Strategy for Use of Genome-Wide Non-Invasive Prenatal Testing for Rare Autosomal Aneuploidies and Unbalanced Structural Chromosomal Anomalies

 ,
,
Abstract
1. Introduction
2. Experimental Section
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, S.; Chang, C.; Cheng, P.; Hsiao, C.; Soong, Y.; Duan, T. First-trimester combined screening is effective for the detection of unbalanced chromosomal translocations at 11 to 12 weeks of gestation. Reprod. Sci. 2014, 21, 594–600. [Google Scholar] [CrossRef][Green Version]
- Torring, N.; Petersen, O.B.; Becher, N.; Vogel, I.; Uldbjerg, N. First trimester screening for other trisomies than trisomy 21, 18, and 13. Prenat. Diagn. 2015, 35, 612–619. [Google Scholar] [CrossRef]
- Lindquist, A.; Poulton, A.; Halliday, J.; Hui, L. Prenatal diagnostic testing and atypical chromosome abnormalities following combined first-trimester screening: Implications for contingent models of non-invasive prenatal testing. Ultrasound Obstet. Gynecol. 2018, 51, 487–492. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Chiu, R.W.K. Sequencing of Circulating Cell-free DNA during Pregnancy. N. Engl. J. Med. 2018, 379, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.; Bono, S.; Pizzuti, F.; Duca, S.; Polverari, A.; Faieta, M.; Baldi, M.; Diano, L.; Spinella, F. The clinical utility of genome-wide non invasive prenatal screening. Prenat. Diagn. 2017, 37, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Lin, Y.; Qiao, F.; Li, H.; Wang, Y.; Zhang, J.; Liu, A.; Ji, X.; Ma, D.; Jiang, T.; et al. Perinatal outcomes following cell-free DNA screening in >32 000 women: Clinical follow-up data from a single tertiary center. Prenat. Diagn. 2018, 38, 755–764. [Google Scholar] [CrossRef]
- Akolekar, R.; Beta, J.; Picciarelli, G.; Ogilvie, C.; D’Antonio, F. Procedure-related risk of miscarriage following amniocentesis and chorionic villus sampling: A systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2015, 45, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Salomon, L.J.; Sotiriadis, A.; Wulff, C.B.; Odibo, A.; Akolekar, R. Risk of miscarriage following amniocentesis or chorionic villus sampling: Systematic review of literature and updated meta-analysis. Ultrasound Obstet. Gynecol. 2019, 54, 442–451. [Google Scholar] [CrossRef]
- Michael, T.; Natalia, S.-L.; James, M.; Jörg, B.; Yuri, N.; Andreas, S.W.; Gregor, S. Mid-trimester preterm premature rupture of membranes (PPROM): Etiology, diagnosis, classification, international recommendations of treatment options and outcome. J. Perinat. Med. 2018, 46, 465–488. [Google Scholar] [CrossRef]
