Preeclampsia Is a Syndrome with a Cascade of Pathophysiologic Events

Abstract
1. Introduction
1.1. Epidemiology of Preeclampsia
1.2. Predisposing Risk Factors
2. Pathophysiology
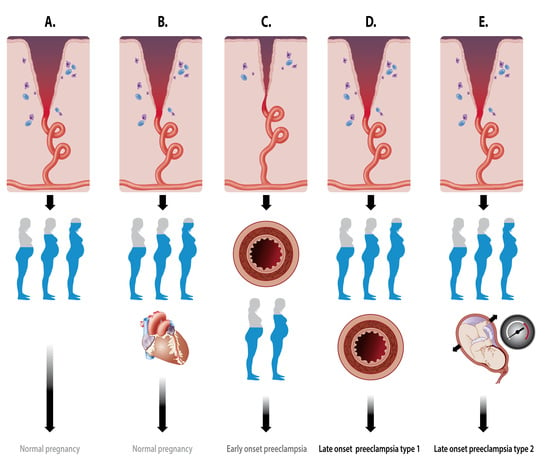
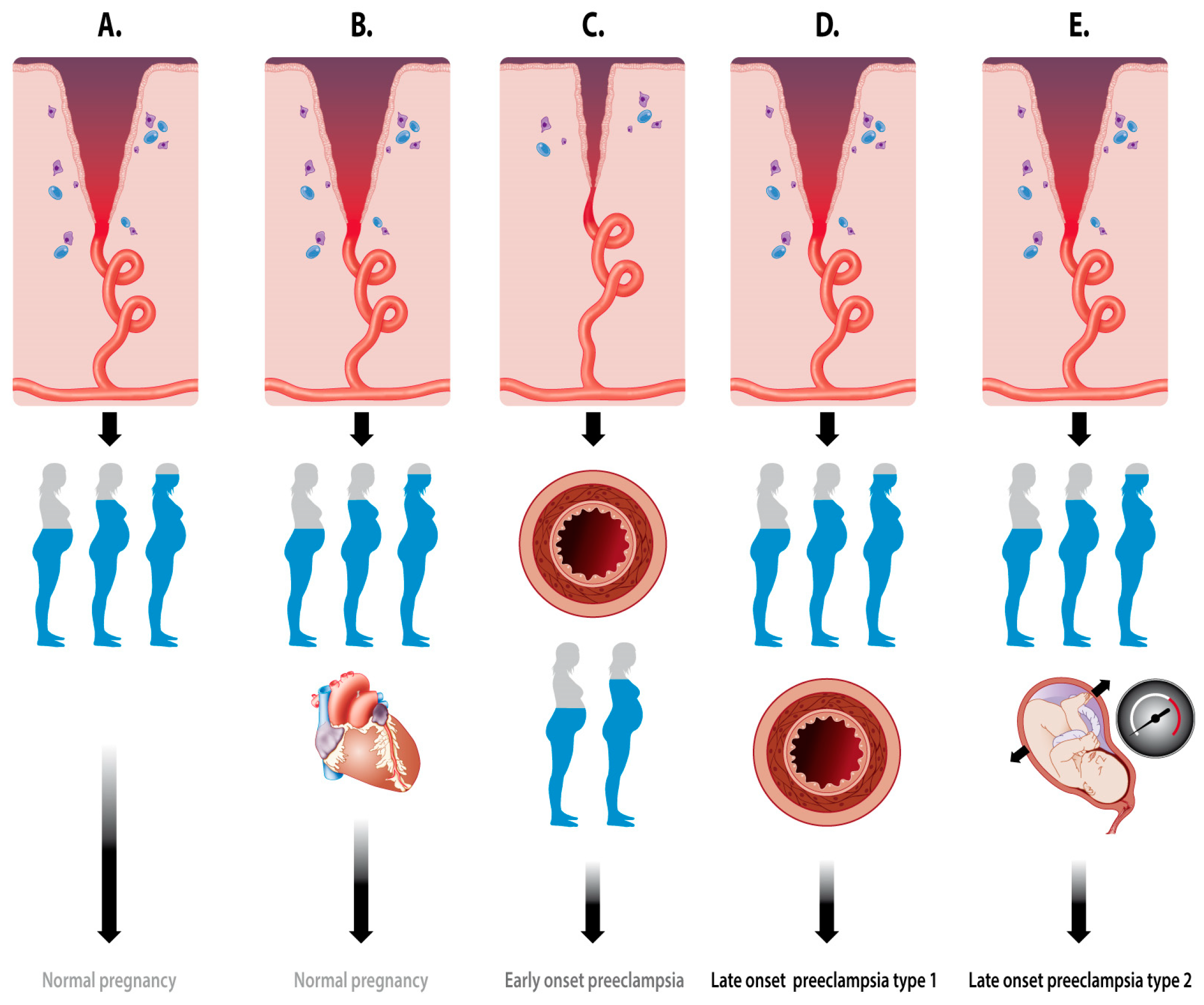
2.1. Types of PE
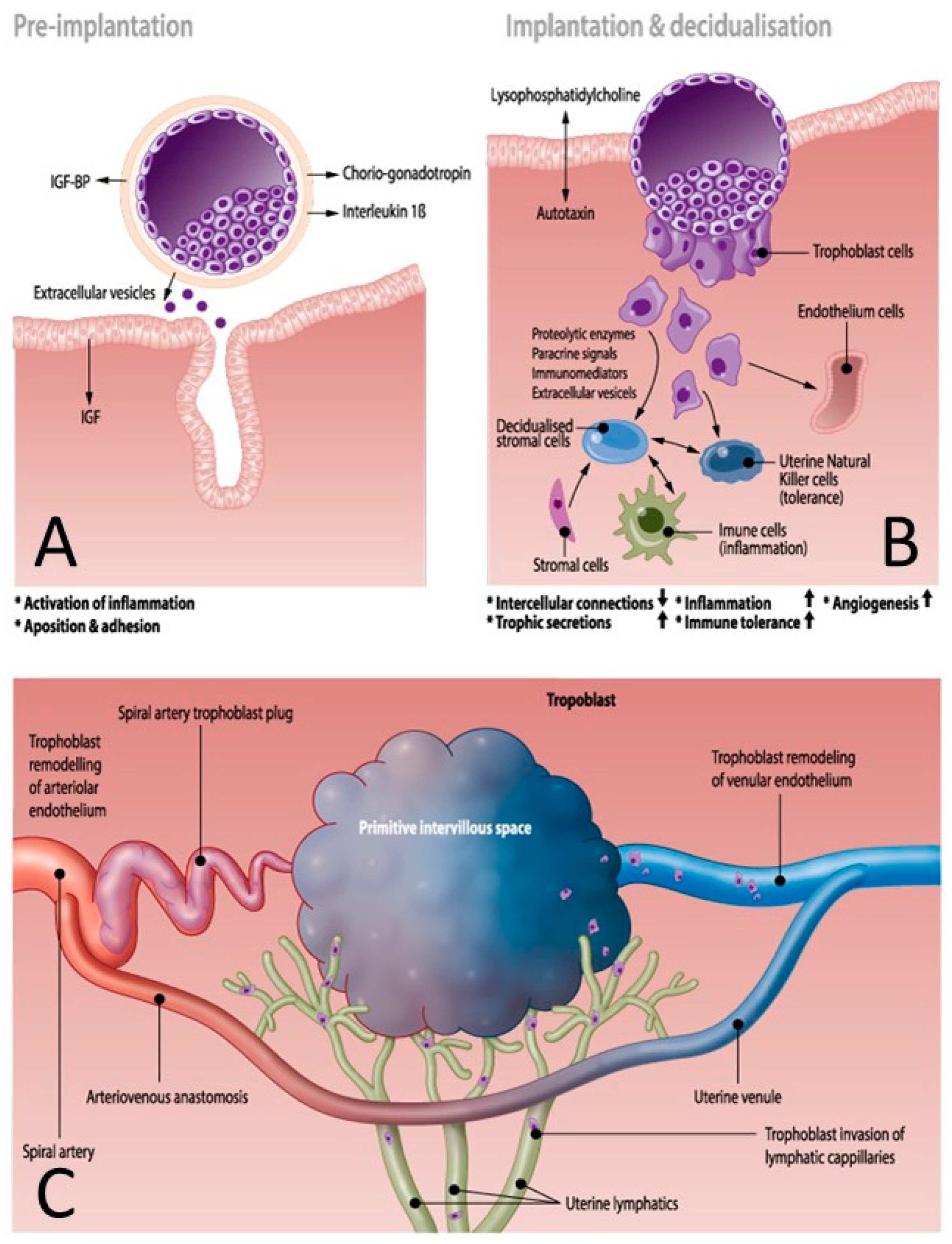
2.2. Placentation Process
2.2.1. Spiral Artery Remodeling in Normal Pregnancy and Preeclampsia
2.2.2. Vascular Uterine Adaptation Involves More than Spiral Artery Remodeling
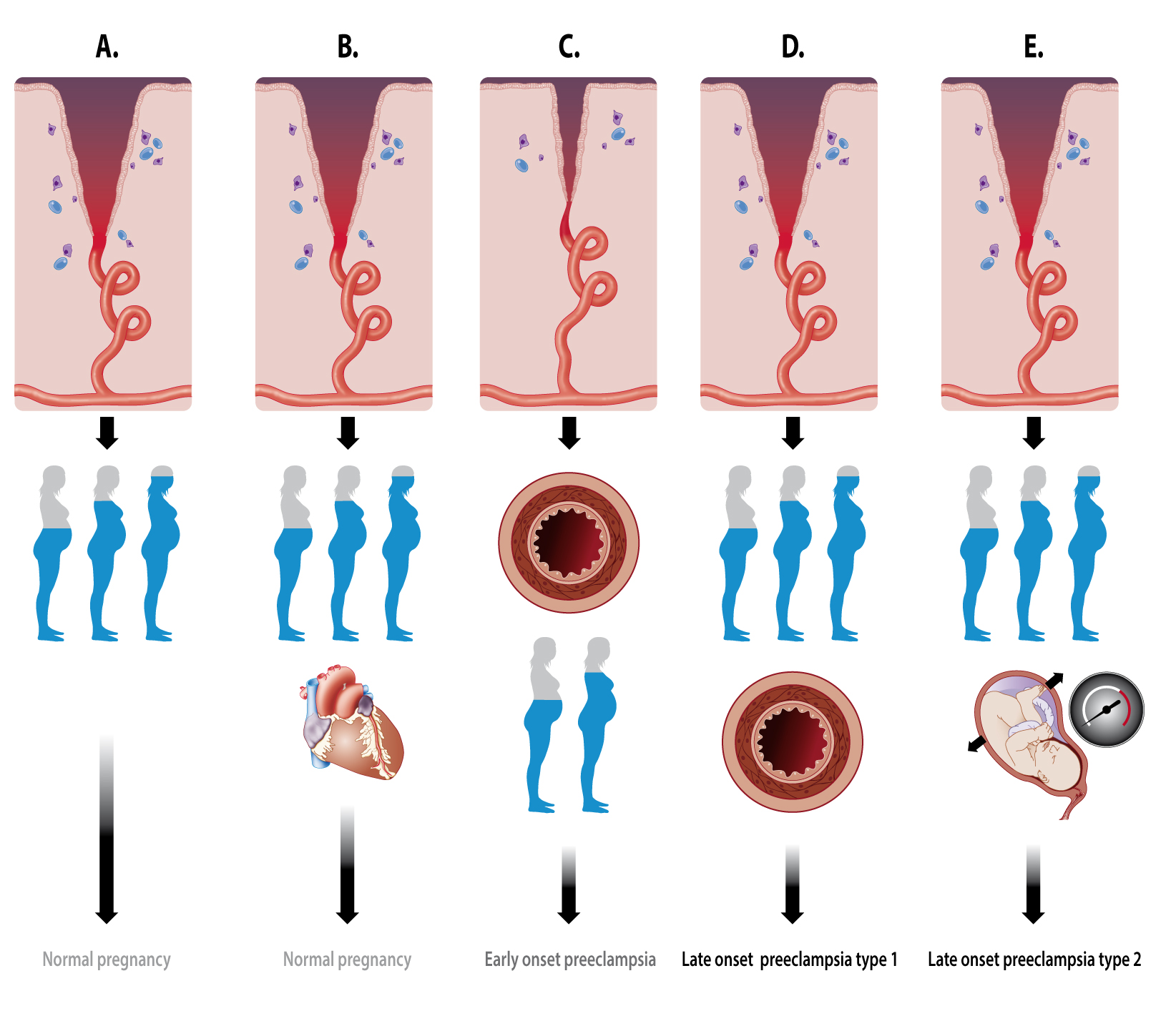
2.2.3. Hemodynamics of the Intervillous Space
- -
- -
- -
- Diversion of the arterial blood from the radial arteries to the uterine venules by arteriovenous anastomoses (Figure 2C). These vascular connections allow direct endothelial cell–cell communication between the uterine venular and arteriolar vessels during the formation of the intervillous space [55,65,66].
- -
- Initiation of the adaptation of the maternal systemic cardiovasculature by causing the drainage of endocrine and trophoblast signals into the maternal circulation. During blockage of the spiral arteries, trophoblast cells invade and dilate the maternal veins [64], a process that also contributes to maintaining the low pressure and flow conditions during the formation of the intervillous space [63].
- -
- In a similar way, a systemic maternal immune response and tolerance are generated by the trophoblast invasion of lymphatic vessels, allowing the drainage and presentation of immunoregulatory signals to immune cells at locations distant from the uterus [64].
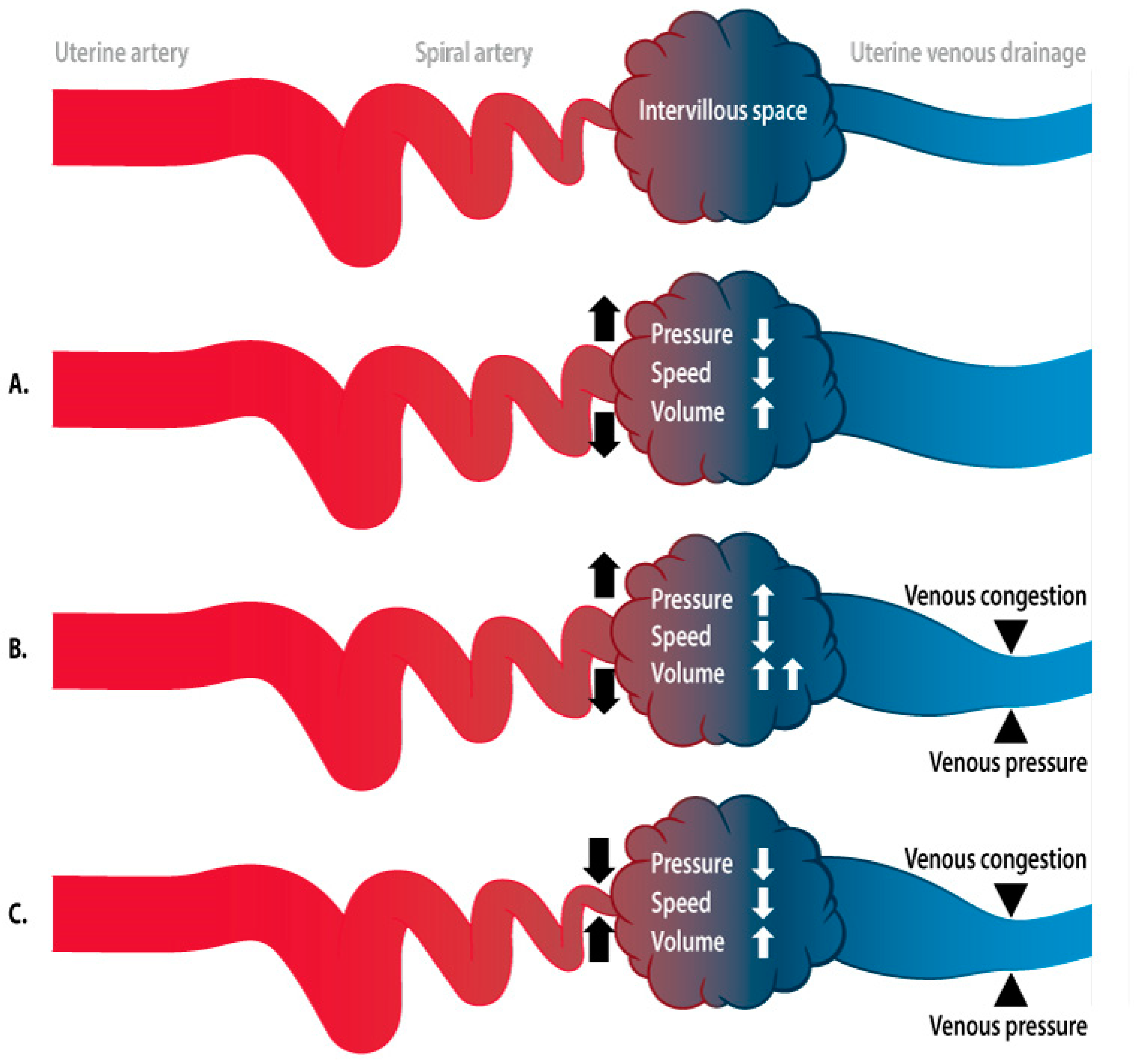
2.3. Immune System, Implantation, and Vascular Remodeling
2.4. Fetal–Maternal Communication
2.4.1. Pre- and Peri-Implantation Signaling
2.4.2. Endocrine, Biological and Chemical Signaling
2.4.3. Syncytiotrophoblast Extracellular Vesicles
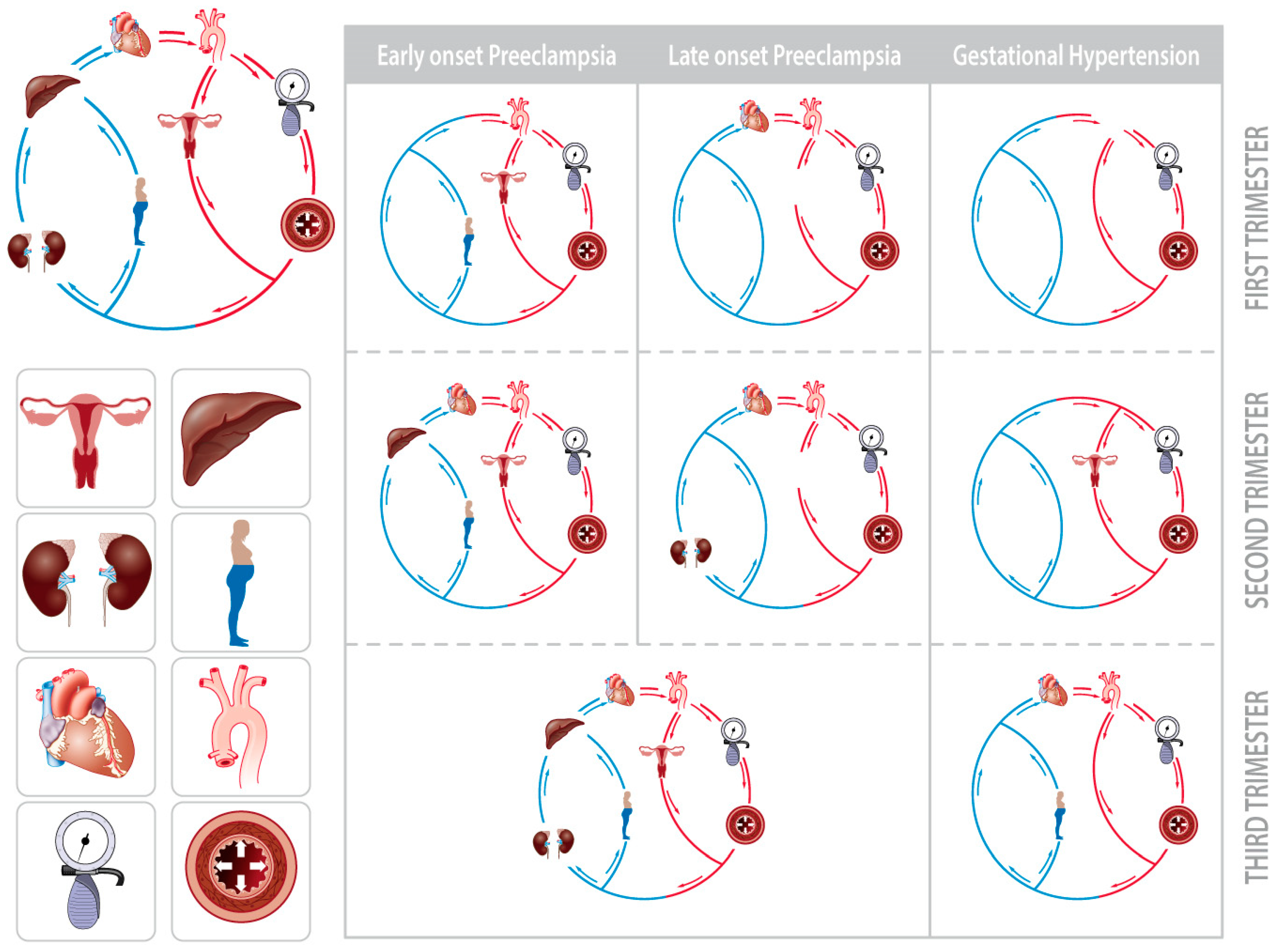
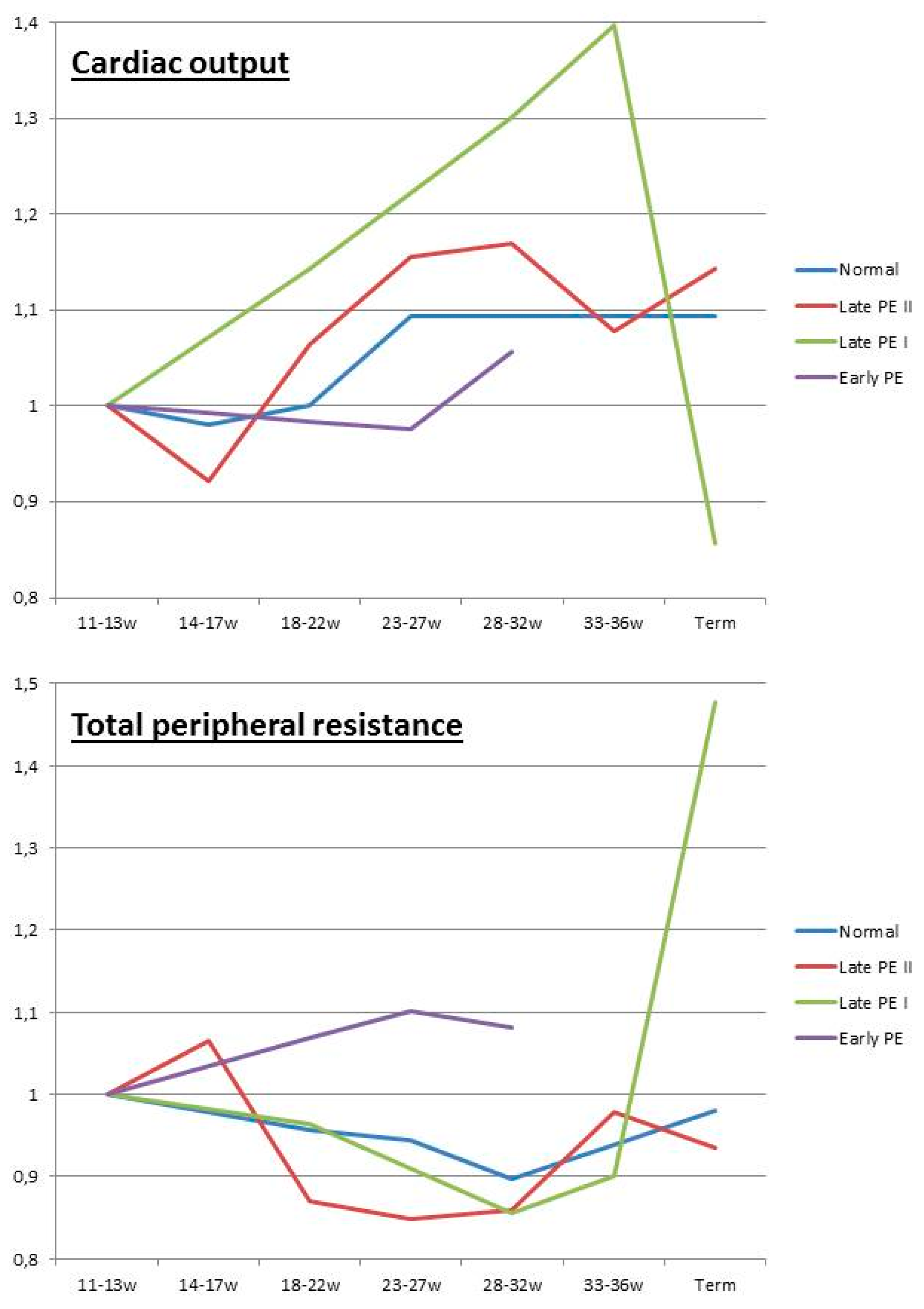
2.5. Maternal Hemodynamics
2.5.1. Peri-Implantation Hemodynamics
2.5.2. Body Water Volume Expansion and (Subclinical) Cardiovascular Dysfunctions
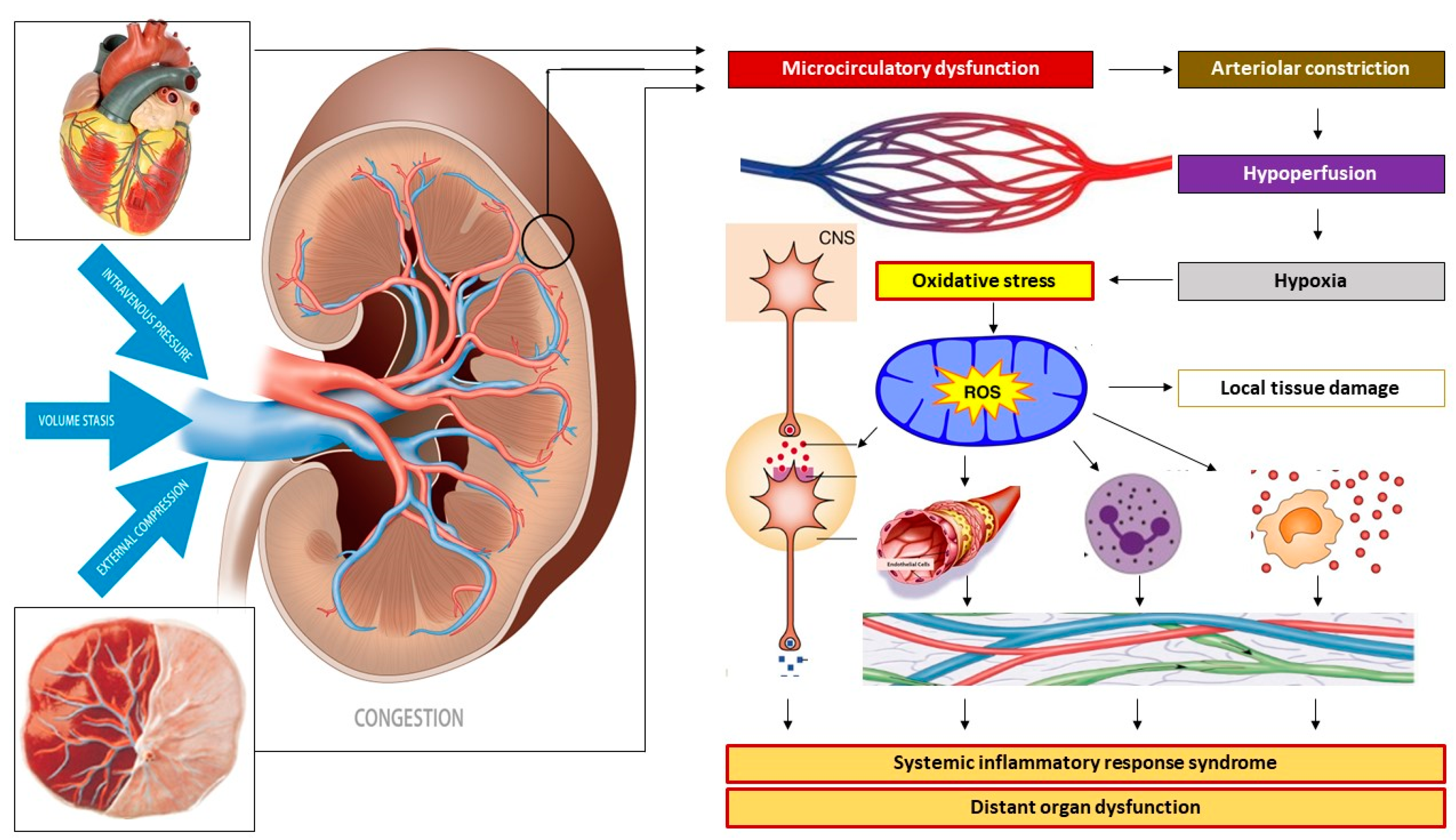
2.5.3. Venous Hemodynamics
2.5.4. Venous Congestion
2.5.5. The Role of Intra-Abdominal Pressure
3. Integrated Pathophysiology of PE
3.1. Immunomodulation
3.2. Maternal-Fetal Crosstalk
3.3. Maternal Hemodynamics and Venous Congestion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chhabra, P. Maternal near miss: An indicator for maternal health and maternal care. Indian J. Community Med. 2014, 39, 132–137. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists. Task Force on Hypertension in Pregnancy Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar]
- Tranquilli, A.L.; Dekker, G.; Magee, L.; Roberts, J.; Sibai, B.M.; Steyn, W.; Zeeman, G.G.; Brown, M.A. The classification, diagnosis and management of the hypertensive disorders of pregnancy: A revised statement from the ISSHP. Pregnancy Hypertens 2014, 4, 97–104. [Google Scholar] [CrossRef]
- Braunthal, S.; Brateanu, A. Hypertension in pregnancy: Pathophysiology and treatment. SAGE Open Med. 2019, 7, 2050312119843700. [Google Scholar] [CrossRef] [PubMed]
- Umesawa, M.; Kobashi, G. Epidemiology of hypertensive disorders in pregnancy: Prevalence, risk factors, predictors and prognosis. Hypertens Res. 2017, 40, 213–220. [Google Scholar] [CrossRef]
- Hutcheon, J.A.; Lisonkova, S.; Joseph, K.S. Epidemiology of pre-eclampsia and the other hypertensive disorders of pregnancy. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Hill, L.D.; Hilliard, D.D.; York, T.P.; Srinivas, S.; Kusanovic, J.P.; Gomez, R.; Elovitz, M.A.; Romero, R.; Strauss, J.F. Fetal ERAP2 variation is associated with preeclampsia in African Americans in a case-control study. BMC Med. Genet. 2011, 11, 12–64. [Google Scholar] [CrossRef] [PubMed]
- Morgan, L.; McGinnis, R.; Steinthorsdottir, V.; Svyatova, G.; Zakhidova, N.; Lee, W.K.; Iversen, A.C.; Magnus, P.; Walker, J.; Casas, J.P.; et al. InterPregGen: Genetic studies of pre-eclampsia in three continents. Nor. Epidemiol. 2014, 24, 141–146. [Google Scholar]
- Galaviz-Hernandez, C.; Sosa-Macias, M.; Teran, E.; Garcia-Ortiz, J.E.; Lazalde-Ramos, B.P. Paternal determinants in preeclampsia. Front. Physiol. 2019, 9, 1870. [Google Scholar] [CrossRef]
- Buurma, A.J.; Turner, R.J.; Driessen, J.H.; Mooyaart, A.L.; Schoones, J.W.; Bruijn, J.A.; Bloemenkamp, K.W.; Dekkers, O.M.; Baelde, H.J. Genetic variants in pre-eclampsia: A meta-analysis. Hum. Reprod. Update 2013, 19, 289–303. [Google Scholar] [CrossRef]
- Ghosh, G.; Grewal, J.; Männistö, T.; Mendola, P.; Chen, Z.; Xie, Y.; Laughon, S.K. Racial/ethnic differences in pregnancy-related hypertensive disease in nulliparous women. Ethn. Dis. 2014, 24, 283–289. [Google Scholar] [PubMed]
