Mitochondrial DNA: Hotspot for Potential Gene Modifiers Regulating Hypertrophic Cardiomyopathy

 , and
, and 
Abstract
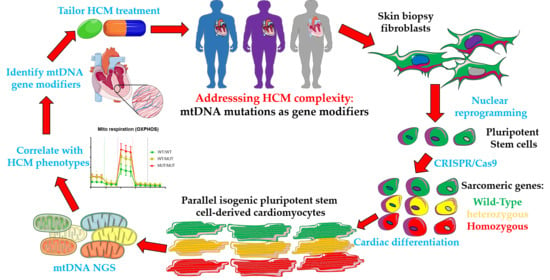
1. Introduction
2. Experimental Section
2.1. Cell Culture
2.2. Ratiometric qPCR
2.3. mtDNA Next Generation Sequencing (NGS) and Analysis
2.4. Seahorse Analysis of Mitochondrial Respiration
2.5. Evaluation of Production of Reactive Oxygen Species (ROS) in Mitochondria
2.6. Statistical Analysis
3. Results
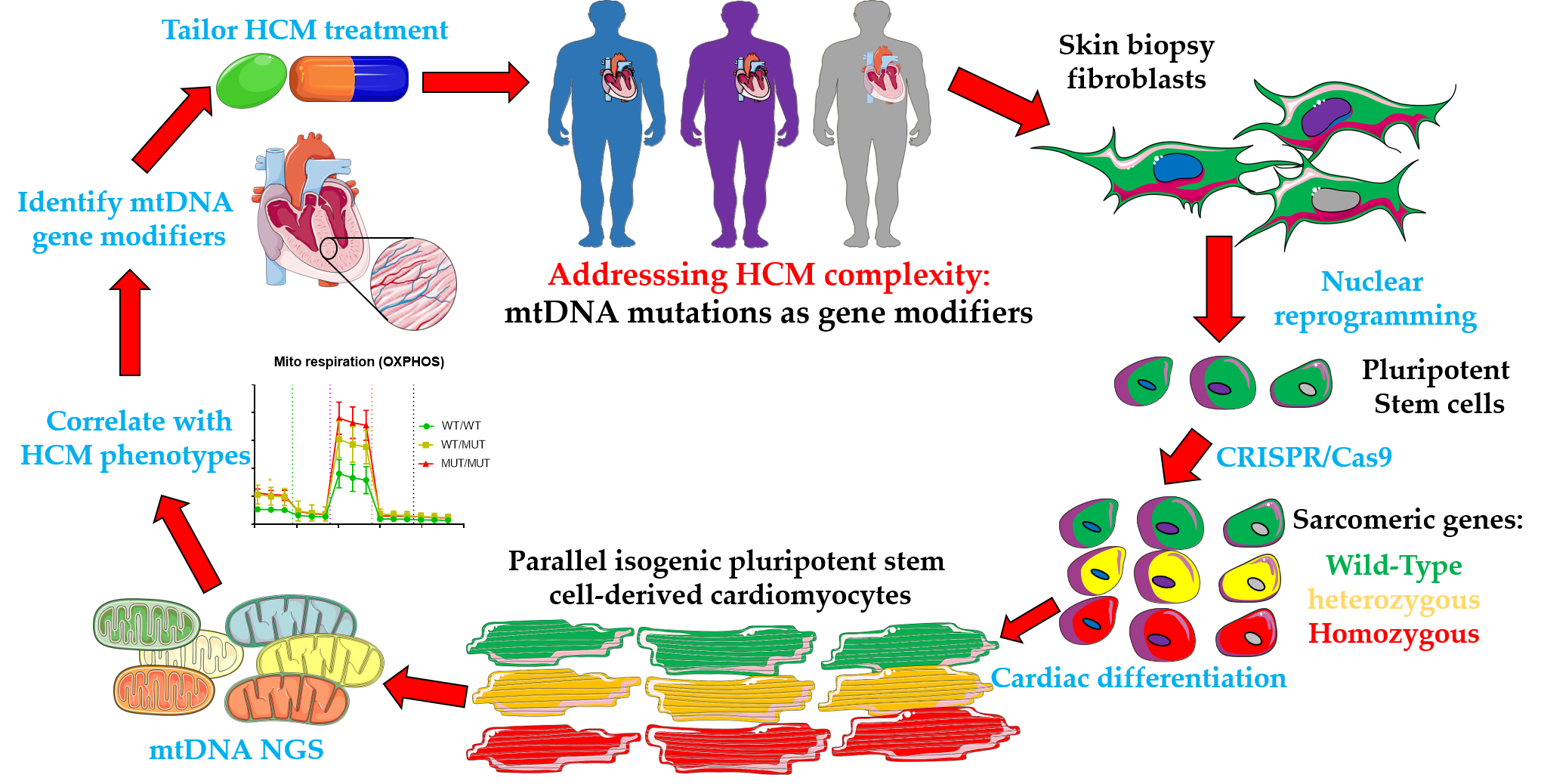
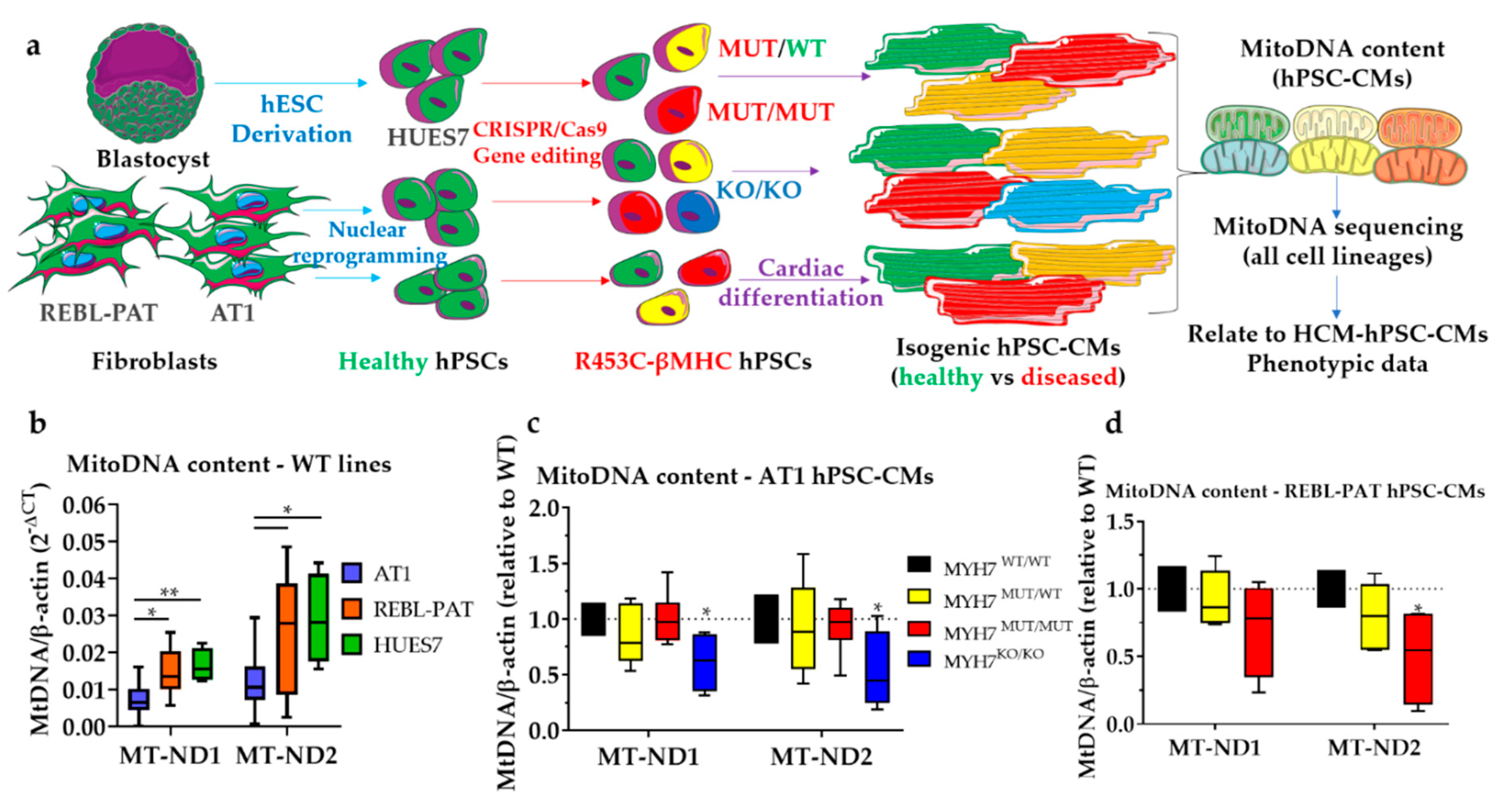
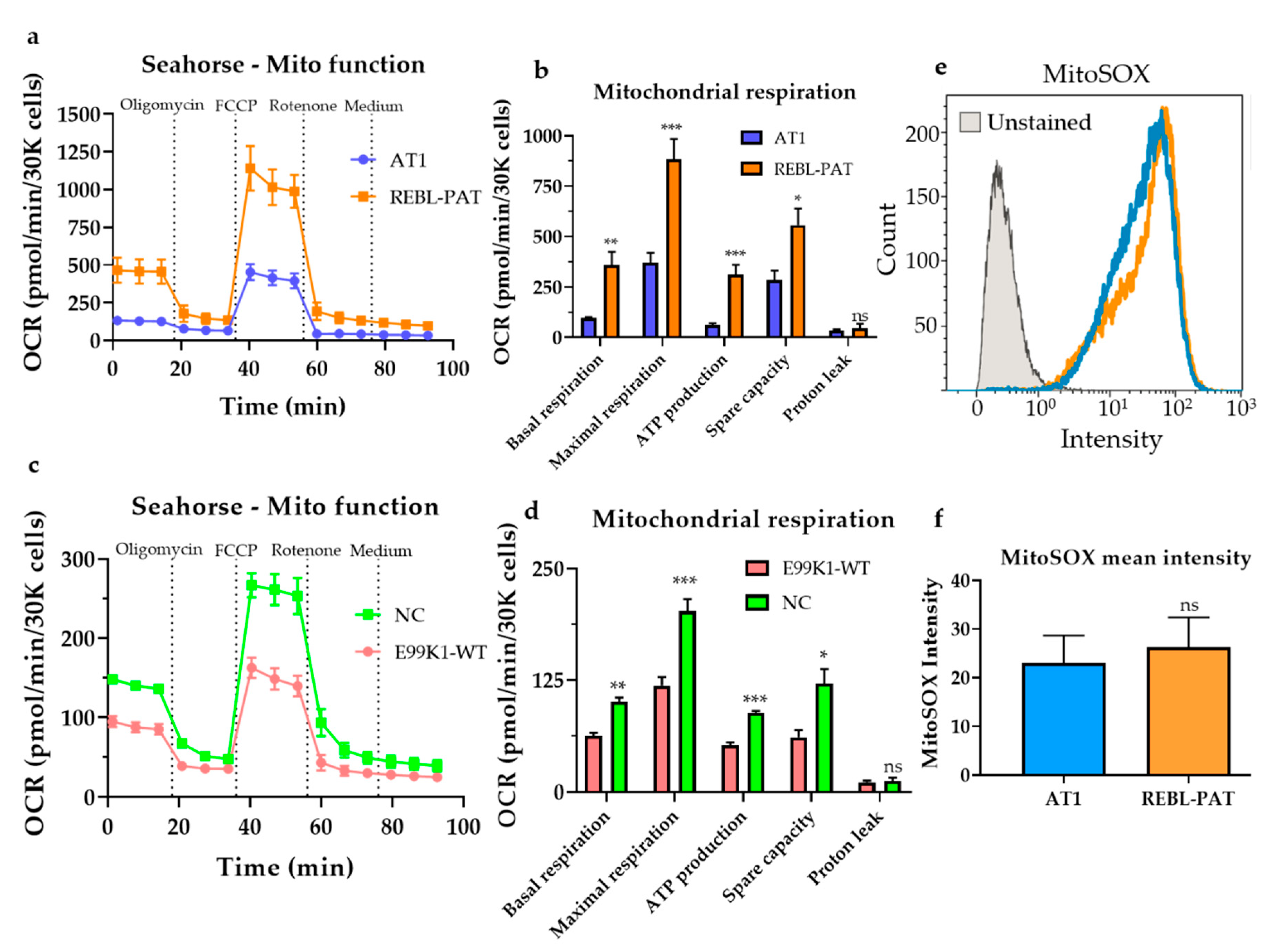
3.1. Revisiting Isogenic hPSC-CM Models to Investigate Varying HCM Mitochondrial Phenotypes
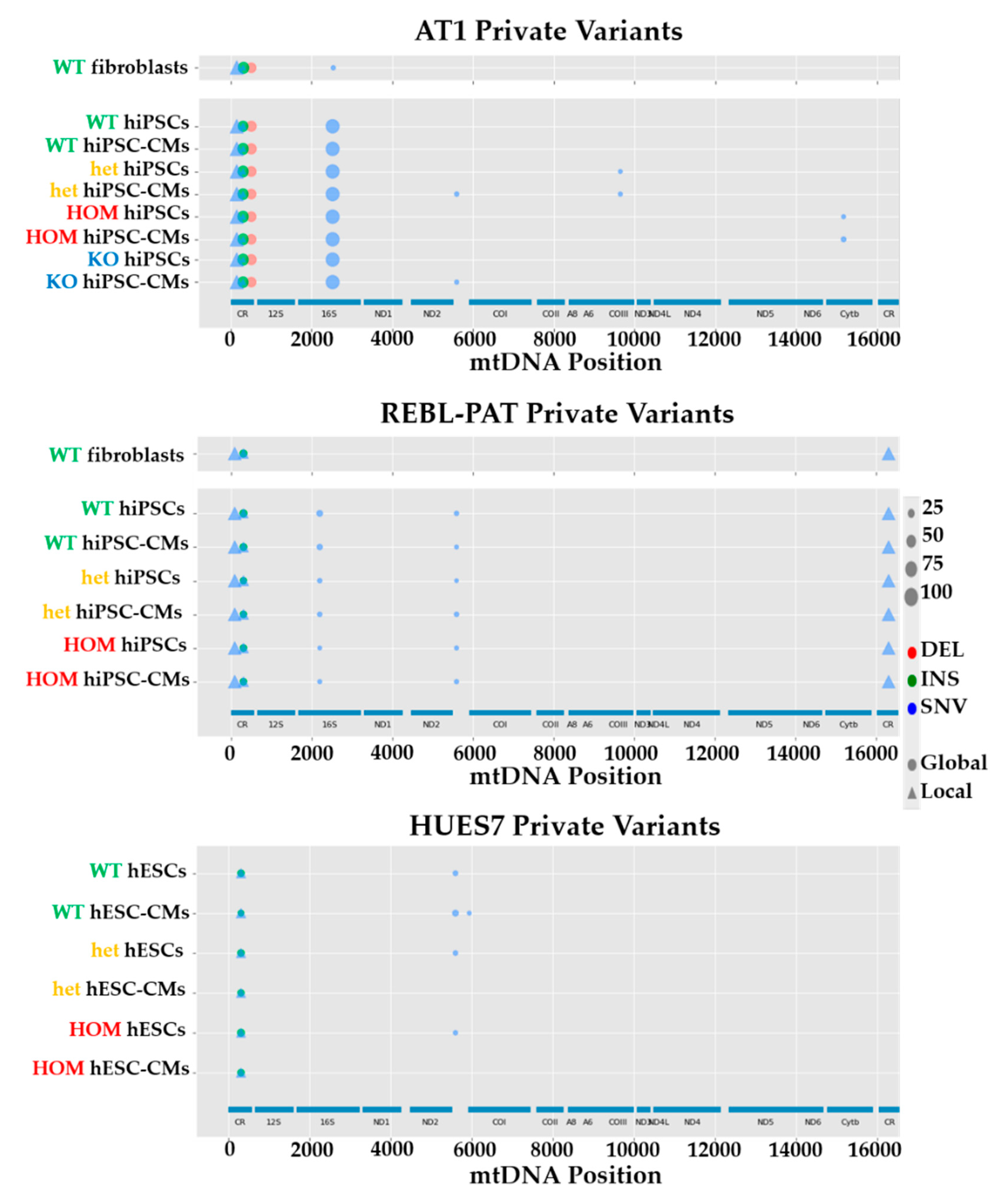
3.2. Comparison of mtDNA Variants between Phenotyped Isogenic Lines Reveals Potential HCM Gene Modifiers
3.3. MtDNA Sequencing Shows Common Variants in Unrelated HCM Patients
3.4. The Identified mtDNA Mutations Are Novel for HCM, But May Be Common in Some Haplogroups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCM | Hypertrophic cardiomyopathy |
| tRNA | Transfer RNA |
| mtDNA | Mitochondrial DNA |
| OXPHOS | Oxidative Phosphorylation |
| MIC | Mitochondrial cardiomyopathies |
| mtRNA | Mitochondrial RNA |
| hPSC | human pluripotent stem cells |
| hiPSC | human induced pluripotent stem cells |
| hESC | human embryonic stem cells |
| hPSC-CM | human pluripotent stem cell-derived cardiomyocyte |
| β-MHC | beta myosin heavy chain |
| NGS | Next generation sequencing |
| HR | Homologous recombination |
| DSB | Double stranded break |
References
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [PubMed]
- Harvey, P.A.; Leinwand, L.A. The cell biology of disease: Cellular mechanisms of cardiomyopathy. J. Cell Biol. 2011, 194, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Elliott, P.M. Genetics of heart failure. Biochim. Biophys. Acta 2013, 1832, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Redwood, C.; Blair, E.; Watkins, H. Hypertrophic cardiomyopathy: A paradigm for myocardial energy depletion. Trends Genet. 2003, 19, 263–268. [Google Scholar] [CrossRef]