- Cederholm, M.; Haglund, B.; Axelsson, O. Maternal complications following amniocentesis and chorionic villus sampling for prenatal karyotyping. BJOG 2003, 110, 392–399. [Google Scholar] [CrossRef]
- Norton, M.E.; Baer, R.J.; Wapner, R.J.; Kuppermann, M.; Jelliffe-Pawlowski, L.L.; Currier, R.J. Cell-free DNA vs sequential screening for the detection of fetal chromosomal abnormalities. Am. J. Obstet. Gynecol. 2016, 214, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Sehnert, A.J.; Rhees, B.; Comstock, D.; de Feo, E.; Heilek, G.; Burke, J.; Rava, R.P. Optimal detection of fetal chromosomal abnormalities by massively parallel DNA sequencing of cell-free fetal DNA from maternal blood. Clin. Chem. 2011, 57, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Ehrich, M.; Deciu, C.; Zwiefelhofer, T.; Tynan, J.A.; Cagasan, L.; Tim, R.; Lu, V.; McCullough, R.; McCarthy, E.; Nygren, A.O.; et al. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: A study in a clinical setting. Am. J. Obstet. Gynecol. 2011, 204, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.J.; Zwiefelhofer, T.; Tim, R.C.; Dzakula, Z.; Kim, S.K.; Mazloom, A.R.; Zhu, Z.; Tynan, J.; Lu, T.; McLennan, G.; et al. High-throughput massively parallel sequencing for fetal aneuploidy detection from maternal plasma. PLoS ONE 2013, 8, e57381. [Google Scholar] [CrossRef]
- Juneau, K.; Bogard, P.E.; Huang, S.; Mohseni, M.; Wang, E.T.; Ryvkin, P.; Kingsley, C.; Struble, C.A.; Oliphant, A.; Zahn, J.M. Microarray-based cell-free DNA analysis improves noninvasive prenatal testing. Fetal Diagn. Ther. 2014, 36, 282–286. [Google Scholar] [CrossRef]
- Zimmermann, B.; Hill, M.; Gemelos, G.; Demko, Z.; Banjevic, M.; Baner, J.; Ryan, A.; Sigurjonsson, S.; Chopra, N.; Dodd, M.; et al. Noninvasive prenatal aneuploidy testing of chromosomes 13, 18, 21, X, and Y, using targeted sequencing of polymorphic loci. Prenat. Diagn. 2012, 32, 1233–1241. [Google Scholar] [CrossRef]
- Pergament, E.; Cuckle, H.; Zimmermann, B.; Banjevic, M.; Sigurjonsson, S.; Ryan, A.; Hall, M.P.; Dodd, M.; Lacroute, P.; Stosic, M.; et al. Single-Nucleotide Polymorphism-Based Noninvasive Prenatal Screening in a High-Risk and Low-Risk Cohort. Obstet. Gynecol. 2014, 124, 210–218. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Analysis of the size distributions of fetal and maternal cell-free DNA by paired-end sequencing. Clin. Chem. 2010, 56, 1279–1286. [Google Scholar] [CrossRef]