- Vanhille, D.L.; Hill, L.D.; Hilliard, D.D.; Lee, E.D.; Teves, M.E.; Srinivas, S.; Kusanovic, J.P.; Gomez, R.; Stratikos, E.; Elovitz, M.A.; et al. A novel ERAP2 haplotype structure in a chilean population: Implications for erap2 protein expression and preeclampsia risk. Mol. Genet. Genom. Med. 2013, 1, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, E.; Medcalf, K.E.; Park, A.L.; Ray, J.G. High Risk of Pre-eclampsia Identification Group. Clinical risk factors for pre-eclampsia determined in early pregnancy: Systematic review and meta-analysis of large cohort studies. BMJ 2016, 353, i1753. [Google Scholar] [CrossRef] [PubMed]
- Abalos, E.; Cuesta, C.; Carroli, G.; Qureshi, Z.; Widmer, M.; Vogel, J.P.; Souza, J.P. WHO multicountry survey on maternal and newborn health research network. Pre-eclampsia, eclampsia and adverse maternal and perinatal outcomes: A secondary analysis of the world health organization multicountry survey on maternal and newborn health. BJOG 2014, 121, 14–24. [Google Scholar] [CrossRef]
- Thomopoulos, C.; Tsioufis, C.; Michalopoulou, H.; Makris, T.; Papademetriou, V.; Stefanadis, C. Assisted reproductive technology and pregnancy-related hypertensive complications: A systematic review. J. Hum. Hypertens 2013, 27, 148–157. [Google Scholar] [CrossRef]
- Storgaard, M.; Loft, A.; Bergh, C.; Wennerholm, U.B.; Söderström-Anttila, V.; Romundstad, L.B.; Aittomaki, K.; Oldereid, N.; Forman, J.; Pinborg, A. Obstetric and neonatal complications in pregnancies conceived after oocyte donation: A systematic review and meta-analysis. BJOG 2017, 124, 561–572. [Google Scholar] [CrossRef]
- Qin, J.; Liu, X.; Sheng, X.; Wang, H.; Gao, S. Assisted reproductive technology and the risk of pregnancy-related complications and adverse pregnancy outcomes in singleton pregnancies: A meta-analysis of cohort studies. Fertil. Steril. 2016, 105, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.; Stayner, L.; Slama, R.; Sørensen, M.; Figueras, F.; Nieuwenhuijsen, M.J.; Raaschou-Nielsen, O.; Dadvand, P. Ambient air pollution and pregnancy-induced hypertensive disorders: A systematic review and meta-analysis. Hypertension 2014, 64, 494–500. [Google Scholar] [CrossRef]
- Wojcik-Baszko, D.; Charkiewicz, K.; Laudanski, P. Role of dyslipidemia in preeclampsia—A review of lipidomic analysis of blood, placenta, syncytiotrophoblast microvesicles and umbilical cord artery from women with preeclampsia. Prostaglandins Other Lipid. Mediat. 2018, 139, 19–23. [Google Scholar] [CrossRef]
- Sanapo, L.; Bublitz, M.H.; Bourjeily, G. Sleep disordered breathing, a novel modifiable risk factor for hypertensive disorders of pregnancy. Curr. Hypertens Rep. 2020, 22, 28. [Google Scholar] [CrossRef]
- Allais, G.; Chiarle, G.; Sinigaglia, S.; Mana, O.; Benedetto, C. Migraine during pregnancy and in the puerperium. Neurol. Sci. 2019, 40, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Schoenaker, D.A.; Soedamah-Muthu, S.S.; Mishra, G.D. The association between dietary factors and gestational hypertension and pre-eclampsia: A systematic review and meta-analysis of observational studies. BMC Med. 2014, 12, 157. [Google Scholar] [CrossRef] [PubMed]
- Akbari, S.; Khodadadi, B.; Ahmadi, S.A.Y.; Abbaszadeh, S.; Shahsavar, F. Association of vitamin D level and vitamin D deficiency with risk of preeclampsia: A systematic review and updated meta-analysis. Taiwan J. Obstet. Gynecol. 2018, 57, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Romagnuolo, I.; Sticchi, E.; Attanasio, M.; Grifoni, E.; Cioni, G.; Cellai, A.P.; Abbate, R.; Fatini, C. Searching for a common mechanism for placenta-mediated pregnancy complications and cardiovascular disease: Role of lipoprotein(a). Fertil. Steril. 2016, 105, 1287–1293. [Google Scholar] [CrossRef]
- Bell, M.J. A historical overview of preeclampsia-eclampsia. J. Obstet. Gynecol. Neonatal. Nurs. 2010, 39, 510–518. [Google Scholar] [CrossRef]
- Robillard, P.Y.; Dekker, G.; Chaouat, G.; Le Bouteiller, P.; Scioscia, M.; Hulsey, T.C. Preeclampsia and the 20th century: “Le siècle des Lumières”. Pregnancy Hypertens 2018, 13, 107–109. [Google Scholar] [CrossRef]
- von Dadelszen, P.; Magee, L.A.; Roberts, J.M. Subclassification of preeclampsia. Hypertens Pregnancy 2003, 22, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Valensise, H.; Vasapollo, B.; Gagliardi, G.; Novelli, G.P. Early and late preeclampsia: Two different maternal hemodynamic states in the latent phase of the disease. Hypertension 2008, 52, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Myatt, L.; Roberts, J.M. Preeclampsia: Syndrome or disease? Curr. Hypertens Rep. 2015, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Phipps, E.; Prasanna, D.; Brima, W.; Jim, B. Preeclampsia: Updates in pathogenesis, definitions, and guidelines. Clin. J. Am. Soc. Nephrol. 2016, 11, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Lees, C.; Ferrazzi, E. Relevance of haemodynamics in treating pre-eclampsia. Curr. Hypertens Rep. 2017, 19, 76. [Google Scholar] [CrossRef]
- Ferrazzi, E.; Stampalija, T.; Monasta, L.; Di Martino, D.; Vonck, S.; Gyselaers, W. Maternal hemodynamics: A method to classify hypertensive disorders of pregnancy. Am. J. Obstet. Gynecol. 2018, 218, 124.e1–124.e11. [Google Scholar] [CrossRef] [PubMed]