- Cahill, T.J.; Ashrafian, H.; Watkins, H. Genetic Cardiomyopathies Causing Heart Failure. Circ. Res. 2013, 113, 660–675. [Google Scholar] [CrossRef]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New Perspectives on the Prevalence of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of Hypertrophic Cardiomyopathy after 20 Years: Clinical Perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Han, Y.; Chen, Y. Different Clinical Presentation and Tissue Characterization in a Monozygotic Twin Pair with MYH7 Mutation-Related Hypertrophic Cardiomyopathy. Int. Heart J. 2019, 60, 477–481. [Google Scholar] [CrossRef]
- Ingles, J.; Burns, C.; Bagnall, R.D.; Lam, L.; Yeates, L.; Sarina, T.; Puranik, R.; Briffa, T.; Atherton, J.J.; Driscoll, T.; et al. Nonfamilial Hypertrophic Cardiomyopathy-Prevalence, Natural History, and Clinical Implications. Circ. Cardiovasc. Genet. 2017, 10, e001620. [Google Scholar] [CrossRef]
- Makavos, G.; Κairis, C.; Tselegkidi, M.-E.; Karamitsos, T.; Rigopoulos, A.G.; Noutsias, M.; Ikonomidis, I. Hypertrophic cardiomyopathy: An updated review on diagnosis, prognosis, and treatment. Heart Fail. Rev. 2019, 24, 439–459. [Google Scholar] [CrossRef]
- Stefania, P.; Romanazzo, S.; Mosqueira, D.; Pinto-do-O., P.; Aoyagi, T.; Forte, G. Adult Stem Cells and Biocompatible Scaffolds as Smart Drug Delivery Tools for Cardiac Tissue Repair. Curr. Med. Chem. 2013, 20, 3429–3447. [Google Scholar]
- DiMauro, S.; Schon, E.A. Mitochondrial Respiratory-Chain Diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 2008, 80, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, A.P.; Dillon, L.M.; Moraes, C.T. Mitochondrial DNA transcription regulation and nucleoid organization. J. Inherit. Metab. Dis. 2011, 34, 941–951. [Google Scholar] [CrossRef]
- Finsterer, J.; Kothari, S. Cardiac manifestations of primary mitochondrial disorders. Int. J. Cardiol. 2014, 177, 754–763. [Google Scholar] [CrossRef]
- Lee, S.R.; Han, J. Mitochondrial Mutations in Cardiac Disorders. Adv. Exp. Med. Biol. 2017, 982, 81–111. [Google Scholar]
- Herrera, A.; Garcia, I.; Gaytan, N.; Jones, E.; Maldonado, A.; Gilkerson, R. Endangered species: Mitochondrial DNA loss as a mechanism of human disease. Front. Biosci. (Sch. Ed.) 2015, 7, 109–124. [Google Scholar]
- Bates, M.G.D.; Bourke, J.P.; Giordano, C.; d’Amati, G.; Turnbull, D.M.; Taylor, R.W. Cardiac involvement in mitochondrial DNA disease: Clinical spectrum, diagnosis, and management. Eur. Heart J. 2012, 33, 3023–3033. [Google Scholar] [CrossRef]
