuang, C.-Y.; Chien, Y.-H.; Stone, L.; Chien, C.-L.; Li, L.-T.; Chiang, S.-C.; Chen, H.-F. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Hum. Mol. Genet. 2011, 20, 4851–4864. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.; Hůlková, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef]
- Pshezhetsky, A.V. Crosstalk between 2 organelles: Lysosomal storage of heparan sulfate causes mitochondrial defects and neuronal death in mucopolysaccharidosis III type C. Rare Dis. 2015, 3, e1049793. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, P.; Li, J.P.; Lannfelt, L.; Lindahl, U.; Zhang, X. Microglial heparan sulfate proteoglycans facilitate the cluster-of-differentiation 14 (CD14)/Toll-like receptor 4 (TLR4)-dependent inflammatory response. J. Biol. Chem. 2015, 290, 14904–14914. [Google Scholar] [CrossRef] [PubMed]
- Figuera-Losada, M.; Rojas, C.; Slusher, B.S. Inhibition of microglia activation as a phenotypic assay in early drug discovery. J. Biomol. Screen. 2014, 19, 17–31. [Google Scholar] [CrossRef]
- Vitner, E.B.; Farfel-Becker, T.; Eilam, R.; Biton, I.; Futerman, A.H. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain 2012, 135, 1724–1735. [Google Scholar] [CrossRef]
- Saha, R.N.; Pahan, K. Signals for the induction of nitric oxide synthase in astrocytes. Neurochem. Int. 2006, 49, 154–163. [Google Scholar] [CrossRef]
- Doll, D.N.; Rellick, S.L.; Barr, T.L.; Ren, X.; Simpkins, J.W. Rapid mitochondrial dysfunction mediates TNF-alpha-induced neurotoxicity. J. Neurochem. 2015, 132, 443–451. [Google Scholar] [CrossRef]
- Baregamian, N.; Song, J.; Bailey, C.E.; Papaconstantinou, J.; Evers, B.M.; Chung, D.H. Tumor necrosis factor-alpha and apoptosis signal-regulating kinase 1 control reactive oxygen species release, mitochondrial autophagy, and c-Jun N-terminal kinase/p38 phosphorylation during necrotizing enterocolitis. Oxid. Med. Cell. Longev. 2009, 2, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Donida, B.; Jacques, C.E.D.; Mescka, C.P.; Rodrigues, D.G.B.; Marchetti, D.P.; Ribas, G.; Giugliani, R.; Vargas, C.R. Oxidative damage and redox in Lysosomal Storage Disorders: Biochemical markers. Clin. Chim. Acta 2017, 466, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Trudel, S.; Trécherel, E.; Gomila, C.; Peltier, M.; Aubignat, M.; Gubler, B.; Morlière, P.; Heard, J.M.; Ausseil, J. Oxidative stress is independent of inflammation in the neurodegenerative Sanfilippo syndrome type B. J. Neurosci. Res. 2015, 93, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Tessitore, A.; Pirozzi, M.; Auricchio, A. Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics 2009, 2, 1–12. [Google Scholar] [CrossRef]
- Boya, P.; González-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Métivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta 2009, 1793, 684–696. [Google Scholar] [CrossRef]
- Pacheco, C.D.; Kunkel, R.; Lieberman, A.P. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 2007, 16, 1495–1503. [Google Scholar] [CrossRef]
- Kuech, E.M.; Brogden, G.; Naim, H.Y. Alterations in membrane trafficking and pathophysiological implications in lysosomal storage disorders. Biochimie 2016, 130, 152–162. [Google Scholar] [CrossRef]
- Jennings, J.J., Jr.; Zhu, J.H.; Rbaibi, Y.; Luo, X.; Chu, C.T.; Kiselyov, K. Mitochondrial aberrations in mucolipidosis Type IV. J. Biol. Chem. 2006, 281, 39041–39050. [Google Scholar] [CrossRef]
- Vergarajauregui, S.; Oberdick, R.; Kiselyov, K.; Puertollano, R. Mucolipin 1 channel activity is regulated by protein kinase A-mediated phosphorylation. Biochem. J. 2008, 410, 417–425. [Google Scholar] [CrossRef]