- Tay, J.; Foo, L.; Masini, G.; Bennett, P.R.; McEniery, C.M.; Wilkinson, I.B.; Lees, C.C. Early and late preeclampsia are characterized by high cardiac output, but in the presence of fetal growth restriction, cardiac output is low: Insights from a prospective study. Am. J. Obstet. Gynecol. 2018, 218, 517.e1–517.e12. [Google Scholar] [CrossRef] [PubMed]
- Secomb, T.W. Hemodynamics. Compr. Physiol. 2016, 6, 975–1003. [Google Scholar] [CrossRef] [PubMed]
- Verlohren, S.; Melchiorre, K.; Khalil, A.; Thilaganathan, B. Uterine artery doppler, birth weight and timing of onset of pre-eclampsia: Providing insights into the dual etiology of late-onset pre-eclampsia. Ultrasound Obstet. Gynecol. 2014, 44, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Lisonkova, S.; Joseph, K.S. Incidence of preeclampsia: Risk factors and outcomes associated with early- versus late-onset disease. Am. J. Obstet. Gynecol. 2013, 209, 544.e1–544.e12. [Google Scholar] [CrossRef]
- Easterling, T.R.; Benedetti, T.J. Preeclampsia: A hyperdynamic disease model. Am. J. Obstet. Gynecol. 1989, 160, 1447–1453. [Google Scholar] [CrossRef]
- Bosio, P.M.; McKenna, P.J.; Conroy, R.; O’Herlihy, C. Maternal central hemodynamics in hypertensive disorders of pregnancy. Obstet. Gynecol. 1999, 94, 978–984. [Google Scholar]
- Rang, S.; van Montfrans, G.A.; Wolf, H. Serial hemodynamic measurement in normal pregnancy, preeclampsia, and intrauterine growth restriction. Am. J. Obstet. Gynecol. 2008, 198, 519-e1. [Google Scholar] [CrossRef]
- Hall, D.; Gebhardt, S.; Theron, G.; Grové, D. Pre-eclampsia and gestational hypertension are less common in HIV infected women. Pregnancy Hypertens 2014, 4, 91–96. [Google Scholar] [CrossRef]
- Foo, F.L.; Mahendru, A.A.; Masini, G.; Fraser, A.; Cacciatore, S.; MacIntyre, D.A.; McEniery, C.M.; Wilkinson, I.B.; Bennett, P.R.; Lees, C.C. Association between prepregnancy cardiovascular function and subsequent preeclampsia or fetal growth restriction. Hypertension 2018, 72, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Vonck, S.; Lanssens, D.; Staelens, A.S.; Tomsin, K.; Oben, J.; Bruckers, L.; Gyselaers, W. Obesity in pregnancy causes a volume overload in third trimester. Eur. J. Clin. Investig. 2019, 49, e13173. [Google Scholar] [CrossRef] [PubMed]
- Vonck, S.; Staelens, A.S.; Lanssens, D.; Tomsin, K.; Oben, J.; Dreesen, P.; Bruckers, L.; Gyselaers, W. Low volume circulation in normotensive women pregnant with neonates small for gestational age. Fetal Diagn. Ther. 2019, 46, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Gyselaers, W.; Thilaganathan, B. Preeclampsia: A gestational cardiorenal syndrome. J. Physiol. 2019, 597, 4695–4714. [Google Scholar] [CrossRef]
- Brosens, I.; Puttemans, P.; Benagiano, G. Placental bed research: I. the placental bed: From spiral arteries remodeling to the great obstetrical syndromes. Am. J. Obstet. Gynecol. 2019, 221, 437–456. [Google Scholar] [CrossRef]
- Ragab, A.; Goda, H.; Raghib, M.; Barakat, R.; El-Samanoudy, A.; Badawy, A. Does immediate postpartum curettage of the endometrium accelerate recovery from preeclampsia-eclampsia? A randomized controlled trial. Arch. Gynecol. Obstet. 2013, 288, 1035–1038. [Google Scholar] [CrossRef]
- Brosens, I.; Brosens, J.J.; Muter, J.; Puttemans, P.; Benagiano, G. Preeclampsia: The role of persistent endothelial cells in uteroplacental arteries. Am. J. Obstet. Gynecol. 2019, 221, 219–226. [Google Scholar] [CrossRef]
- Falco, M.L.; Sivanathan, J.; Laoreti, A.; Thilaganathan, B.; Khalil, A. Placental histopathology associated with pre-eclampsia: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2017, 50, 295–301. [Google Scholar] [CrossRef] [PubMed]
- James, J.L.; Chamley, L.W.; Clark, A.R. Feeding your baby in utero: How the uteroplacental circulation impacts pregnancy. Physiology (Bethesda) 2017, 32, 234–245. [Google Scholar] [CrossRef]
- He, N.; van Iperen, L.; de Jong, D.; Szuhai, K.; Helmerhorst, F.M.; van der Westerlaken, L.A.; Chuva de Sousa Lopes, S.M. Human extravillous trophoblasts penetrate decidual veins and lymphatics before remodeling spiral arteries during early pregnancy. PLoS ONE 2017, 12, e0169849. [Google Scholar] [CrossRef]
- Huppertz, B.; Weiss, G.; Moser, G. Trophoblast invasion and oxygenation of the placenta: Measurements versus presumptions. J. Reprod. Immunol. 2014, 101–102, 74–79. [Google Scholar] [CrossRef]
- Kakogawa, J.; Sumimoto, K.; Kawamura, T.; Minoura, S.; Kanayama, N. Transabdominal measurement of placental oxygenation by near-infrared spectroscopy. Am. J. Perinatol. 2010, 27, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.J.; Haeussner, E.; Ruebelmann, T.; Koch, F.V.; Schmitz, C.; Frank, H.G.; Wall, W.A. Dynamic modeling of uteroplacental blood flow in IUGR indicates vortices and elevated pressure in the intervillous space—A pilot study. Sci. Rep. 2017, 7, 40771. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.S.; Brownbill, P.; Jones, N.W.; Abrahams, V.M.; Baker, P.N.; Sibley, C.P.; Crocker, I.P. Utero-placental haemodynamics in the pathogenesis of pre-eclampsia. Placenta 2009, 30, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Gyselaers, W.; Peeters, L. Physiological implications of arteriovenous anastomoses and venous hemodynamic dysfunction in early gestational uterine circulation: A review. J. Matern. Fetal Neonatal Med. 2013, 26, 841–846. [Google Scholar] [CrossRef]