- Mosqueira, D.; Mannhardt, I.; Bhagwan, J.R.; Lis-Slimak, K.; Katili, P.; Scott, E.; Hassan, M.; Prondzynski, M.; Harmer, S.C.; Tinker, A.; et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur. Heart J. 2018, 39, 3879–3892. [Google Scholar] [CrossRef]
- Smith, J.G.W.; Owen, T.; Bhagwan, J.R.; Mosqueira, D.; Scott, E.; Mannhardt, I.; Patel, A.; Barriales-Villa, R.; Monserrat, L.; Hansen, A.; et al. Isogenic Pairs of hiPSC-CMs with Hypertrophic Cardiomyopathy/LVNC-Associated ACTC1 E99K Mutation Unveil Differential Functional Deficits. Stem. Cell Rep. 2018, 11, 1226–1243. [Google Scholar] [CrossRef]
- Bhagwan, J.R.; Mosqueira, D.; Chairez-Cantu, K.; Mannhardt, I.; Bodbin, S.E.; Bakar, M.; Smith, J.G.W.; Denning, C. Isogenic models of hypertrophic cardiomyopathy unveil differential phenotypes and mechanism-driven therapeutics. J. Mol. Cell. Cardiol. 2020, 145, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Bhagwan, J.; Collins, E.; Mosqueira, D.; Bakar, M.; Johnson, B.; Thompson, A.; Smith, J.; Denning, C. Variable expression and silencing of CRISPR-Cas9 targeted transgenes identifies the AAVS1 locus as not an entirely safe harbour. F1000 Research 2019, 8, 1991. [Google Scholar] [CrossRef]
- Breckwoldt, K.; Letuffe-Breniere, D.; Mannhardt, I.; Schulze, T.; Ulmer, B.; Werner, T.; Benzin, A.; Klampe, B.; Reinsch, M.C.; Laufer, S.; et al. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat. Protoc. 2017, 12, 1177–1197. [Google Scholar] [CrossRef]
- Mosqueira, D.; Lis-Slimak, K.; Denning, C. High-Throughput Phenotyping Toolkit for Characterizing Cellular Models of Hypertrophic Cardiomyopathy In Vitro. Methods Protoc. 2019, 2, 83. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Perales-Clemente, E.; Cook, A.N.; Evans, J.M.; Roellinger, S.; Secreto, F.; Emmanuele, V.; Oglesbee, D.; Mootha, V.K.; Hirano, M.; Schon, E.A.; et al. Natural underlying mtDNA heteroplasmy as a potential source of intra-person hiPSC variability. EMBO J. 2016, 35, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Van Oven, M.; Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mut. 2009, 30, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Kloss-Brandstätter, A.; Pacher, D.; Schönherr, S.; Weissensteiner, H.; Binna, R.; Specht, G.; Kronenberg, F. HaploGrep: A fast and reliable algorithm for automatic classification of mitochondrial DNA haplogroups. Hum. Mut. 2011, 32, 25–32. [Google Scholar] [CrossRef]