- Lo, Y.M.; Chan, K.C.; Sun, H.; Chen, E.Z.; Jiang, P.; Lun, F.M.; Zheng, Y.W.; Leung, T.Y.; Lau, T.K.; Cantor, C.R.; et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2010, 2, 61ra91. [Google Scholar] [CrossRef]
- Palomaki, G.E.; Kloza, E.M. Prenatal cell-free DNA screening test failures: A systematic review of failure rates, risks of Down syndrome, and impact of repeat testing. Genet. Med. 2018, 20, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Ren, J.; Chen, F.; Zhou, Y.; Xie, J.; Dan, S.; Su, Y.; Xie, J.; Yin, B.; Su, W.; et al. Noninvasive Fetal Trisomy (NIFTY) test: An advanced noninvasive prenatal diagnosis methodology for fetal autosomal and sex chromosomal aneuploidies. BMC Med. Genom. 2012, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Yin, A.H.; Peng, C.F.; Fu, F.; Yang, J.X.; Li, R.; Chen, Y.Y.; Luo, D.H.; Zhang, Y.L.; Ou, Y.M.; et al. Noninvasive prenatal diagnosis of common aneuploidies by semiconductor sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 7415–7420. [Google Scholar] [CrossRef] [PubMed]
- Stumm, M.; Entezami, M.; Haug, K.; Blank, C.; Wustemann, M.; Schulze, B.; Raabe-Meyer, G.; Hempel, M.; Schelling, M.; Ostermayer, E.; et al. Diagnostic accuracy of random massively parallel sequencing for non-invasive prenatal detection of common autosomal aneuploidies: A collaborative study in Europe. Prenat. Diagn. 2014, 34, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Samango-Sprouse, C.; Banjevic, M.; Ryan, A.; Sigurjonsson, S.; Zimmermann, B.; Hill, M.; Hall, M.P.; Westemeyer, M.; Saucier, J.; Demko, Z.; et al. SNP-based non-invasive prenatal testing detects sex chromosome aneuploidies with high accuracy. Prenat. Diagn. 2013, 33, 643–649. [Google Scholar] [CrossRef]
- Mazloom, A.R.; Dzakula, Z.; Oeth, P.; Wang, H.; Jensen, T.; Tynan, J.; McCullough, R.; Saldivar, J.S.; Ehrich, M.; van den Boom, D.; et al. Noninvasive prenatal detection of sex chromosomal aneuploidies by sequencing circulating cell-free DNA from maternal plasma. Prenat. Diagn. 2013, 33, 591–597. [Google Scholar] [CrossRef]
- Helgeson, J.; Wardrop, J.; Boomer, T.; Almasri, E.; Paxton, W.B.; Saldivar, J.S.; Dharajiya, N.; Monroe, T.J.; Farkas, D.H.; Grosu, D.S.; et al. Clinical outcome of subchromosomal events detected by whole-genome noninvasive prenatal testing. Prenat. Diagn. 2015, 35, 999–1004. [Google Scholar] [CrossRef]