- Arishe, O.O.; Ebeigbe, A.B.; Webb, R.C. Mechanotransduction and uterine blood flow in preeclampsia: The role of mechanosensing piezo 1 ion channels. Am. J. Hypertens 2020, 33, 1–9. [Google Scholar] [CrossRef]
- James, J.L.; Whitley, G.S.; Cartwright, J.E. Shear stress and spiral artery remodelling: The effects of low shear stress on trophoblast-induced endothelial cell apoptosis. Cardiovasc. Res. 2011, 90, 130–139. [Google Scholar] [CrossRef]
- Sánchez-Aranguren, L.C.; Prada, C.E.; Riaño-Medina, C.E.; Lopez, M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front. Physiol. 2014, 5, 372. [Google Scholar] [CrossRef] [PubMed]
- Kalapotharakos, G.; Murtoniemi, K.; Åkerström, B.; Hämäläinen, E.; Kajantie, E.; Räikkönen, K.; Villa, P.; Laivuori, H.; Hansson, S.R. Plasma heme scavengers alpha-1-microglobulin and hemopexin as biomarkers in high-risk pregnancies. Front Physiol. 2019, 10, 300. [Google Scholar] [CrossRef]
- Apicella, C.; Ruano, C.S.M.; Méhats, C.; Miralles, F.; Vaiman, D. The role of epigenetics in placental development and the etiology of preeclampsia. Int. J. Mol. Sci. 2019, 20, 2837. [Google Scholar] [CrossRef]
- Lv, Y.; Lu, C.; Ji, X.; Miao, Z.; Long, W.; Ding, H.; Lv, M. Roles of microRNAs in preeclampsia. J. Cell Physiol. 2019, 234, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Li, J. Role of circular RNAs in preeclampsia. Dis. Markers 2019, 2019, 7237495. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Sundl, M.; Glasner, A.; Huppertz, B.; Moser, G. The trophoblast plug during early pregnancy: A deeper insight. Histochem. Cell Biol. 2016, 146, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Moser, G.; Windsperger, K.; Pollheimer, J.; de Sousa Lopes, S.C.; Huppertz, B. Human trophoblast invasion: New and unexpected routes and functions. Histochem. Cell Biol. 2018, 150, 361–370. [Google Scholar] [CrossRef] [PubMed]
- James, J.L.; Saghian, R.; Perwick, R.; Clark, A.R. Trophoblast plugs: Impact on utero-placental haemodynamics and spiral artery remodelling. Hum. Reprod. 2018, 33, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- Ko, N.L.; Mandalà, M.; John, L.; Gelinne, A.; Osol, G. Venoarterial communication mediates arterial wall shear stress-induced maternal uterine vascular remodeling during pregnancy. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H709–H717. [Google Scholar] [CrossRef]
- Zhang, S.; Lin, H.; Kong, S.; Wang, S.; Wang, H.; Wang, H.; Armant, D.R. Physiological and molecular determinants of embryo implantation. Mol. Aspects Med. 2013, 34, 939–980. [Google Scholar] [CrossRef]
- Schumacher, A.; Zenclussen, A.C. Human chorionic gonadotropin-mediated immune responses that facilitate embryo implantation and placentation. Front. Immunol. 2019, 10, 2896. [Google Scholar] [CrossRef]
- Boeldt, D.S.; Bird, I.M. Vascular adaptation in pregnancy and endothelial dysfunction in preeclampsia. J. Endocrinol. 2017, 232, R27–R44. [Google Scholar] [CrossRef]
- Makrigiannakis, A.; Vrekoussis, T.; Zoumakis, E.; Kalantaridou, S.N.; Jeschke, U. The role of hcg in implantation: A mini-review of molecular and clinical evidence. Int. J. Mol. Sci. 2017, 18, 1305. [Google Scholar] [CrossRef]
- Ivanov, P.; Tsvyatkovska, T.; Konova, E.; Komsa-Penkova, R. Inherited thrombophilia and IVF failure: The impact of coagulation disorders on implantation process. Am. J. Reprod. Immunol. 2012, 68, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Boij, R.; Svensson, J.; Nilsson-Ekdahl, K.; Sandholm, K.; Lindahl, T.L.; Palonek, E.; Garle, M.; Berg, G.; Ernerudh, J.; Jenmalm, M.; et al. Biomarkers of coagulation, inflammation, and angiogenesis are independently associated with preeclampsia. Am. J. Reprod. Immunol. 2012, 68, 258–270. [Google Scholar] [CrossRef]
- Du Clos, T.W.; Mold, C. Pentraxins (CRP, SAP) in the process of complement activation and clearance of apoptotic bodies through Fcγ receptors. Curr. Opin. Organ Transplant. 2011, 16, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Herrler, A.; von Rango, U.; Beier, H.M. Embryo-maternal signalling: How the embryo starts talking to its mother to accomplish implantation. Reprod. Biomed. Online 2003, 6, 244–256. [Google Scholar] [CrossRef]
- Ashary, N.; Tiwari, A.; Modi, D. embryo implantation: War in times of love. Endocrinology 2018, 159, 1188–1198. [Google Scholar] [CrossRef]
- Maliqueo, M.; Echiburú, B.; Crisosto, N. Sex steroids modulate uterine-placental vasculature: Implications for obstetrics and neonatal outcomes. Front. Physiol. 2016, 7, 152. [Google Scholar] [CrossRef]
- Berkane, N.; Liere, P.; Oudinet, J.P.; Hertig, A.; Lefèvre, G.; Pluchino, N.; Schumacher, M.; Chabbert-Buffet, N. From pregnancy to preeclampsia: A key role for estrogens. Endocr. Rev. 2017, 38, 123–144. [Google Scholar] [CrossRef]
- Chamley, L.W.; Holland, O.J.; Chen, Q.; Viall, C.A.; Stone, P.R.; Abumaree, M. Review: Where is the maternofetal interface? Placenta 2014, 35, S74–S80. [Google Scholar] [CrossRef]