- Vergarajauregui, S.; Puertollano, R. Mucolipidosis type IV: The importance of functional lysosomes for efficient autophagy. Autophagy 2008, 4, 832–834. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jezela-Stanek, A.; Ciara, E.; Stepien, K.M. Neuropathophysiology, genetic profile, and clinical manifestation of mucolipidosis IV—A review and case series. Int. J. Mol. Sci. 2020, 21, 4564. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, F.; Signorile, A.; Tamma, G.; Ranieri, M.; Emma, F.; De Rasmo, D. Impact of atypical mitochondrial cyclic-AMP level in nephropathic cystinosis. Cell. Mol. Life Sci. 2018, 75, 3411–3422. [Google Scholar] [CrossRef]
- De Rasmo, D.; Signorile, A.; De Leo, E.; Polishchuk, E.V.; Ferretta, A.; Raso, R.; Russo, S.; Polishchuk, R.; Emma, F.; Bellomo, F. Mitochondrial dynamics of proximal tubular epithelial cells in nephropathic cystinosis. Int. J. Mol. Sci. 2019, 21, 192. [Google Scholar] [CrossRef] [PubMed]
- Hashem, S.I.; Perry, C.N.; Bauer, M.; Han, S.; Clegg, S.D.; Ouyang, K.; Deacon, D.C.; Spinharney, M.; Panopoulos, A.D.; Izpisua Belmonte, J.C. Brief report: Oxidative stress mediates cardiomyocyte apoptosis in a human model of Danon disease and heart failure. Stem Cells 2015, 33, 2343–2350. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lüllmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef]
- Plotegher, N.; Duchen, M.R. Mitochondrial dysfunction and neurodegeneration in lysosomal storage disorders. Trends Mol. Med. 2017, 23, 116–134. [Google Scholar] [CrossRef]
- Hamano, K.; Hayashi, M.; Shioda, K.; Fukatsu, R.; Mizutani, S. Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: Analysis of human brain tissue. Acta Neuropathol. 2008, 115, 547–559. [Google Scholar] [CrossRef]
- De Pablo-Latorre, R.; Saide, A.; Polishhuck, E.V.; Nusco, E.; Fraldi, A.; Ballabio, A. Impaired parkin-mediated mitochondrial targeting to autophagosomes differentially contributes to tissue pathology in lysosomal storage diseases. Hum. Mol. Genet. 2012, 21, 1770–1781. [Google Scholar] [CrossRef]
- Boonen, M.; van Meel, E.; Oorschot, V.; Klumperman, J.; Kornfeld, S. Vacuolization of mucolipidosis type II mouse exocrine gland cells represents accumulation of autolysosomes. Mol. Biol. Cell 2011, 22, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Otomo, T.; Higaki, K.; Nanba, E.; Ozono, K.; Sakai, N. Inhibition of autophagosome formation restores mitochondrial function in mucolipidosis II and III skin fibroblasts. Mol. Genet. Metab. 2009, 98, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Otomo, T.; Higaki, K.; Nanba, E.; Ozono, K.; Sakai, N. Lysosomal storage causes cellular dysfunction in mucolipidosis II skin fibroblasts. J. Biol. Chem. 2011, 286, 35283–35290. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Takahashi-Fujigasaki, J.; Fukuda, T.; Sakurai, K.; Shimada, Y.; Nomura, K.; Ariga, M.; Ohashi, T.; Eto, Y.; Otomo, T.; et al. Pathology of the first autopsy case diagnosed as mucolipidosis type III α/β suggesting autophagic dysfunction. Mol. Genet. Metab. 2011, 102, 170–175. [Google Scholar] [CrossRef]
- Elleder, M.; Sokolova, J.; Hřebíček, M. Follow-up study of subunit c of mitochondrial ATP synthase (SCMAS) in Batten disease and in unrelated lysosomal disorders. Acta Neuropathol. 1997, 93, 379–390. [Google Scholar] [CrossRef]
- Takamura, A.; Higaki, K.; Kajimaki, K.; Otsuka, S.; Ninomiya, H.; Matsuda, J.; Ohno, K.; Suzuki, Y.; Nanba, E. Enhanced autophagy and mitochondrial aberrations in murine GM1-gangliosidosis. Biochem. Biophys. Res. Commun. 2008, 367, 616–622. [Google Scholar] [CrossRef]