- Martin, K.; Iyengar, S.; Kalyan, A.; Lan, C.; Simon, A.L.; Stosic, M.; Kobara, K.; Ravi, H.; Truong, T.; Ryan, A.; et al. Clinical experience with a single-nucleotide polymorphism-based non-invasive prenatal test for five clinically significant microdeletions. Clin. Genet. 2018, 93, 293–300. [Google Scholar] [CrossRef]
- van der Meij, K.R.M.; Sistermans, E.A.; Macville, M.V.E.; Stevens, S.J.C.; Bax, C.J.; Bekker, M.N.; Bilardo, C.M.; Boon, E.M.J.; Boter, M.; Diderich, K.E.M.; et al. TRIDENT-2: National Implementation of Genome-wide Non-invasive Prenatal Testing as a First-Tier Screening Test in the Netherlands. Am. J. Hum. Genet. 2019, 105, 1091–1101. [Google Scholar] [CrossRef]
- Pertile, M.D.; Halks-Miller, M.; Flowers, N.; Barbacioru, C.; Kinnings, S.L.; Vavrek, D.; Seltzer, W.K.; Bianchi, D.W. Rare autosomal trisomies, revealed by maternal plasma DNA sequencing, suggest increased risk of feto-placental disease. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Pescia, G.; Guex, N.; Iseli, C.; Brennan, L.; Osteras, M.; Xenarios, I.; Farinelli, L.; Conrad, B. Cell-free DNA testing of an extended range of chromosomal anomalies: Clinical experience with 6388 consecutive cases. Genet. Med. 2017, 19, 169–175. [Google Scholar] [CrossRef]
- Agence de la BioMédecine. Rapport médical et scientifique de l’assistance médicale à la procréation et de la génétique humaines en France. In Prenatal Diagnosis and Preimplantation Genetic Diagnosis; 2017. Available online: https://rams.agence-biomedecine.fr/diagnostic-sur-lembryon-et-le-foetus (accessed on 23 September 2019).
- Bayindir, B.; Dehaspe, L.; Brison, N.; Brady, P.; Ardui, S.; Kammoun, M.; Van der Veken, L.; Lichtenbelt, K.; Van den Bogaert, K.; Van Houdt, J.; et al. Noninvasive prenatal testing using a novel analysis pipeline to screen for all autosomal fetal aneuploidies improves pregnancy management. Eur. J. Hum. Genet. 2015, 23, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Van Opstal, D.; van Maarle, M.C.; Lichtenbelt, K.; Weiss, M.M.; Schuring-Blom, H.; Bhola, S.L.; Hoffer, M.J.V.; Huijsdens-van Amsterdam, K.; Macville, M.V.; Kooper, A.J.A.; et al. Origin and clinical relevance of chromosomal aberrations other than the common trisomies detected by genome-wide NIPS: Results of the TRIDENT study. Genet. Med. 2018, 20, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.; Bonifacio, M.; Sandow, R.; Ellis, K.; Smet, M.E.; McLennan, A. Rare autosomal trisomies: Important and not so rare. Prenat. Diagn. 