- Rafaeli-Yehudai, T.; Imterat, M.; Douvdevani, A.; Tirosh, D.; Benshalom-Tirosh, N.; Mastrolia, S.A.; Beer-Weisel, R.; Klaitman, V.; Riff, R.; Greenbaum, S.; et al. Maternal total cell-free DNA in preeclampsia and fetal growth restriction: Evidence of differences in maternal response to abnormal implantation. PLoS ONE 2018, 13, e0200360. [Google Scholar] [CrossRef]
- Shirasuna, K.; Karasawa, T.; Takahashi, M. Role of the NLRP3 Inflammasome in Preeclampsia. Front. Endocrinol. (Lausanne) 2020, 11, 80. [Google Scholar] [CrossRef]
- Umapathy, A.; Chamley, L.W.; James, J.L. Reconciling the distinct roles of angiogenic/anti-angiogenic factors in the placenta and maternal circulation of normal and pathological pregnancies. Angiogenesis 2019. [Google Scholar] [CrossRef]
- Rebelo, F.; Schlüssel, M.M.; Vaz, J.S.; Franco-Sena, A.B.; Pinto, T.J.; Bastos, F.I.; Adegboye, A.R.; Kac, G. C-reactive protein and later preeclampsia: Systematic review and meta-analysis taking into account the weight status. J. Hypertens 2013, 31, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Zhang, F.; Zhang, C.; Zheng, L.R.; Yang, J. The biomarkers for acute myocardial infarction and heart failure. Biomed. Res. Int. 2020, 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kleinrouweler, C.E.; Wiegerinck, M.M.; Ris-Stalpers, C.; Bossuyt, P.M.; van der Post, J.A.; von Dadelszen, P.; Mol, B.W.; Pajkrt, E.; EBM CONNECT Collaboration. Accuracy of circulating placental growth factor, vascular endothelial growth factor, soluble fms-like tyrosine kinase 1 and soluble endoglin in the prediction of pre-eclampsia: A systematic review and meta-analysis. BJOG 2012, 119, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.Q.; Wang, Y.; Han, X.Q.; Zhu, Y.; Liu, N.F. Association of genetic polymorphisms in vascular endothelial growth factor with susceptibility to coronary artery disease: A meta-analysis. BMC Med. Genet. 2018, 19, 108. [Google Scholar] [CrossRef] [PubMed]
- Draker, N.; Torry, D.S.; Torry, R.J. Placenta growth factor and sFlt-1 as biomarkers in ischemic heart disease and heart failure: A review. Biomarks Med. 2019, 13, 785–799. [Google Scholar] [CrossRef]
- Ikemoto, T.; Hojo, Y.; Kondo, H.; Takahashi, N.; Hirose, M.; Nishimura, Y.; Katsuki, T.; Shimada, K.; Kario, K. Plasma endoglin as a marker to predict cardiovascular events in patients with chronic coronary artery diseases. Heart Vessel. 2012, 27, 344–351. [Google Scholar] [CrossRef]
- Kolla, V.; Jenö, P.; Moes, S.; Lapaire, O.; Hoesli, I.; Hahn, S. Quantitative proteomic (iTRAQ) analysis of 1st trimester maternal plasma samples in pregnancies at risk for preeclampsia. J. Biomed. Biotechnol. 2012, 2012, 305964. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Erbel, C.; Linden, F.; Domschke, G.; Akhavanpoor, M.; Helmes, C.M.; Doesch, A.O.; Kleber, M.E.; Katus, H.A.; Maerz, W. Galectin-3 binding protein, coronary artery disease and cardiovascular mortality: Insights from the LURIC study. Atherosclerosis 2017, 260, 121–129. [Google Scholar] [CrossRef]
- Muttukrishna, S.; North, R.A.; Morris, J.; Schellenberg, J.C.; Taylor, R.S.; Asselin, J.; Ledger, W.; Groome, N.; Redman, C.W. Serum inhibin A and activin A are elevated prior to the onset of pre-eclampsia. Hum. Reprod. 2000, 15, 1640–1645. [Google Scholar] [CrossRef]
- Sugatani, T. Systemic activation of activin a signaling causes chronic kidney disease-mineral bone disorder. Int. J. Mol. Sci. 2018, 19, 2490. [Google Scholar] [CrossRef]
- Taylor, B.D.; Ness, R.B.; Olsen, J.; Hougaard, D.M.; Skogstrand, K.; Roberts, J.M.; Haggerty, C.L. Serum leptin measured in early pregnancy is higher in women with preeclampsia compared with normotensive pregnant women. Hypertension 2015, 65, 594–599. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Banach, M. Leptin, cardiovascular diseases and type 2 diabetes mellitus. Acta Pharm. Sin. 2018, 39, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Bersinger, N.A.; Smárason, A.K.; Muttukrishna, S.; Groome, N.P.; Redman, C.W. Women with preeclampsia have increased serum levels of pregnancy-associated plasma protein A (PAPP-A), inhibin A, activin A and soluble E-selectin. Hypertens Pregnancy 2003, 22, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Roldán, V.; Marín, F.; Lip, G.Y.; Blann, A.D. Soluble E-selectin in cardiovascular disease and its risk factors. A review of the literature. Thromb. Haemost. 2003, 90, 1007–1020. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbiny, W.; Nasr, A.; Soliman, A. Metalloprotease (ADAM12-S) as a predictor of preeclampsia: Correlation with severity, maternal complications, fetal outcome, and doppler parameters. Hypertens Pregnancy 2012, 31, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chow, F.L.; Oka, T.; Hao, L.; Lopez-Campistrous, A.; Kelly, S.; Cooper, S.; Odenbach, J.; Finegan, B.A.; Schulz, R.; et al. Matrix metalloproteinase-7 and ADAM-12 (a disintegrin and metalloproteinase-12) define a signaling axis in agonist-induced hypertension and cardiac hypertrophy. Circulation 2009, 119, 2480–2489. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Shixia, C.; Duan, T. First-trimester maternal serum levels of sflt1, pgf and adma predict preeclampsia. PLoS ONE 2015, 10, e0124684. [Google Scholar] [CrossRef]