- Castellana, S.; Fusilli, C.; Mazzoccoli, G.; Biagini, T.; Capocefalo, D.; Carella, M.; Vescovi, A.L.; Mazza, T. High-confidence assessment of functional impact of human mitochondrial non-synonymous genome variations by APOGEE. PLoS Comput. Biol. 2017, 13, e1005628. [Google Scholar] [CrossRef]
- Sonney, S.; Leipzig, J.; Lott, M.T.; Zhang, S.; Procaccio, V.; Wallace, D.C.; Sondheimer, N. Predicting the pathogenicity of novel variants in mitochondrial tRNA with MitoTIP. PLoS Comput. Biol. 2017, 13, e1005867. [Google Scholar] [CrossRef]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS. React. Oxyg. Species (Apex. N.C.) 2016, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Kondrashov, A.; Duc Hoang, M.; Smith, J.G.W.; Bhagwan, J.R.; Duncan, G.; Mosqueira, D.; Munoz, M.B.; Vo, N.T.N.; Denning, C. Simplified Footprint-Free Cas9/CRISPR Editing of Cardiac-Associated Genes in Human Pluripotent Stem Cells. Stem Cells Dev. 2018, 27, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Bris, C.; Goudenege, D.; Desquiret-Dumas, V.; Charif, M.; Colin, E.; Bonneau, D.; Amati-Bonneau, P.; Lenaers, G.; Reynier, P.; Procaccio, V. Bioinformatics Tools and Databases to Assess the Pathogenicity of Mitochondrial DNA Variants in the Field of Next Generation Sequencing. Front. Genet. 2018, 9, 632. [Google Scholar] [CrossRef] [PubMed]
- Muftuoglu, M.; Mori, M.P.; Souza-Pinto, N.C.d. Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion 2014, 17, 164–181. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Procaccio, V.; Lebre, A.S.; Jardel, C.; Chaussenot, A.; Hoarau, C.; Maoulida, H.; Charrier, N.; Gai, X.; Xie, H.M.; et al. Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J. Med. Genet. 2013, 50, 704. [Google Scholar] [CrossRef]
- Govindaraj, P.; Khan, N.A.; Rani, B.; Rani, D.S.; Selvaraj, P.; Jyothi, V.; Bahl, A.; Narasimhan, C.; Rakshak, D.; Premkumar, K.; et al. Mitochondrial DNA variations associated with hypertrophic cardiomyopathy. Mitochondrion 2014, 16, 65–72. [Google Scholar] [CrossRef]
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. mtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr. Protoc. Bioinform. 2013, 44, 1–23. [Google Scholar] [CrossRef]