2018, 38, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Chatron, N.; Till, M.; Abel, C.; Bardel, C.; Ramond, F.; Sanlaville, D.; Schluth-Bolard, C. Detection of rare autosomal trisomies through non-invasive prenatal testing: Benefits for pregnancy management. Ultrasound Obstet. Gynecol. 2019, 53, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 2012, 18, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Grati, F.R. Chromosomal Mosaicism in Human Feto-Placental Development: Implications for Prenatal Diagnosis. J. Clin. Med. 2014, 3, 809–837. [Google Scholar] [CrossRef]
- Gil, M.M.; Accurti, V.; Santacruz, B.; Plana, M.N.; Nicolaides, K.H. Analysis of cell-free DNA in maternal blood in screening for aneuploidies: Updated meta-analysis. Ultrasound Obstet. Gynecol. 2017, 50, 302–314. [Google Scholar] [CrossRef]
- de Wergifosse, S.; Bevilacqua, E.; Mezela, I.; El Haddad, S.; Gounongbe, C.; de Marchin, J.; Maggi, V.; Conotte, S.; Badr, D.A.; Fils, J.F.; et al. Cell-free DNA analysis in maternal blood: Comparing genome-wide versus targeted approach as a first-line screening test. J. Matern. Fetal Neonatal Med. 2019. [Google Scholar] [CrossRef]
- Grati, F.R.; Ferreira, J.; Benn, P.; Izzi, C.; Verdi, F.; Vercellotti, E.; Dalpiaz, C.; D’Ajello, P.; Filippi, E.; Volpe, N.; et al. Outcomes in pregnancies with a confined placental mosaicism and implications for prenatal screening using cell-free DNA. Genet. Med. 2020, 22, 309–316. [Google Scholar] [CrossRef]
- Gadsbøll, K.; Petersen, O.B.; Gatinois, V.; Strange, H.; Jacobsson, B.; Wapner, R.; Vermeesch, J.R.; Group, T.N.-m.S.; Vogel, I. Current use of noninvasive prenatal testing in Europe, Australia and the USA: A graphical presentation. Acta Obstet. Gynecol. Scand. 2020, 99, 722–730. [Google Scholar] [CrossRef]
- Benachi, A.; Caffrey, J.; Calda, P.; Carreras, E.; Jani, J.C.; Kilby, M.D.; Klein, H.G.; Rizzo, G.; Yaron, Y. Understanding attitudes and behaviors towards cell-free DNA-based noninvasive prenatal testing (NIPT): A survey of European health-care providers. Eur. J. Med. Genet. 2020, 63, 103616. [Google Scholar] [CrossRef] [PubMed]
- Ehrich, M.; Tynan, J.; Mazloom, A.; Almasri, E.; McCullough, R.; Boomer, T.; Grosu, D.; Chibuk, J. Genome-wide cfDNA screening: Clinical laboratory experience with the first 10,000 cases. Genet. Med. 2017, 19, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Cram, D.S.; Tan, H.; Linpeng, S.; Liu, Y.; Sun, H.; Zhang, Y.; Tian, F.; Zhu, H.; Xu, M.; et al. Clinical utility of noninvasive prenatal screening for expanded chromosome disease syndromes. Genet. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhao, G.; Li, H.; Zhang, Q.; Lu, J.; Yu, B.; Wang, T. Non-invasive prenatal testing to detect chromosome aneuploidies in 57,204 pregnancies. Mol. Cytogenet. 2019, 12, 29. [Google Scholar] [CrossRef] [PubMed]