- Sibal, L.; Agarwal, S.C.; Home, P.D.; Boger, R.H. The role of asymmetric dimethylarginine (ADMA) in endothelial dysfunction and cardiovascular disease. Curr. Cardiol. Rev. 2010, 6, 82–90. [Google Scholar] [CrossRef]
- Gagnon, A.; Wilson, R.D. Society of Obstetricians and Gynaecologists of Canada Genetics Committee. Obstetrical complications associated with abnormal maternal serum markers analytes. J. Obstet. Gynaecol. Can. 2008, 30, 918–932. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, C.; Zhou, X.; Li, L.; Hui, R. Pregnancy-associated plasma protein a predicts adverse vascular events in patients with coronary heart disease: A systematic review and meta-analysis. Arch. Med. Sci. 2013, 9, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Matson, B.C.; Caron, K.M. Adrenomedullin and endocrine control of immune cells during pregnancy. Cell Mol. Immunol. 2014, 11, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.A.; Baxter, G.F. Adrenomedullin: Regulator of systemic and cardiac homeostasis in acute myocardial infarction. Pharm. Ther. 2005, 105, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Vokalova, L.; Balogh, A.; Toth, E.; Van Breda, S.V.; Schäfer, G.; Hoesli, I.; Lapaire, O.; Hahn, S.; Than, N.G.; Rossi, S.W. Placental protein 13 (Galectin-13) polarizes neutrophils toward an immune regulatory phenotype. Front Immunol. 2020, 11, 145. [Google Scholar] [CrossRef]
- Berkane, N.; Liere, P.; Lefevre, G.; Alfaidy, N.; Nahed, R.A.; Vincent, J.; Oudinet, J.P.; Pianos, A.; Cambourg, A.; Rozenberg, P.; et al. Abnormal steroidogenesis and aromatase activity in preeclampsia. Placenta 2018, 69, 40–49. [Google Scholar] [CrossRef]
- Duvekot, J.J.; Cheriex, E.C.; Pieters, F.A.; Menheere, P.P.; Peeters, L.H. Early pregnancy changes in hemodynamics and volume homeostasis are consecutive adjustments triggered by a primary fall in systemic vascular tone. Am. J. Obstet. Gynecol. 1993, 169, 1382–1392. [Google Scholar] [CrossRef]
- Lanssens, D.; Smeets, C.J.P.; Vandervoort, P.; Grieten, L.; Gyselaers, W. Intrathoracic fluid changes from preconception to postpartum as measured by bio-impedance monitoring. J. Matern. Fetal Neonatal Med. 2020, 33, 1625–1627. [Google Scholar] [CrossRef]
- Tiralongo, G.M.; Lo Presti, D.; Pisani, I.; Gagliardi, G.; Scala, R.L.; Novelli, G.P.; Vasapollo, B.; Andreoli, A.; Valensise, H. Assessment of total vascular resistance and total body water in normotensive women during the first trimester of pregnancy. A key for the prevention of preeclampsia. Pregnancy Hypertens 2015, 5, 193–197. [Google Scholar] [CrossRef]
- de Haas, S.; Ghossein-Doha, C.; van Kuijk, S.M.; van Drongelen, J.; Spaanderman, M.E. Physiological adaptation of maternal plasma volume during pregnancy: A systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2017, 49, 177–187. [Google Scholar] [CrossRef]
- Kalafat, E.; Thilaganathan, B. Cardiovascular origins of preeclampsia. Curr. Opin. Obstet. Gynecol. 2017, 29, 383–389. [Google Scholar] [CrossRef]
- Gyselaers, W.; Vonck, S.; Staelens, A.S.; Lanssens, D.; Tomsin, K.; Oben, J.; Dreesen, P.; Bruckers, L. Body fluid volume homeostasis is abnormal in pregnancies complicated with hypertension and/or poor fetal growth. PLoS ONE 2018, 13, e0206257. [Google Scholar] [CrossRef] [PubMed]
- Tobias, A.; Mohiuddin, S.S. Physiology, water balance. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK541059/ (accessed on 30 May 2020).
- Melchiorre, K.; Sharma, R.; Khalil, A.; Thilaganathan, B. Maternal cardiovascular function in normal pregnancy: Evidence of maladaptation to chronic volume overload. Hypertension 2016, 67, 754–762. [Google Scholar] [CrossRef]
- Gyselaers, W.; Vonck, S.; Staelens, A.S.; Lanssens, D.; Tomsin, K.; Oben, J.; Dreesen, P.; Bruckers, L. Gestational hypertensive disorders show unique patterns of circulatory deterioration with ongoing pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R210–R221. [Google Scholar] [CrossRef] [PubMed]
- Vonck, S.; Staelens, A.S.; Lanssens, D.; Tomsin, K.; Oben, J.; Bruckers, L.; Gyselaers, W. Development of a biophysical screening model for gestational hypertensive diseases. J. Biomed. Sci. 2019, 26, 38. [Google Scholar] [CrossRef]
- Gyselaers, W.; Tomsin, K.; Staelens, A.; Mesens, T.; Oben, J.; Molenberghs, G. Maternal venous hemodynamics in gestational hypertension and preeclampsia. BMC Pregnancy Childbirth 2014, 14, 212. [Google Scholar] [CrossRef]
- Gyselaers, W.; Staelens, A.; Mesens, T.; Tomsin, K.; Oben, J.; Vonck, S.; Verresen, L.; Molenberghs, G. Maternal venous doppler characteristics are abnormal in pre-eclampsia but not in gestational hypertension. Ultrasound Obstet. Gynecol. 2015, 45, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Vonck, S.; Staelens, A.S.; Mesens, T.; Tomsin, K.; Gyselaers, W. Hepatic hemodynamics and fetal growth: A relationship of interest for further research. PLoS ONE 2014, 9, e115594. [Google Scholar] [CrossRef] [PubMed]