- Hagen, C.M.; Aidt, F.H.; Hedley, P.L.; Jensen, M.K.; Havndrup, O.; Kanters, J.K.; Moolman-Smook, J.C.; Larsen, S.O.; Bundgaard, H.; Christiansen, M. Mitochondrial Haplogroups Modify the Risk of Developing Hypertrophic Cardiomyopathy in a Danish Population. PLoS ONE 2013, 8, e71904. [Google Scholar] [CrossRef]
- Mosqueira, D.; Smith, J.G.W.; Bhagwan, J.R.; Denning, C. Modeling Hypertrophic Cardiomyopathy: Mechanistic Insights and Pharmacological Intervention. Trends Mol. Med. 2019, 25, 775–790. [Google Scholar] [CrossRef]
- Denning, C.; Borgdorff, V.; Crutchley, J.; Firth, K.S.A.; George, V.; Kalra, S.; Kondrashov, A.; Hoang, M.D.; Mosqueira, D.; Patel, A.; et al. Cardiomyocytes from human pluripotent stem cells: From laboratory curiosity to industrial biomedical platform. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 1728–1748. [Google Scholar] [CrossRef]
- Sala, L.; Bellin, M.; Mummery, C.L. Integrating cardiomyocytes from human pluripotent stem cells in safety pharmacology: Has the time come? Br. J. Pharm. 2017, 174, 3749–3765. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet 2000, 355, 58–60. [Google Scholar] [CrossRef]
- Lopes, L.R.; Rahman, M.S.; Elliott, P.M. A systematic review and meta-analysis of genotype–phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 2013, 99, 1800–1811. [Google Scholar] [CrossRef] [PubMed]
- Naue, J.; Hörer, S.; Sänger, T.; Strobl, C.; Hatzer-Grubwieser, P.; Parson, W.; Lutz-Bonengel, S. Evidence for frequent and tissue-specific sequence heteroplasmy in human mitochondrial DNA. Mitochondrion 2015, 20, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.-P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef]
- Hagen, C.M.; Elson, J.L.; Hedley, P.L.; Aidt, F.H.; Havndrup, O.; Jensen, M.K.; Kanters, J.K.; Atherton, J.J.; McGaughran, J.; Bundgaard, H.; et al. Evolutionary dissection of mtDNA hg H: A susceptibility factor for hypertrophic cardiomyopathy. Mitochondrial DNA Part A 2020, 1–7. [Google Scholar] [CrossRef]
- Arbustini, E.; Fasani, R.; Morbini, P.; Diegoli, M.; Grasso, M.; Dal, B.; Marangoni, E.; Banfi, P.; Banchieri, N.; Bellini, O.; et al. Coexistence of mitochondrial DNA and β myosin heavy chain mutations in hypertrophic cardiomyopathy with late congestive heart failure. Heart 1998, 80, 548–558. [Google Scholar] [CrossRef][Green Version]