- Ivanova, M.M.; Changsila, E.; Iaonou, C.; Goker-Alpan, O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE 2019, 14, e0210617. [Google Scholar] [CrossRef]
- Ferreira, N.S.; Goldschmidt-Arzi, M.; Sabanay, H.; Storch, J.; Levade, T.; Ribeiro, M.G.; Addadi, L.; Futerman, A.H. Accumulation of ordered ceramide-cholesterol domains in farber disease fibroblasts. JIMD Rep. 2014, 12, 71–77. [Google Scholar] [CrossRef]
- Ivankovic, D.; Chau, K.Y.; Schapira, A.H.; Gegg, M.E. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J. Neurochem. 2016, 136, 388–402. [Google Scholar] [CrossRef]
- Das, A.M.; Von Harlem, R.; Feist, M.; Lücke, T.; Kohlschotter, A. Altered levels of high-energy phosphate compoundsin fibroblasts from different forms of neuronal ceroid lipofuscinoses: Further evidence for mitochondria) involvement. Eur. J. Paediatr. Neurol. 2001, 5, 143–146. [Google Scholar] [CrossRef]
- Gegg, M.E.; Schapira, A.H. Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiol. Dis. 2016, 90, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Perocheau, D.; Ferrazza, R.; Massaro, G.; Bhosale, G.; Zambon, F.; Rahim, A.A.; Guella, G.; Waddington, S.N.; Szabadkai, G.; et al. Impaired cellular bioenergetics caused by GBA1 depletion sensitizes neurons to calcium overload. Cell Death Differ. 2020, 27, 1588–1603. [Google Scholar] [CrossRef] [PubMed]
- Pchelina, S.N.; Nuzhnyi, E.P.; Emelyanov, A.K.; Boukina, T.M.; Usenko, T.S.; Nikolaev, M.A.; Salogub, G.N.; Yakimovskii, A.F.; Zakharova, E.Y. Increased plasma oligomeric alpha-synuclein in patients with lysosomal storage diseases. Neurosci. Lett. 2014, 583, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Voccoli, V.; Tonazzini, I.; Signore, G.; Caleo, M.; Cecchini, M. Role of extracellular calcium and mitochondrial oxygen species in psychosine-induced oligodendrocyte cell death. Cell Death Dis. 2014, 5, e1529. [Google Scholar] [CrossRef]
- Jolly, R.D.; Brown, S.; Das, A.M.; Walkley, S.U. Mitochondrial dysfunction in the neuronal ceroid-lipofuscinoses (Batten disease). Neurochem. Int. 2002, 40, 565–571. [Google Scholar] [CrossRef]
- Koike, M.; Shibata, M.; Waguri, S.; Yoshimura, K.; Tanida, I.; Kominami, E.; Gotow, T.; Peters, C.; von Figura, K.; Mizushima, N.; et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). Am. J. Pathol. 2005, 167, 1713–1728. [Google Scholar] [CrossRef]
- Vines, D.J.; Warburton, M.J. Classical late infantile neuronal ceroid lipofuscinosis fibroblasts are deficient in lysosomal tripeptidyl peptidase I. FEBS Lett. 1999, 443, 131–135. [Google Scholar] [CrossRef]
- Desnick, R.J. Fabry disease: α-galactosidase A deficiency. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease; Rosenberg, R.N., Pascual, J.M., Eds.; Elsevier: London, UK, 2015; pp. 419–430. [Google Scholar]
- Shen, J.-S.; Meng, X.-L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef]
- Biancini, G.B.; Vanzin, C.S.; Rodrigues, D.B.; Deon, M.; Ribas, G.S.; Barschak, A.G.; Manfredini, V.; Netto, C.B.; Jardim, L.B.; Giugliani, R. Globotriaosylceramide is correlated with oxidative stress and inflammation in Fabry patients treated with enzyme replacement therapy. Biochim. Biophys. Acta 2012, 1822, 226–232. [Google Scholar] [CrossRef]
- Rozenfeld, P.; Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef]