- Gyselaers, W.; Mesens, T.; Tomsin, K.; Molenberghs, G.; Peeters, L. Maternal renal interlobar vein impedance index is higher in early—Than in late-onset pre-eclampsia. Ultrasound Obstet. Gynecol. 2010, 36, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Gyselaers, W.; Mullens, W.; Tomsin, K.; Mesens, T.; Peeters, L. Role of dysfunctional maternal venous hemodynamics in the pathophysiology of pre-eclampsia: A review. Ultrasound Obstet. Gynecol. 2011, 38, 123–129. [Google Scholar] [CrossRef]
- Siegmund, A.S.; Kampman, M.A.M.; Oudijk, M.A.; Mulder, B.J.M.; Sieswerda, G.T.J.; Koenen, S.V.; Hummel, Y.M.; de Laat, M.W.M.; Sollie-Szarynska, K.M.; Groen, H.; et al. Maternal right ventricular function, uteroplacental circulation in first trimester and pregnancy outcome in women with congenital heart disease. Ultrasound Obstet. Gynecol. 2019, 54, 359–366. [Google Scholar] [CrossRef]
- Bokslag, A.; Franssen, C.; Alma, L.J.; Kovacevic, I.; Kesteren, F.V.; Teunissen, P.W.; Kamp, O.; Ganzevoort, W.; Hordijk, P.L.; Groot, C.J.M.; et al. Early-onset preeclampsia predisposes to preclinical diastolic left ventricular dysfunction in the fifth decade of life: An observational study. PLoS ONE 2018, 13, e0198908. [Google Scholar] [CrossRef] [PubMed]
- Gyselaers, W. Maternal venous hemodynamic dysfunction in proteinuric gestational hypertension: Evidence and implications. J. Clin. Med. 2019, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C. The two-stage placental model of preeclampsia: An update. J. Reprod. Immunol. 2019, 134–135, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Andreas, M.; Kuessel, L.; Kastl, S.P.; Wirth, S.; Gruber, K.; Rhomberg, F.; Gomari-Grisar, F.A.; Franz, M.; Zeisler, H.; Gottsauner-Wolf, M. Bioimpedance cardiography in pregnancy: A longitudinal cohort study on hemodynamic pattern and outcome. BMC Pregnancy Childbirth 2016, 16, 128. [Google Scholar] [CrossRef] [PubMed]
- Staelens, A.S.; Van Cauwelaert, S.; Tomsin, K.; Mesens, T.; Malbrain, M.L.; Gyselaers, W. Intra-abdominal pressure measurements in term pregnancy and postpartum: An observational study. PLoS ONE 2014, 9, e104782. [Google Scholar] [CrossRef]
- Lozada, M.J.; Goyal, V.; Levin, D.; Walden, R.L.; Osmundson, S.S.; Pacheco, L.D.; Malbrain, M.L.N.G. Management of peripartum intra-abdominal hypertension and abdominal compartment syndrome. Acta Obstet. Gynecol. Scand. 2019, 98, 1386–1397. [Google Scholar] [CrossRef]
- Malbrain, M.L.; Cheatham, M.L. Definitions and pathophysiological implications of intra-abdominal hypertension and abdominal compartment syndrome. Am. Surg. 2011, 77, S6–S11. [Google Scholar]
- Regli, A.; De Keulenaer, B.; De Laet, I.; Roberts, D.; Dabrowski, W.; Malbrain, M.L. Fluid therapy and perfusional considerations during resuscitation in critically ill patients with intra-abdominal hypertension. Anaesthesiol. Intensive Ther. 2015, 47, 45–53. [Google Scholar] [CrossRef]
- Dreesen, P.; Schoutteten, M.K.; Velde, N.V.; Kaminski, I.; Heylen, L.; Moor, B.; Malbrain, M.L.N.G.; Gyselaers, W. Increased intra-abdominal pressure during laparoscopic pneumoperitoneum enhances albuminuria via renal venous congestion, illustrating pathophysiological aspects of high output preeclampsia. J. Clin. Med. 2020, 9, 487. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hypertension (HT): | Systolic blood pressure ≥ 140 mm Hg and/or | |||||
| diastolic blood pressure ≥ 90 mm Hg | ||||||
| Chronic hypertension (CH): | hypertension before conception or | |||||
| diagnosed ≤ 20 weeks of gestation | ||||||
| GH (GH): | de novo hypertension after 20 weeks of gestation, without any signs of organ dysfunction | |||||
| PE (PE): | de novo HT ≥ 20 weeks of gestation, with ≥1 of following signs: | |||||
| ACOG 2019 | ISSHP 2018 | |||||
| HT 2 x ≥ 4 h apart | HT 2 x over few hours | |||||
| Proteinuria ≥ 300 mg/24 h | ||||||
| Protein/creatinine ≥ 30 mg/mmol | ||||||
| Dipstick ≥ 1+ | Dipstick ≥ 2+ (> 1g/L) | |||||
| Liver transaminases > 2 × normal | ||||||
| Platelets < 100 E9/L | Platelets < 150 E9/L | |||||
| Creatinine > 100 µmol/L | Creatinine ≥ 90 µmol/L | |||||
| Subjective signs | ||||||
| Uteroplacental dysfunction | ||||||
| ACOG | HT Canada | ESC | SOGC | ISSHP | SOMANZ | RCOG |
| 2019 | 2018 | 2018 | 2014 | 2018 | 2014 | 2011 |
| CH | CH | CH | CH ± Sympt | CH 1°/2° | CH 1°/2° | CH |
| GH | GH | GH | GH ± Sympt | GH | GH | GH |
| PE/Ecl | PE/Ecl/HELLP | PE | PE | PE, CH→PE | PE/Ecl | PE |
| CH→PE | CH→PE | Transient HT | Transient HT | CH→PE | ||
| UHT | Wh Coat HT | Wh Coat HT | Wh Coat | |||
| Masked HT | Masked HT | |||||
| HELLP | ||||||
| Ecl | ||||||
| Mild | Mild | |||||
| 140-159/ | 140-149/ | |||||
| 90-109 | 90-99 | |||||
| Moderate | ||||||
| 150-159/ | ||||||
| 100-109 | ||||||
| Severe | Severe | Severe | Severe | Severe | Severe | Severe |
| ≥160/110 | ≥160/110 | ≥160/110 | ≥160/110 | ≥160/110 | ≥160/110 | ≥160/100 |
| Emergent | ≥15 min | ≥15 min | over few h | |||