- Sebastiani, M.; Giordano, C.; Nediani, C.; Travaglini, C.; Borchi, E.; Zani, M.; Feccia, M.; Mancini, M.; Petrozza, V.; Cossarizza, A.; et al. Induction of Mitochondrial Biogenesis Is a Maladaptive Mechanism in Mitochondrial Cardiomyopathies. J. Am. Coll. Cardiol. 2007, 50, 1362–1369. [Google Scholar] [CrossRef]
- Scheubel, R.J.; Tostlebe, M.; Simm, A.; Rohrbach, S.; Prondzinsky, R.; Gellerich, F.N.; Silber, R.-E.; Holtz, J. Dysfunction of mitochondrial respiratory chain complex I in human failing myocardium is not due to disturbed mitochondrial gene expression. J. Am. Coll. Cardiol. 2002, 40, 2174–2181. [Google Scholar] [CrossRef]
- Jang, Y.-h.; Lim, K.-i. Recent Advances in Mitochondria-Targeted Gene Delivery. Molecules 2018, 23, 2316. [Google Scholar] [CrossRef]
- Patananan, A.N.; Wu, T.H.; Chiou, P.Y.; Teitell, M.A. Modifying the Mitochondrial Genome. Cell Metab. 2016, 23, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.B.; Rasmussen, M.; Rasmussen, L.J. Nuclear and mitochondrial DNA repair: Similar pathways? Mitochondrion 2005, 5, 89–108. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Moraes, C.T. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum. Mol. Genet. 2001, 10, 3093–3099. [Google Scholar] [CrossRef]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Peralta, S.; Moraes, C.T. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013, 19, 1111–1113. [Google Scholar] [CrossRef]
- Gammage, P.A.; Rorbach, J.; Vincent, A.I.; Rebar, E.J.; Minczuk, M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014, 6, 458–466. [Google Scholar] [CrossRef]
- Weissig, V. Targeted drug delivery to mammalian mitochondria in living cells. Expert Opin. Drug Deliv. 2005, 2, 89–102. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isogenic hPSC-CM Set(s) | Locus | mtDNA Mutation | Percent Heteroplasmy (WT-Het-HOM-KO) | In silico Prediction (Score) | Potential HCM Effect |
|---|---|---|---|---|---|
| AT1 | MT-HV2, MT-OHR | m.152T > C | 99.9 (all lines) | N/A (non-coding) | Aggravator (present only in AT1s) |
| REBL-PAT + HUES7 | MT-HV2, MT-OHR, MT-CSB2 | m.309_310insCCT | REBL-PAT: 28.9–26.4–27.2 HUES7: 17.4–13.8–15.6 | N/A (non-coding) | Protective (absent in AT1) |
| HUES7 | MT-HV2, MT-OHR, MT-CSB2 | m.309_310insCCCT | 11.7–11.6–13.3 | N/A (non-coding) | Protective (absent in AT1) |