- Tseng, W.-L.; Chou, S.-J.; Chiang, H.-C.; Wang, M.-L.; Chien, C.-S.; Chen, K.-H.; Leu, H.-B.; Wang, C.-Y.; Chang, Y.-L.; Liu, Y.-Y. Imbalanced production of reactive oxygen species and mitochondrial antioxidant SOD2 in Fabry disease-specific human induced pluripotent stem cell-differentiated vascular endothelial cells. Cell Transplant. 2017, 26, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Namdar, M.; Gebhard, C.; Studiger, R.; Shi, Y.; Mocharla, P.; Schmied, C.; Brugada, P.; Lüscher, T.F.; Camici, G.G. Globotriaosylsphingosine accumulation and not alpha-galactosidase—A deficiency causes endothelial dysfunction in Fabry disease. PLoS ONE 2012, 7, e36373. [Google Scholar] [CrossRef]
- Bodary, P.F.; Shen, Y.; Vargas, F.B.; Bi, X.; Ostenso, K.A.; Gu, S.; Shayman, J.A.; Eitzman, D.T. α-galactosidase A deficiency accelerates atherosclerosis in mice with apolipoprotein E deficiency. Circulation 2005, 111, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.F.; Scott, L.T.; Gladwin, M.T.; Altarescu, G.; Kaneski, C.; Suzuki, K.; Pease-Fye, M.; Ferri, R.; Brady, R.O.; Herscovitch, P. Regional cerebral hyperperfusion and nitric oxide pathway dysregulation in Fabry disease: Reversal by enzyme replacement therapy. Circulation 2001, 104, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, C.; Scopelliti, F.; Vulpis, E.; Tafani, M.; Villanova, L.; Verardo, R.; De Paulis, R.; Russo, M.A.; Frustaci, A. Increased oxidative stress contributes to cardiomyocyte dysfunction and death in patients with Fabry disease cardiomyopathy. Hum. Pathol. 2015, 46, 1760–1768. [Google Scholar] [CrossRef]
- Shu, L.; Vivekanandan-Giri, A.; Pennathur, S.; Smid, B.E.; Aerts, J.M.; Hollak, C.E.; Shayman, J.A. Establishing 3-nitrotyrosine as a biomarker for the vasculopathy of Fabry disease. Kidney Int. 2014, 86, 58–66. [Google Scholar] [CrossRef]
- Askari, H.; Kaneski, C.R.; Semino-Mora, C.; Desai, P.; Ang, A.; Kleiner, D.E.; Perlee, L.T.; Quezado, M.; Spollen, L.E.; Wustman, B.A.; et al. Cellular and tissue localization of globotriaosylceramide in Fabry disease. Virchows Arch. 2007, 451, 823–834. [Google Scholar] [CrossRef]
- Ponnuswamy, P.; Ostermeier, E.; Schröttle, A.; Chen, J.; Huang, P.L.; Ertl, G.; Nieswandt, B.; Kuhlencordt, P.J. Oxidative stress and compartment of gene expression determine proatherosclerotic effects of inducible nitric oxide synthase. Am. J. Pathol. 2009, 174, 2400–2410. [Google Scholar] [CrossRef]
- Lücke, T.; Höppner, W.; Schmidt, E.; Illsinger, S.; Das, A.M. Fabry disease: Reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol. Genet. Metab. 2004, 82, 93–97. [Google Scholar] [CrossRef]
- Das, A.M.; Naim, H.Y. Biochemical basis of Fabry disease with emphasis on mitochondrial function and protein trafficking. Adv. Clin. Chem. 2009, 49, 57–71. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, C.; Chico, L.; Concolino, D.; Sestito, S.; Fancellu, L.; Boadu, W.; Sechi, G.; Feliciani, C.; Gnarra, M.; Zampetti, A. Mitochondrial DNA haplogroups may influence Fabry disease phenotype. Neurosci. Lett. 2016, 629, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Halliday, G.M.; Simuni, T. Calcium, mitochondrial dysfunction and slowing the progression of Parkinson’s disease. Exp. Neurol. 2017, 298, 202–209. [Google Scholar] [CrossRef] [PubMed]
- De la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; Garrido-Maraver, J.; Cordero, M.D.; Villanueva Paz, M.; Delgado Pavón, A.; Alcocer-Gómez, E.; de Lavera, I.; Ybot-González, P.; et al. Pharmacological chaperones and coenzyme Q10 treatment improves mutant β-glucocerebrosidase activity and mitochondrial function in neuronopathic forms of gaucher disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef]
- A Study to Evaluate N-Acetyl-L-Leucine for Niemann-Pick Disease, Type C (NPC). Available online: https://www.mayo.edu/research/clinical-trials/cls-20467313 (accessed on 10 August 2020).