| ≥170/110 | ||||||
| Pathophysiologic Process | Figure | Predisposing Factors/Mechanisms | Clinics [references] | |
|---|---|---|---|---|
| Inflammation | Preimplantation fetal signaling | 2A | Genetic predisposition | Paternal preeclampsia [8,9] |
| Maternal response | 2B | Preconceptional paternal antigen immune memory | Primipaternity [6,9] | |
| Local adaptation | Local maternal immune tolerance | 2B | Maternal immunologic precondition | Auto-immune disease [13,14] |
| Local inflammation | 2B | Maternal immunologic precondition | Auto-immune disease [13,14], immune suppression [40] | |
| Local vascular adaptation | 2B | Maternal cardiovascular/endothelial precondition | CV disease [13,14], endoth dysfunction [18] | |
| Systemic adaptation | Systemic maternal immune tolerance | 2C | Maternal immunologic precondition | Auto-immune disease [13,14] |
| Systemic cardiovascular adaptation | 2C | Maternal cardiovascular/endothelial precondition | CV disease [13,14], endoth dysfunction [18] | |
| Early oxidative stress | Local tissue damage & inflammation | 4 | Maternal (immunologic) precondition | Auto-immune disease [13,14] |
| Intercellular mediators, chemokines | 4 | Genetic/immunologic precondition | Genetic diseases [7.8.10.11], auto-immune diseases [13,14] | |
| IVS hemodynamics | Veno-lymphatic remodelling & Arterial plugging | 2C | Venous/ body volume dysfunction | (Subclinical) CV disease [41], body constitution [42,43] |
| 2C | Maternal immunologic dysfunction | Auto-immune disease [13,14] | ||
| Spiral artery remodelling | 1 | Maternal arterial/endothelial dysfunction | CV disease [13,14], endoth dysfunction [18] | |
| 1 | Arterial response to poor venous adaptation | CV disease [13,14], endoth dysfunction [18] | ||
| Systemic Hemodynamics | Body water volume | 3 | Preconceptional low/high body water volume | Maternal body constitution [83.93], renal disease [13.14] |
| Body water expansion upon early CV dysfunction | 3 | Extravasation/poor intravascular expansion | CV disease [13,14], renal disease [13.14], endoth dysfunction [18] | |
| Volume induced endoth dysfunction | 3 | Maternal constitutional/CV/endoth precondition | CV disease [13,14], renal disease [13.14], endoth dysfunction [18] | |
| Raised venous tone/venous hypertension | 4 | Maternal precondition | Aut NS [44], endoth dysfunction [18] | |
| External venous compression | 4 | Preconceptional raised intra-abdominal pressure | Maternal body constitution [83.93], renal disease [13.14] | |
| 4 | Increased gestational rise of intra-abdominal pressure | Large uterine volume [13,14] | ||
| Late oxidative stress | Intercellular mediators, chemokines | 4 | Genetic/immunologic precondition | Genetic [7.8.10.11], maternal disease [13.14], endoth [18], Aut NS [44] |
| Systemic inflammation | 4 | Genetic/immunologic precondition | Genetic [7.8.10.11], maternal disease [13.14], endoth [18], Aut NS [44] | |
| 4 | Developing during pregnancy | Genetic [7.8.10.11], maternal disease [13.14], endoth [18], Aut NS [44] |
| Physiologic Effects | PE | Non Pregnant | Type CVD | Ref | |
|---|---|---|---|---|---|
| CRP | Immunomodulation | ↑ | [82] | ||
| ↑ | CHD, HF | [83] | |||
| VEGF | Pro-angiogenic | ↓ | [84] | ||
| Pro-vasculogenic | Polymorphisms | CHD | [85] | ||
| sFlt-1 | Anti-angiogenic | ↑ | [84] | ||
| ↑ | CHD, HF | [86] | |||
| sEng | Anti-angiogenic | ↑ | [84] | ||
| ↑ | CHD | [87] | |||
| Gal-3BP | Immunomodulation | ↑ | [88] | ||
| ↑ | CHD | [89] | |||
| Activin A | Immunomodulation | ↑ | [90] | ||
| Apoptosis | ↑ | HT, CHD | [91] | ||
| Leptin | Immunomodulation | ↑ | [92] | ||
| angiogenetic | ↑ | CHD | [93] | ||
| sE-selectin | Immunomodulation | ↑ | [94] | ||
| ↑ | HT | [95] | |||
| ADAM 12 | angiogenetic | ↑ | [96] | ||
| immunomodulation | ↑ | HT | [97] | ||
| ADMA | vasodilatation | ↑ | [98] | ||
| ↑ | CHD, HF, HT | [99] | |||
| PLGF | Pro-angiogenic | ↓ | [84] | ||
| ↑ | CHD, HF | [86] | |||
| PAPP-A | Proteolysis IGF-BP | ↓ → ↑ | [94,100] | ||
| ↑ | CHD | [101] | |||
| ADM | Pro-angiogenic | ↓ | [102] | ||
| Vasodilatation | ↑ | AMI | [103] | ||
| PP13 | Immunomodulation | ↓ | [104] | ||
| Inhibin A | Undetermined in pregnancy | ↑ | [90] | ||
| E3 | Undetermined in pregnancy | ↓ | [105] | ||
| AFP | Undetermined in pregnancy | ↑ | [100] | ||
| HbF | A1M Immunomodulation | ↑ | [59] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gyselaers, W. Preeclampsia Is a Syndrome with a Cascade of Pathophysiologic Events. J. Clin. Med. 2020, 9, 2245. https://doi.org/10.3390/jcm9072245
Gyselaers W. Preeclampsia Is a Syndrome with a Cascade of Pathophysiologic Events. Journal of Clinical Medicine. 2020; 9(7):2245. https://doi.org/10.3390/jcm9072245
Chicago/Turabian StyleGyselaers, Wilfried. 2020. "Preeclampsia Is a Syndrome with a Cascade of Pathophysiologic Events" Journal of Clinical Medicine 9, no. 7: 2245. https://doi.org/10.3390/jcm9072245
APA StyleGyselaers, W. (2020). Preeclampsia Is a Syndrome with a Cascade of Pathophysiologic Events. Journal of Clinical Medicine, 9(7), 2245. https://doi.org/10.3390/jcm9072245