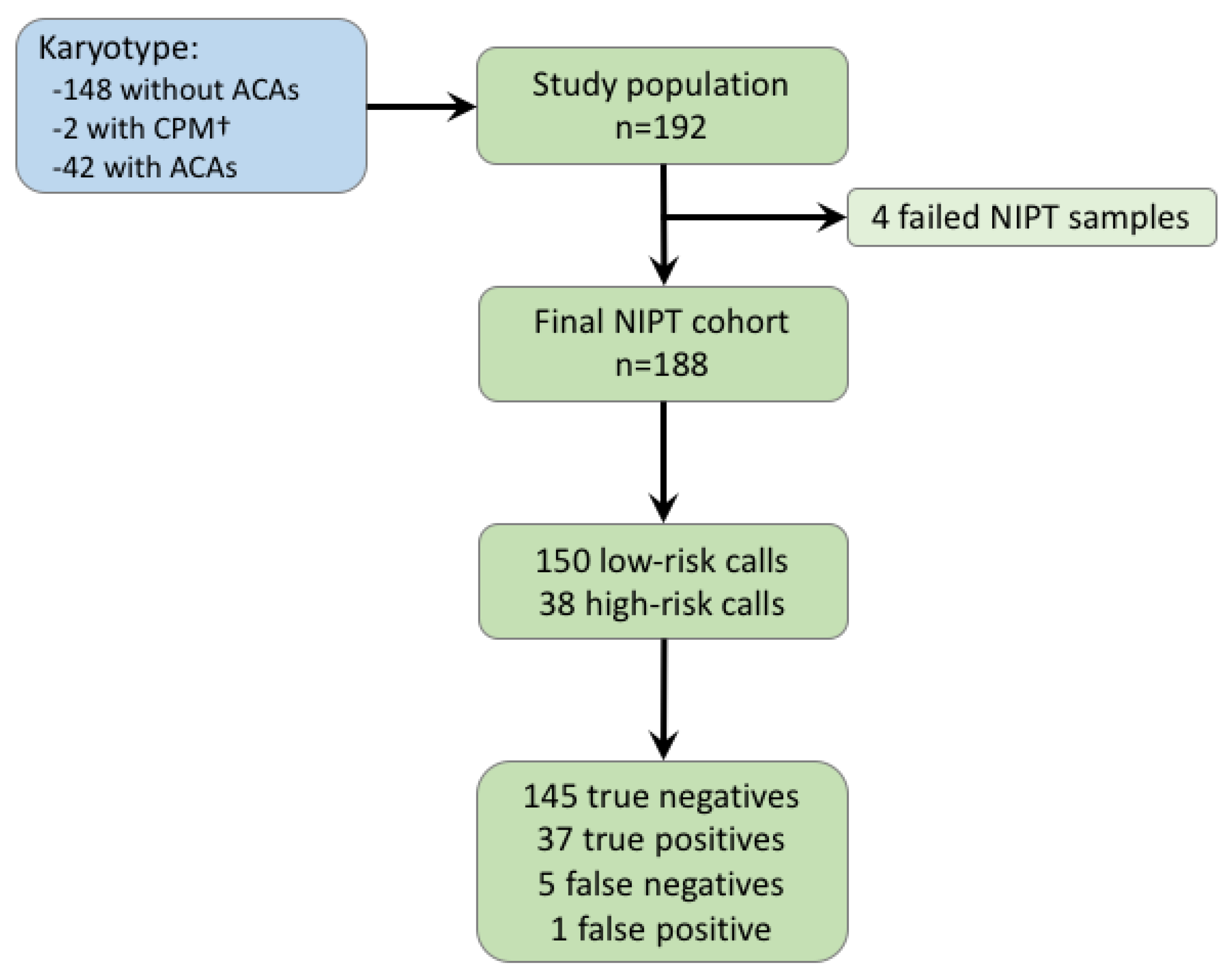{kind=link}
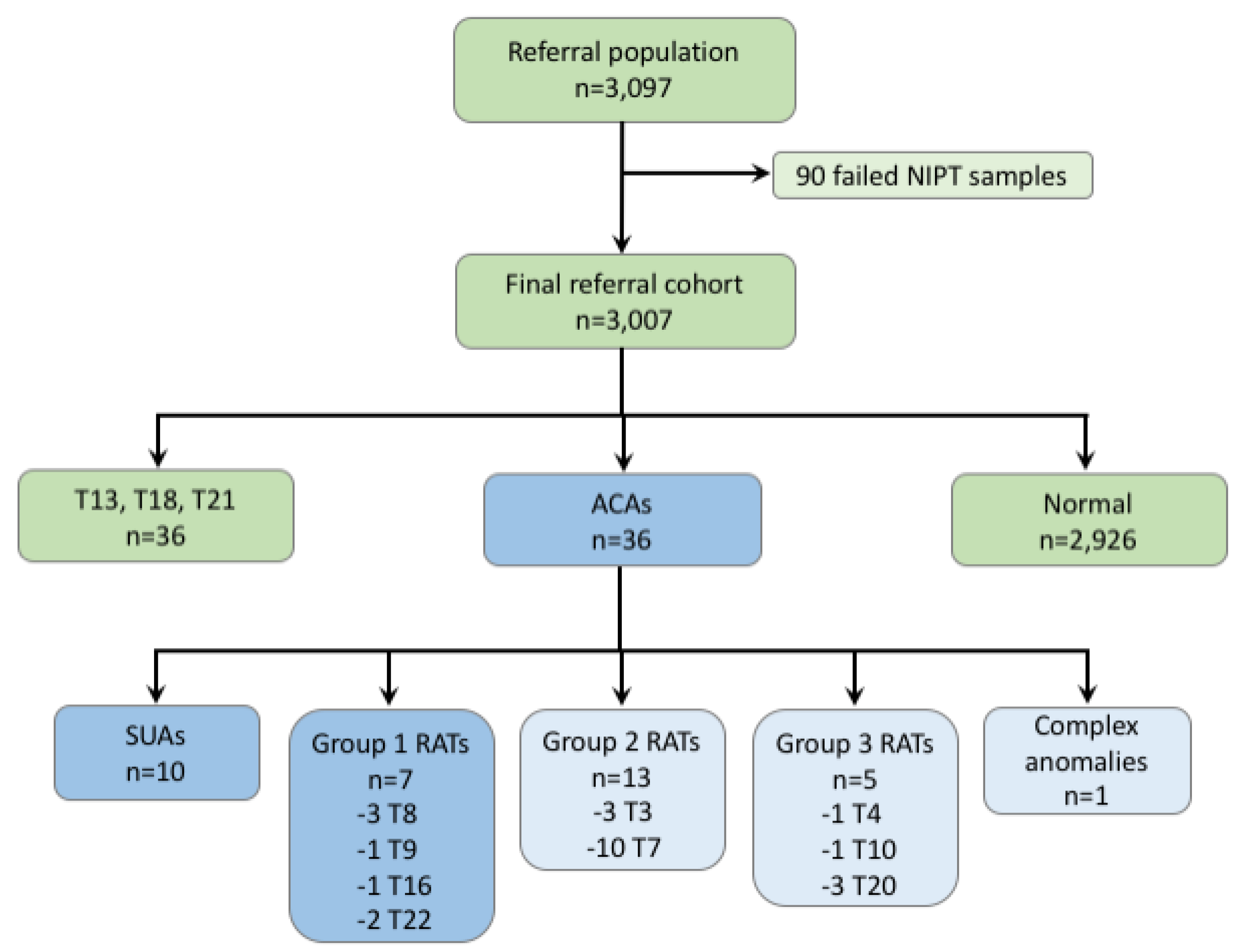{kind=link}
| Sample | Fetal Fraction | Karyotype | Size (Mb) | Comments |
|---|---|---|---|---|
| 1 | 10% | 46,XX,del(4)(p16.3).ish del(4)(WHS-,D4S3359-) | 5–8, based on the karyotype | Possible size < 7 Mb |
| 2 | 15% | arr[GRCh37] 12p13.33q11(173786_37876500)x3 | 37.7 | Suspected mosaicism |
| 3 | 9% | 46,XX,der(8)?add(8)(p?)?dup(8)(q22q23)dn.ish der(8)(qter->?::?->qter)(D8S504, VIJyRM2053+,wcp8+,VIJyRM2053+).arr[GRCh38] 8p23.3p23.1(158048_6935930)x1,8p23.1p11.23(12585435_38267493)x3, 8p11.22(38314367_39246760)x3, 8p11.22(39247087_39386852)x1,8p11.22(39389765_40264413)x3, 8q22.3q23.2(104688373_111952230)x3, 8q24.3(144972747_146295771)x3 | Total deletion = 6.9 Total duplication = 36.2 | Chromoanasynthesis |
| 4 | 3% | 46,XX,add(4)(qter).ish add(4)(wcp4-).arr[GRCh37] 5q31.2q35.3(138522878_180715096)x3 | 41.2 | |
| 5 | 4% | 46,XY,i(18)(q10) |
| Sample | NIPT | Array |
|---|---|---|
| 1 | 18.2 | 12.8 |
| 2 | 9.7 | 9.8 |
| 3 | 11.5 | 11.1 |
| 4 | 28.8 | 29.9 |
| 5 | Trisomy 11 | 2.7 |
| 6 | 8.1 | 6.7 |
| 7 | 10.7 | 11.5 |
| 8 | 11.3 | 17.4 |
| 9 | 26 | 26.6 |
| 10 | 17.2 | 7.9 |
| 11 | 18.3 | 18.3 |
| 12 | 9 | 8.8 |
| 13 | 12.5 | 12.5 |
| 14 | 13.7 | 11.8 |
| 15 | 60.1 | 59.9 |
| Type of Rearrangement | Observed on Karyotype, n | Detected by NIPT, n | Detection Rate, % (95% CI) |
|---|---|---|---|
| Deletion | 13 | 11 | 84.6 (54.6–98.1) |
| Duplication | 28 | 24 | 85.7 (67.3–96.0) |
| Interstitial | 5 | 5 | 100 (47.8–100) |
| Terminal | 36 | 28 | 77.8 (60.9–89.9) |
| Measurement | General Population | MSS Score 1/51–1/1000 | MSS Score >1/1000 | MSS Score 1/51–1/300 | MSS Score >1/300 |
|---|---|---|---|---|---|
| Prevalence 1 | 0.10% | 0.37% | 0.61% | 1.01% | 1.40% |
| PPV | 11% | 32% | 44% | 56% | 64% |
| Population Type | Failed | No Anomalies | Common Trisomies | ACAs | Total, n (%) |
|---|---|---|---|---|---|
| MSS ≥ 1/1000 | 71 | 2596 | 35 | 29 | 2731 (88) |
| MSS < 1/1000 | 9 | 139 | 0 | 1 | 149 (5) |
| Parental Robertsonian translocation | 0 | 2 | 0 | 0 | 2 (0) |
| Previous history of fetal trisomy | 2 | 51 | 0 | 1 | 54 (2) |
| First-tier screening | 8 | 147 | 1 | 5 | 161 (5) |
| Total | 90 | 2935 | 36 | 36 | 3097 |