- Matalonga, L.; Arias, A.; Coll, M.J.; Garcia-Villoria, J.; Gort, L.; Ribes, A. Treatment effect of coenzyme Q(10) and an antioxidant cocktail in fibroblasts of patients with Sanfilippo disease. J. Inherit. Metab. Dis. 2014, 37, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Pietrzynowska, K.; Gaffke, L.; Hac, A.; Mantej, J.; Niedzialek, N.; Brokowska, J.; Węgrzyn, G. Correction of Huntington’s disease phenotype by genistein-induced autophagy in the cellular model. Neuromuscul. Med. 2018, 20, 112–123. [Google Scholar]
- Lotfi, P.; Tse, D.Y.; Di Ronza, A.; Seymour, M.L.; Martano, G.; Cooper, J.D.; Pereira, F.A.; Passafaro, M.; Wu, S.M.; Sardiello, M. Trehalose reduces retinal degeneration, neuroinflammation and storage burden caused by a lysosomal hydrolase deficiency. Autophagy 2018, 14, 1419–1434. [Google Scholar] [CrossRef]
- Pietrzynowska, K.; Gaffke, L.; Podlacha, M.; Brokowska, J.; Wegrzyn, G. Mucopolysaccharidosis and autophagy: Controversies on the contribution of the process to the pathogenesis and possible therapeutic appliations. NeuroMol. Med. 2020, 22, 25–30. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Khan, A.; Zheng, H.; Yuan, C.; Jiang, H. Advances in drug therapy for mitochondrial diseases. Ann. Transl. Med. 2020, 8, 17. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorder | Enzyme Deficient/Protein Affected | Genetic Defect | Storage Material/Pathologic Hallmark | Domain of Mitochondrial Dysfunction | References |
|---|---|---|---|---|---|
| Lysosomal Storage Disorders (LSDs) Associated with Integral Lysosomal Membrane Proteins | |||||
| Niemann Pick C | Glycosylated transmembrane protein of the lysosomal membrane | NPC1 and NPC2 gene | Intracellular accumulation of cholesterol and other lipids like glycosphingolipids, sphingomyelin and sphingosine in the lysosomes and late endosomes | Autophagosomes accumulation and autophagy dysfunction in disease pathogenesis. | [57,58,59] |
| Induction of autophagy. | |||||
| Lysosomal cholesterol and lipid export, foam cells in visceral organs and neuronal storage. | |||||
| Mucolipidosis IV | TRPML-I (ion channel- mucolipin 1) | MCOLN1 gene | Lipofuscin in lysosomes, lipids | Autophagosomes accumulation and authophagy dysfunction in disease pathogenesis. | [57,60,61,62,63] |
| Block of authophagosome-lysosome maturation. | |||||
| Cystinosis | Reduced or absent function of the specific carrier cystinosin | CTNS gene | Cystine in lysosomes | Mitochondrial fusion is mediated by three key regulatory fusion proteins: the dynamin-related GTPases mitofusin 1 (MFN1) and mitofusin 2 (MFN2), and the dynamin-related GTPases optic atrophy 1 (OPA1); altered endosomal trafficking, impaired autophagy, and cell oxidation | [64,65] |
| Danon disease | LAMP-2 lysosomal membrane protein | LAMP-2 gene | Glycogen | Autophagic flux, which results in excessive oxidative stress, and subsequent cardiomyocyte apoptosis; autophagosome accumulation, autophagy dysfunction in disease pathogenesis, block of autophagosome–lysosome maturation. | [57,66,67] |
| LSDs Associated with Nonmembrane-Bound Lysosomal Hydrolases | |||||
| Pompe | alpha-Glucosidase | GAA gene | Glycogen | Autophagosomes accumulation and autophagy dysfunction in disease pathogenesis; | [30,57,59] |
| (Autophagic) accumulation in type II muscle fibers | |||||
| Mucopolysaccharidosis: III A | heparan N-sulfatase | SGSH gene | Heparan sulphate | Autophagosomes accumulation, | [44,56,57,68,69] |
| N-acetyl-alpha-D-glucosaminidase | |||||
| IIIB | acetyl-CoA: aglucosaminide acetyltransferase | NAGLU gene | autophagy dysfunction in disease pathogenesis, | ||
| block of autophagosome–lysosome maturation; | |||||
| IIIC | N-acetylglucosamine-6-sulfate sulfatase | HGSNAT gene | Accumulation, Fragmentation/swelling, Selective reduction of OXPHOS complexes, | ||
| IIID | GNS gene | Decreased coenzyme Q10 | |||