| AT1 + REBL-PAT + HUES7 | MT-HV2, MT-OHR, MT-CSB2 | m.310T > C | AT1: 15.0–17.1–14.5–17.0; REBL-PAT: 61.2–63.2–61.9; HUES7: 61.6–56.4–57.6 | N/A (non-coding) | Protective (less prevalent in AT1) |
| AT1 + REBL-PAT + HUES7 | MT-HV2, MT-OHR, MT-CSB2 | m.310_311insC | AT1: 67.8–64.2–65.6–63.9 REBL-PAT: 30.1–26.7–27.2 HUES7: 26.1–32.0–33.3 | N/A (non-coding) | Aggravator (more prevalent in AT1s) |
| AT1 | MT-HV3 | m.514_515delCA | 60.1–53.5–53.6–53.7; | N/A (non-coding) | Aggravator only present in AT1s) |
| REBL-PAT | MT-RNR2 | m.2203G > A | 14.5% | N/A (non-coding) | None: acquired during reprogramming |
| AT1 | MT-RNR2 | m.2525C > T | Fibroblasts: 12.1% iPSCs + iPSC-CMs: 99.9% | N/A (non-coding) | None: increased upon nuclear reprogramming |
| AT1 + REBL-PAT + HUES7 | MT-TA | m.5597A > C | AT1: 5.4–12.4–6.5–11.4; REBL-PAT: 11.1–14.3–13.2; HUES7: 24.8–7.8–8.8 | Possibly pathogenic (73.80%) § | None: acquired during reprogramming/culture |
| HUES7 | MT-CO1 | m.5938A > C | 12.2–1.2–1.1 | Probably damaging (1.000) * Deleterious (1) † Neutral (0.45) ‡ | None: acquired during culture |
| REBL-PAT | MT-HV1 | m.16319G > A | 99.7–99.7–99.6 | N/A (non-coding) | Protective (absent in AT1) |
| Isogenic hPSC-CM Set(s) | Locus | mtDNA Mutation | Percent Heteroplasmy (WT-Het) | In silico Prediction (Score) | Potential HCM Effect |
|---|---|---|---|---|---|
| E99K1 | MT-HV2, MT-OHR | m.152T > C | 99.3–99.9 | N/A (non-coding) | Aggravator (present only in E99K1) |
| E99K1 | MT-HV2, MT-OHR, MT-CSB2 | m.309_310insCT | 15.3–25.3 | N/A (non-coding) | Aggravator (present only in E99K1) |
| E99K1 | MT-HV2, MT-OHR, MT-CSB2 | m.309_310insCCT | 18.8–9.3 | N/A (non-coding) | Aggravator (present only in E99K1) |
| E99K1 + NC-EDIT-E99K + E99K2 | MT-HV2, MT-OHR, MT-CSB2 | m.310T > C | E99K1: 60.6–59.9 NC-EDIT-E99K: 17.0–16.3 E99K2: 17.5–16.4 | N/A (non-coding) | Aggravator (more prevalent in E99K1) |
| E99K1 + NC-EDIT-E99K + E99K2 | MT-HV2, MT-OHR, MT-CSB2 | m.310_311insC | E99K1: 26.6–27.2 NC-EDIT-E99K: 61.6–62.7 E99K2: 63.6–61.7 | N/A (non-coding) | Protective (less prevalent in E99K1) |
| E99K1 + NC-EDIT-E99K + E99K2 | MT-TA | m.5597A > C | E99K1: 9.6–9.6 NC-EDIT-E99K: 7.5–6.9 E99K2: 10.2–7.1 | Possibly pathogenic (73.80%) § | None (present in all lines) |