| Measurement | Biobank Samples (Cohort A) (n = 189) | Normal Biobank Samples from Cohort A (n = 147) | Abnormal Biobank Samples from Cohort A (n = 42) | Referral Samples (Cohort B) (n = 3007) |
|---|---|---|---|---|
| Average | 12.27% | 12.40% | 11.76% | 11% |
| Median | 11% | 11% | 10.5% | 10% |
| Range | 3–35% | 4–35% | 3–24% | 2–35% |
| Population Type | Total, Excluding Failures | Group 1 + SUAs | Group 2 | Group 3 | Prevalence of All ACAs | Prevalence of Group 1 + SUAs |
|---|---|---|---|---|---|---|
| MSS ≥ 1/1000 | 2660 | 14 | 10 | 5 | 1.09% | 0.53% |
| MSS < 1/1000 | 140 | 1 | 0 | 0 | 0.71% | 0.71% |
| First-tier screening | 153 | 1 | 3 | 1 | 3.27% | 0.65% |
| Previous history of fetal trisomy or parental Robertsonian translocation | 54 | 1 | 0 | 0 | 1.85% | 1.85% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleinfinger, P.; Lohmann, L.; Luscan, A.; Trost, D.; Bidat, L.; Debarge, V.; Castaigne, V.; Senat, M.-V.; Brechard, M.-P.; Guilbaud, L.; et al. Strategy for Use of Genome-Wide Non-Invasive Prenatal Testing for Rare Autosomal Aneuploidies and Unbalanced Structural Chromosomal Anomalies. J. Clin. Med. 2020, 9, 2466. https://doi.org/10.3390/jcm9082466
Kleinfinger P, Lohmann L, Luscan A, Trost D, Bidat L, Debarge V, Castaigne V, Senat M-V, Brechard M-P, Guilbaud L, et al. Strategy for Use of Genome-Wide Non-Invasive Prenatal Testing for Rare Autosomal Aneuploidies and Unbalanced Structural Chromosomal Anomalies. Journal of Clinical Medicine. 2020; 9(8):2466. https://doi.org/10.3390/jcm9082466
Chicago/Turabian StyleKleinfinger, Pascale, Laurence Lohmann, Armelle Luscan, Detlef Trost, Laurent Bidat, Véronique Debarge, Vanina Castaigne, Marie-Victoire Senat, Marie-Pierre Brechard, Lucie Guilbaud, and et al. 2020. "Strategy for Use of Genome-Wide Non-Invasive Prenatal Testing for Rare Autosomal Aneuploidies and Unbalanced Structural Chromosomal Anomalies" Journal of Clinical Medicine 9, no. 8: 2466. https://doi.org/10.3390/jcm9082466
APA StyleKleinfinger, P., Lohmann, L., Luscan, A., Trost, D., Bidat, L., Debarge, V., Castaigne, V., Senat, M.-V., Brechard, M.-P., Guilbaud, L., Le Guyader, G., Satre, V., Laurichesse Delmas, H., Lallaoui, H., Manca-Pellissier, M.-C., Boughalem, A., Valduga, M., Hodeib, F., Benachi, A., & Costa, J. M. (2020). Strategy for Use of Genome-Wide Non-Invasive Prenatal Testing for Rare Autosomal Aneuploidies and Unbalanced Structural Chromosomal Anomalies. Journal of Clinical Medicine, 9(8), 2466. https://doi.org/10.3390/jcm9082466