| Mucopolysaccharidosis VI (Maroteaux–Lamy) | N-acetylgalactosamine-4-sulfatase | ARSB gene | Dermatan sulphate | Autophagosome–lysosome fusion is not completely blocked; accumulation of dysfunctional mitochondria; increased levels of autophagic proteins, increased polyubiquitination and abnormal mitochondrial function | [54] |
| Multiple Sulphatase Deficiency | Sulphate esters (At least 7 lysosomal sulfatases and a microsomal sulfatase) | SUMF1 gene | - | Autophagosomes accumulation, autophagy dysfunction in disease pathogenesis, induction of autophagy. Accumulation, Fragmentation, Decreased ATP content | [56,57,68,70] |
| Mucolipidoses II | N acetylglucosaminylphosphotransferase resulting in multiple enzyme deficiencies | GNPTAB gene | Mucopolysaccharides, lipids, glycoproteins | Autophagic and mitochondrial impairment. | [71,72,73] |
| Mucolipidosis III | Dysfunctional autophagosome, vacuolization (accumulation of autolysosomes) | [74] | |||
| Gangliosidosis: GM1 | beta-galactosidase | GLB1 gene | Gangiosides | Autophagosomes accumulation, autophagy dysfunction is disease pathogenesis, induction of autophagy. | [57,75,76] |
| GM2 | beta-hexosaminidase | HEXB gene | |||
| Fabry disease | alpha-galactosidase A | GLA gene | globotriasylceramide (Gb3) | Increased vacuoles level, reduction of mitochondrial activity, Lipid storage in endothelial and smooth muscle cells of blood vessels | [59,77] |
| Farber disease | Ceramidase | ASAH1 gene | Ceramides, mucopolysaccharides, gangliosides | Ceremide/cholesterol structures accumulate in late endosomes and lysosomes, in mitochondria and the plasma membrane | [78] |
| Gaucher disease | glucocerebrosidase (GBA1) | GBA1 gene | Glycosphingolipids, Glucosylceramide, GM1, GM2, GM3, GD3, Glucosylsphingosine | Impaired autophagy and proteasomal degradation pathways and mitochondrial dysfunction; pathological [Ca2+]c responses and delayed calcium deregulation, collapse of mitochondrial membrane potential and an irreversible fall in the ATP/ADP ratio; lipid storing macrophages | [7,8,79,80,81,82] |
| Krabbe disease | Galactosylceramidase | GALC gene | galactosyl-ceramide and galactosyl-sphingosine (psychosine) | Mitochondrial morphology ad accumulation, | [68,83,84] |
| calcium signaling, oxidative stress and macromolecules accumulation in mitochondria; | |||||
| Elevated ROS/reduced GSH, | |||||
| Ca2+ overload, | |||||
| Cytochrome c release | |||||
| LSDs Associated with Other Defects | |||||
| Neuronal Ceroid Lipofuscinosis (Batten’s disease) | Lysosome protease deficiencies | CLN1–CLN12 gene | Lipofuscin | Active regulation of ATP synthase by Ca2+ is maintained in fibroblasts from CLN 2 and CLN3 but absent in those from CLN1. Autophagosome accumulation, autophagy dysfunction in disease pathogenesis, induction of authophagy. | [57,68,85,86,87] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stepien, K.M.; Roncaroli, F.; Turton, N.; Hendriksz, C.J.; Roberts, M.; Heaton, R.A.; Hargreaves, I. Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. J. Clin. Med. 2020, 9, 2596. https://doi.org/10.3390/jcm9082596
Stepien KM, Roncaroli F, Turton N, Hendriksz CJ, Roberts M, Heaton RA, Hargreaves I. Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. Journal of Clinical Medicine. 2020; 9(8):2596. https://doi.org/10.3390/jcm9082596
Chicago/Turabian StyleStepien, Karolina M., Federico Roncaroli, Nadia Turton, Christian J. Hendriksz, Mark Roberts, Robert A. Heaton, and Iain Hargreaves. 2020. "Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review" Journal of Clinical Medicine 9, no. 8: 2596. https://doi.org/10.3390/jcm9082596
APA StyleStepien, K. M., Roncaroli, F., Turton, N., Hendriksz, C. J., Roberts, M., Heaton, R. A., & Hargreaves, I. (2020). Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. Journal of Clinical Medicine, 9(8), 2596. https://doi.org/10.3390/jcm9082596