| NC-EDIT-E99K + E99K2 | MT-ATP6 | m.8952T > C | NC-EDIT-E99K: 99.9–99.9 E99K2:100–99.9 | N/A (redundant) | Protective (absent in E99K1); |
| E99K1 | MT-ATP6 | m.9116T > C | 100–99.5 | Benign (0.000) * Neutral (0.8) † Neutral (0.42) ‡ | None (benign mutation) |
| E99K1 | MT-ND4 | m.11176G > A | 99.9–99.2 | N/A (redundant) | Aggravator (only present in E99K1) |
| NC-EDIT-E99K + E99K2 | MT-ND5 | m.12715A > G | NC-EDIT-E99K: 99.8–99.8 E99K2: 99.9–99.7 | Benign (0.1) * Neutral (0.47) † Neutral (0.28) ‡ | Protective (absent in E99K1) |
| mtDNA Mutation | Variant Type | Percent Variant Frequency (Full Length Sequences) | Percent Variant Frequency (Control Region Sequences) | Found at Haplogroups at 50% or Higher |
|---|---|---|---|---|
| m.152T > C | Local | 26.5 | 17.6 | Yes |
| m.309_310insCT | Global | 26.4 | 16.4 | Yes |
| m.309_310insCCT | Global | 7.1 | 4.9 | Yes |
| m.309_310insCCCT | Global | 0.1 | 0.2 | No |
| m.310T > C | Local | 40.7 | 27.8 | Yes |
| m.310_311_insC | Global | 0.0 | 0.0 | No |
| m.514_515delCA | Global | 24.2 | 1.7 | No |
| m.8952T > C | Local | 0.0 | N/A | No |
| m.12715A > G | Local | 0.1 | N/A | No |
| m.16319G > A | Local | 5.9 | 8.0 | Yes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kargaran, P.K.; Evans, J.M.; Bodbin, S.E.; Smith, J.G.W.; Nelson, T.J.; Denning, C.; Mosqueira, D. Mitochondrial DNA: Hotspot for Potential Gene Modifiers Regulating Hypertrophic Cardiomyopathy. J. Clin. Med. 2020, 9, 2349. https://doi.org/10.3390/jcm9082349
Kargaran PK, Evans JM, Bodbin SE, Smith JGW, Nelson TJ, Denning C, Mosqueira D. Mitochondrial DNA: Hotspot for Potential Gene Modifiers Regulating Hypertrophic Cardiomyopathy. Journal of Clinical Medicine. 2020; 9(8):2349. https://doi.org/10.3390/jcm9082349
Chicago/Turabian StyleKargaran, Parisa K., Jared M. Evans, Sara E. Bodbin, James G. W. Smith, Timothy J. Nelson, Chris Denning, and Diogo Mosqueira. 2020. "Mitochondrial DNA: Hotspot for Potential Gene Modifiers Regulating Hypertrophic Cardiomyopathy" Journal of Clinical Medicine 9, no. 8: 2349. https://doi.org/10.3390/jcm9082349
APA StyleKargaran, P. K., Evans, J. M., Bodbin, S. E., Smith, J. G. W., Nelson, T. J., Denning, C., & Mosqueira, D. (2020). Mitochondrial DNA: Hotspot for Potential Gene Modifiers Regulating Hypertrophic Cardiomyopathy. Journal of Clinical Medicine, 9(8), 2349. https://doi.org/10.3390/jcm9082349